NF2/Merlin Inactivation and Potential Therapeutic Targets in Mesothelioma

Abstract
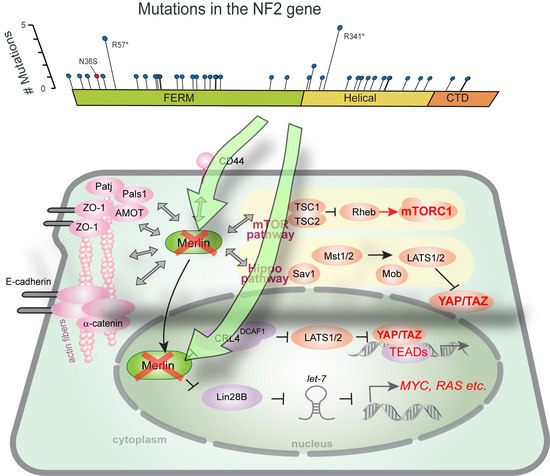
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Domain Organization and Functions of Merlin
2.1. NF2 Transcript Variants
2.2. Domain Organization
2.3. Molecular Conformation and Phosphorylation
2.4. NF2 Inactivation in Mesothelioma
2.5. Loss of Contact Inhibition in NF2-Deficient Cells
2.6. Subcellular Localization
3. Proteins and Signaling Related to Merlin’s Functions
3.1. Hippo Signaling Pathway
3.2. DCAF1
3.3. PI3K/AKT/mTOR Signaling Pathway
3.4. Lin28B and let-7 miRNAs
3.5. TRAF7
4. Potential Molecular Targets in Merlin-Negative Mesothelioma
4.1. FAK Inhibitors
4.2. YAP Inhibitors
4.3. mTOR Inhibitors
4.4. Statins
4.5. COX2 Inhibitors
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AMOT | angiomotin |
| BAP1 | BRCA1 Associated Protein 1 |
| CCND2 | cyclin D2 |
| CDK | cyclin-dependent kinase |
| COX-2 | cyclooxygenase 2 |
| CPI-17 | protein kinase C-potentiated inhibitor protein of 17 kDa |
| CRL4 | Cullin-RING ubiquitin ligase 4 |
| CTD | C-terminal domain |
| CUL4 | Cullin 4 |
| DCAF1 | DDB1- and CUL4-associated factor 1 |
| DDB1 | damaged DNA binding protein 1 |
| ERM | ezrin/radixin/moesin |
| FAK | focal adhesion kinase |
| FDA | Food and Drug Administration |
| FERM | band 4.1/ezrin/radixin/moesin |
| FLIP | caspase-like apoptosis regulatory protein |
| FOXM1 | forkhead box M1 |
| LATS1/2 | large tumor suppressor kinase 1/2 |
| Lin28B | lin-28 homolog B |
| MAPK | mitogen-activated protein kinase |
| mLST8 | mammalian lethal with Sec13 protein 8 |
| MM | malignant mesothelioma |
| Mob | Mps one binder kinase activator-like protein |
| mTOR | mechanistic target of rapamycin |
| mTORC | mTOR complex |
| MST1/2 | mammalian Ste20-like protein kinase 1/2 |
| MYPT1-PP1δ | myosin phosphatase target subunit 1-protein phosphatase 1 δ |
| NAE | NEDD8-activating enzyme |
| NF2 | neurofibromatosis type 2 |
| PAK | p21-activated kinase |
| Pals1 | protein associated with Lin7-1 |
| Patj | Pals1-associated tight junction protein |
| PI3K | phosphatidylinositol 3-kinase |
| PKA | protein kinase A |
| PLCB4 | phospholipase C beta 4 |
| PTEN | phosphatase and tensin homolog |
| Rheb | Ras homolog enriched in brain |
| ROS | reactive oxygen species |
| SAV1 | Salvador Family WW Domain Containing Protein 1 |
| siRNA | small interfering RNA |
| TAZ | WW domain-containing transcription regulator 1 |
| TEAD | TEA domain transcription factor |
| TNF | tumor necrosis factor |
| TRAF | TNF receptor-associated factor |
| TRAIL | TNF-related apoptosis inducing ligand |
| TSC1/2 | tuberous sclerosis complex subunit 1/2 |
| VprBP | viral protein R (VPR)-binding protein |
| YAP | Yes-associated protein 1 |
| ZO-1 | Zonula occludens-1 |
References
- Trofatter, J.A.; MacCollin, M.M.; Rutter, J.L.; Murrell, J.R.; Duyao, M.P.; Parry, D.M.; Eldridge, R.; Kley, N.; Menon, A.G.; Pulaski, K.; et al. A novel moesin-, ezrin-, radixin-like gene is a candidate for the neurofibromatosis 2 tumor suppressor. Cell 1993, 72, 791–800. [Google Scholar] [CrossRef]
- Altomare, D.A.; Vaslet, C.A.; Skele, K.L.; De Rienzo, A.; Devarajan, K.; Jhanwar, S.C.; McClatchey, A.I.; Kane, A.B.; Testa, J.R. A mouse model recapitulating molecular features of human mesothelioma. Cancer Res. 2005, 65, 8090–8095. [Google Scholar] [CrossRef] [PubMed]
- Stemmer-Rachamimov, A.O.; Xu, L.; Gonzalez-Agosti, C.; Burwick, J.A.; Pinney, D.; Beauchamp, R.; Jacoby, L.B.; Gusella, J.F.; Ramesh, V.; Louis, D.N. Universal absence of merlin, but not other ERM family members, in schwannomas. Am. J. Pathol. 1997, 151, 1649–1654. [Google Scholar] [PubMed]
- Ruttledge, M.H.; Sarrazin, J.; Rangaratnam, S.; Phelan, C.M.; Twist, E.; Merel, P.; Delattre, O.; Thomas, G.; Nordenskjold, M.; Collins, V.P.; et al. Evidence for the complete inactivation of the NF2 gene in the majority of sporadic meningiomas. Nat. Genet. 1994, 6, 180–184. [Google Scholar] [CrossRef] [PubMed]
- Sekido, Y.; Pass, H.I.; Bader, S.; Mew, D.J.; Christman, M.F.; Gazdar, A.F.; Minna, J.D. Neurofibromatosis type 2 (NF2) gene is somatically mutated in mesothelioma but not in lung cancer. Cancer Res. 1995, 55, 1227–1231. [Google Scholar] [PubMed]
- Bianchi, A.B.; Mitsunaga, S.I.; Cheng, J.Q.; Klein, W.M.; Jhanwar, S.C.; Seizinger, B.; Kley, N.; Klein-Szanto, A.J.; Testa, J.R. High frequency of inactivating mutations in the neurofibromatosis type 2 gene (NF2) in primary malignant mesotheliomas. Proc. Natl. Acad. Sci. USA 1995, 92, 10854–10858. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Cooper, J.; Karajannis, M.A.; Giancotti, F.G. Merlin: A tumour suppressor with functions at the cell cortex and in the nucleus. EMBO Rep. 2012, 13, 204–215. [Google Scholar] [CrossRef] [PubMed]
- Petrilli, A.M.; Fernandez-Valle, C. Role of Merlin/NF2 inactivation in tumor biology. Oncogene 2016, 35, 537–548. [Google Scholar] [CrossRef] [PubMed]
- McClatchey, A.I.; Saotome, I.; Ramesh, V.; Gusella, J.F.; Jacks, T. The NF2 tumor suppressor gene product is essential for extraembryonic development immediately prior to gastrulation. Genes Dev. 1997, 11, 1253–1265. [Google Scholar] [CrossRef] [PubMed]
- Giovannini, M.; Robanus-Maandag, E.; van der Valk, M.; Niwa-Kawakita, M.; Abramowski, V.; Goutebroze, L.; Woodruff, J.M.; Berns, A.; Thomas, G. Conditional biallelic NF2 mutation in the mouse promotes manifestations of human neurofibromatosis type 2. Genes Dev. 2000, 14, 1617–1630. [Google Scholar] [PubMed]
- McClatchey, A.I.; Saotome, I.; Mercer, K.; Crowley, D.; Gusella, J.F.; Bronson, R.T.; Jacks, T. Mice heterozygous for a mutation at the NF2 tumor suppressor locus develop a range of highly metastatic tumors. Genes Dev. 1998, 12, 1121–1133. [Google Scholar] [CrossRef] [PubMed]
- Thurneysen, C.; Opitz, I.; Kurtz, S.; Weder, W.; Stahel, R.A.; Felley-Bosco, E. Functional inactivation of NF2/merlin in human mesothelioma. Lung Cancer 2009, 64, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Jongsma, J.; van Montfort, E.; Vooijs, M.; Zevenhoven, J.; Krimpenfort, P.; van der Valk, M.; van de Vijver, M.; Berns, A. A conditional mouse model for malignant mesothelioma. Cancer Cell 2008, 13, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Poulikakos, P.I.; Xiao, G.H.; Gallagher, R.; Jablonski, S.; Jhanwar, S.C.; Testa, J.R. Re-expression of the tumor suppressor NF2/merlin inhibits invasiveness in mesothelioma cells and negatively regulates FAK. Oncogene 2006, 25, 5960–5968. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Sato, T.; Yokoi, K.; Sekido, Y. E-cadherin expression is correlated with focal adhesion kinase inhibitor resistance in Merlin-negative malignant mesothelioma cells. Oncogene 2017, 36, 5522–5531. [Google Scholar] [CrossRef] [PubMed]
- Xiao, G.H.; Gallagher, R.; Shetler, J.; Skele, K.; Altomare, D.A.; Pestell, R.G.; Jhanwar, S.; Testa, J.R. The NF2 tumor suppressor gene product, merlin, inhibits cell proliferation and cell cycle progression by repressing cyclin D1 expression. Mol. Cell. Biol. 2005, 25, 2384–2394. [Google Scholar] [CrossRef] [PubMed]
- Rouleau, G.A.; Merel, P.; Lutchman, M.; Sanson, M.; Zucman, J.; Marineau, C.; Hoang-Xuan, K.; Demczuk, S.; Desmaze, C.; Plougastel, B.; et al. Alteration in a new gene encoding a putative membrane-organizing protein causes neuro-fibromatosis type 2. Nature 1993, 363, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.S.; Akhmametyeva, E.M.; Wu, Y.; Zhu, L.; Welling, D.B. Multiple transcription initiation sites, alternative splicing, and differential polyadenylation contribute to the complexity of human neurofibromatosis 2 transcripts. Genomics 2002, 79, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Gutmann, D.H.; Sherman, L.; Seftor, L.; Haipek, C.; Hoang Lu, K.; Hendrix, M. Increased expression of the NF2 tumor suppressor gene product, merlin, impairs cell motility, adhesionand spreading. Hum. Mol. Genet. 1999, 8, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Sherman, L.; Xu, H.M.; Geist, R.T.; Saporito-Irwin, S.; Howells, N.; Ponta, H.; Herrlich, P.; Gutmann, D.H. Interdomain binding mediates tumor growth suppression by the NF2 gene product. Oncogene 1997, 15, 2505–2509. [Google Scholar] [CrossRef] [PubMed]
- Lallemand, D.; Saint-Amaux, A.L.; Giovannini, M. Tumor-suppression functions of merlin are independent of its role as an organizer of the actin cytoskeleton in Schwann cells. J. Cell Sci. 2009, 122, 4141–4149. [Google Scholar] [CrossRef] [PubMed]
- Zoch, A.; Mayerl, S.; Schulz, A.; Greither, T.; Frappart, L.; Rubsam, J.; Heuer, H.; Giovannini, M.; Morrison, H. Merlin Isoforms 1 and 2 Both Act as Tumour Suppressors and Are Required for Optimal Sperm Maturation. PLoS ONE 2015, 10, e0129151. [Google Scholar] [CrossRef] [PubMed]
- Sher, I.; Hanemann, C.O.; Karplus, P.A.; Bretscher, A. The tumor suppressor merlin controls growth in its open state, and phosphorylation converts it to a less-active more-closed state. Dev. Cell 2012, 22, 703–705. [Google Scholar] [CrossRef] [PubMed]
- Bueno, R.; Stawiski, E.W.; Goldstein, L.D.; Durinck, S.; De Rienzo, A.; Modrusan, Z.; Gnad, F.; Nguyen, T.T.; Jaiswal, B.S.; Chirieac, L.R.; et al. Comprehensive genomic analysis of malignant pleural mesothelioma identifies recurrent mutations, gene fusions and splicing alterations. Nat. Genet. 2016, 48, 407–416. [Google Scholar] [CrossRef] [PubMed]
- McClatchey, A.I.; Giovannini, M. Membrane organization and tumorigenesis—The NF2 tumor suppressor, Merlin. Genes Dev. 2005, 19, 2265–2277. [Google Scholar] [CrossRef] [PubMed]
- Morrison, H.; Sherman, L.S.; Legg, J.; Banine, F.; Isacke, C.; Haipek, C.A.; Gutmann, D.H.; Ponta, H.; Herrlich, P. The NF2 tumor suppressor gene product, merlin, mediates contact inhibition of growth through interactions with CD44. Genes Dev. 2001, 15, 968–980. [Google Scholar] [CrossRef] [PubMed]
- Sainio, M.; Zhao, F.; Heiska, L.; Turunen, O.; den Bakker, M.; Zwarthoff, E.; Lutchman, M.; Rouleau, G.A.; Jaaskelainen, J.; Vaheri, A.; et al. Neurofibromatosis 2 tumor suppressor protein colocalizes with ezrin and CD44 and associates with actin-containing cytoskeleton. J. Cell Sci. 1997, 110 Pt 18, 2249–2260. [Google Scholar] [PubMed]
- Murthy, A.; Gonzalez-Agosti, C.; Cordero, E.; Pinney, D.; Candia, C.; Solomon, F.; Gusella, J.; Ramesh, V. NHE-RF, a regulatory cofactor for Na+-H+ exchange, is a common interactor for merlin and ERM (MERM) proteins. J. Biol. Chem. 1998, 273, 1273–1276. [Google Scholar] [CrossRef] [PubMed]
- Reczek, D.; Berryman, M.; Bretscher, A. Identification of EBP50: A PDZ-containing phosphoprotein that associates with members of the ezrin-radixin-moesin family. J. Cell Biol. 1997, 139, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Lallemand, D.; Curto, M.; Saotome, I.; Giovannini, M.; McClatchey, A.I. NF2 deficiency promotes tumorigenesis and metastasis by destabilizing adherens junctions. Genes Dev. 2003, 17, 1090–1100. [Google Scholar] [CrossRef] [PubMed]
- Fehon, R.G.; McClatchey, A.I.; Bretscher, A. Organizing the cell cortex: The role of ERM proteins. Nat. Rev. Mol. Cell Biol. 2010, 11, 276–287. [Google Scholar] [CrossRef] [PubMed]
- Mani, T.; Hennigan, R.F.; Foster, L.A.; Conrady, D.G.; Herr, A.B.; Ip, W. FERM domain phosphoinositide binding targets merlin to the membrane and is essential for its growth-suppressive function. Mol. Cell. Biol. 2011, 31, 1983–1996. [Google Scholar] [CrossRef] [PubMed]
- Hamada, K.; Shimizu, T.; Matsui, T.; Tsukita, S.; Hakoshima, T. Structural basis of the membrane-targeting and unmasking mechanisms of the radixin FERM domain. EMBO J. 2000, 19, 4449–4462. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Seto, A.; Maita, N.; Hamada, K.; Tsukita, S.; Tsukita, S.; Hakoshima, T. Structural basis for neurofibromatosis type 2. Crystal structure of the merlin FERM domain. J. Biol. Chem. 2002, 277, 10332–10336. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.M.; Gutmann, D.H. Merlin differentially associates with the microtubule and actin cytoskeleton. J. Neurosci. Res. 1998, 51, 403–415. [Google Scholar] [CrossRef]
- LaJeunesse, D.R.; McCartney, B.M.; Fehon, R.G. Structural analysis of Drosophila merlin reveals functional domains important for growth control and subcellular localization. J. Cell Biol. 1998, 141, 1589–1599. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.C.; Kissil, J.L.; Fry, J.L.; Jacks, T. Cellular transformation by a FERM domain mutant of the NF2 tumor suppressor gene. Oncogene 2002, 21, 5990–5997. [Google Scholar] [CrossRef] [PubMed]
- Surace, E.I.; Haipek, C.A.; Gutmann, D.H. Effect of merlin phosphorylation on neurofibromatosis 2 (NF2) gene function. Oncogene 2004, 23, 580–587. [Google Scholar] [CrossRef] [PubMed]
- Rong, R.; Surace, E.I.; Haipek, C.A.; Gutmann, D.H.; Ye, K. Serine 518 phosphorylation modulates merlin intramolecular association and binding to critical effectors important for NF2 growth suppression. Oncogene 2004, 23, 8447–8454. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Sperka, T.; Herrlich, P.; Morrison, H. Tumorigenic transformation by CPI-17 through inhibition of a merlin phosphatase. Nature 2006, 442, 576–579. [Google Scholar] [CrossRef] [PubMed]
- Shaw, R.J.; Paez, J.G.; Curto, M.; Yaktine, A.; Pruitt, W.M.; Saotome, I.; O’Bryan, J.P.; Gupta, V.; Ratner, N.; Der, C.J.; et al. The NF2 tumor suppressor, merlin, functions in Rac-dependent signaling. Dev. Cell 2001, 1, 63–72. [Google Scholar] [CrossRef]
- Kissil, J.L.; Wilker, E.W.; Johnson, K.C.; Eckman, M.S.; Yaffe, M.B.; Jacks, T. Merlin, the product of the NF2 tumor suppressor gene, is an inhibitor of the p21-activated kinase, Pak1. Mol. Cell 2003, 12, 841–849. [Google Scholar] [CrossRef]
- Xiao, G.H.; Beeser, A.; Chernoff, J.; Testa, J.R. p21-activated kinase links Rac/Cdc42 signaling to merlin. J. Biol. Chem. 2002, 277, 883–886. [Google Scholar] [CrossRef] [PubMed]
- Alfthan, K.; Heiska, L.; Gronholm, M.; Renkema, G.H.; Carpen, O. Cyclic AMP-dependent protein kinase phosphorylates merlin at serine 518 independently of p21-activated kinase and promotes merlin-ezrin heterodimerization. J. Biol. Chem. 2004, 279, 18559–18566. [Google Scholar] [CrossRef] [PubMed]
- Okada, T.; López-Lago, M.; Giancotti, F.G. Merlin/NF-2 mediates contact inhibition of growth by suppressing recruitment of Rac to the plasma membrane. J. Cell Biol. 2005, 171, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Laulajainen, M.; Muranen, T.; Carpen, O.; Gronholm, M. Protein kinase A-mediated phosphorylation of the NF2 tumor suppressor protein merlin at serine 10 affects the actin cytoskeleton. Oncogene 2008, 27, 3233–3243. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Jang, S.W.; Wang, X.; Liu, Z.; Bahr, S.M.; Sun, S.Y.; Brat, D.; Gutmann, D.H.; Ye, K. AKT phosphorylation regulates the tumour-suppressor merlin through ubiquitination and degradation. Nat. Cell Biol. 2007, 9, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Yi, C.; Troutman, S.; Fera, D.; Stemmer-Rachamimov, A.; Avila, J.L.; Christian, N.; Persson, N.L.; Shimono, A.; Speicher, D.W.; Marmorstein, R.; et al. A tight junction-associated Merlin-angiomotin complex mediates Merlin’s regulation of mitogenic signaling and tumor suppressive functions. Cancer Cell 2011, 19, 527–540. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, R.; Reczek, D.; Bretscher, A. Hierarchy of merlin and ezrin N- and C-terminal domain interactions in homo- and heterotypic associations and their relationship to binding of scaffolding proteins EBP50 and E3KARP. J. Biol. Chem. 2001, 276, 7621–7629. [Google Scholar] [CrossRef] [PubMed]
- Hennigan, R.F.; Foster, L.A.; Chaiken, M.F.; Mani, T.; Gomes, M.M.; Herr, A.B.; Ip, W. Fluorescence resonance energy transfer analysis of merlin conformational changes. Mol. Cell. Biol. 2010, 30, 54–67. [Google Scholar] [CrossRef] [PubMed]
- Gutmann, D.H.; Hirbe, A.C.; Haipek, C.A. Functional analysis of neurofibromatosis 2 (NF2) missense mutations. Hum. Mol. Genet. 2001, 10, 1519–1529. [Google Scholar] [CrossRef] [PubMed]
- Singhi, A.D.; Krasinskas, A.M.; Choudry, H.A.; Bartlett, D.L.; Pingpank, J.F.; Zeh, H.J.; Luvison, A.; Fuhrer, K.; Bahary, N.; Seethala, R.R.; et al. The prognostic significance of BAP1, NF2, and CDKN2A in malignant peritoneal mesothelioma. Mod. Pathol. 2016, 29, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Bosco, E.E.; Nakai, Y.; Hennigan, R.F.; Ratner, N.; Zheng, Y. NF2-deficient cells depend on the Rac1-canonical Wnt signaling pathway to promote the loss of contact inhibition of proliferation. Oncogene 2010, 29, 2540–2549. [Google Scholar] [CrossRef] [PubMed]
- Gladden, A.B.; Hebert, A.M.; Schneeberger, E.E.; McClatchey, A.I. The NF2 tumor suppressor, Merlin, regulates epidermal development through the establishment of a junctional polarity complex. Dev. Cell 2010, 19, 727–739. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Agosti, C.; Xu, L.; Pinney, D.; Beauchamp, R.; Hobbs, W.; Gusella, J.; Ramesh, V. The merlin tumor suppressor localizes preferentially in membrane ruffles. Oncogene 1996, 13, 1239–1247. [Google Scholar] [PubMed]
- Scherer, S.S.; Gutmann, D.H. Expression of the neurofibromatosis 2 tumor suppressor gene product, merlin, in Schwann cells. J. Neurosci. Res. 1996, 46, 595–605. [Google Scholar] [CrossRef]
- Yokoyama, T.; Osada, H.; Murakami, H.; Tatematsu, Y.; Taniguchi, T.; Kondo, Y.; Yatabe, Y.; Hasegawa, Y.; Shimokata, K.; Horio, Y.; et al. YAP1 is involved in mesothelioma development and negatively regulated by Merlin through phosphorylation. Carcinogenesis 2008, 29, 2139–2146. [Google Scholar] [CrossRef] [PubMed]
- Harvey, K.F.; Zhang, X.; Thomas, D.M. The Hippo pathway and human cancer. Nat. Rev. Cancer 2013, 13, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP/TAZ at the Roots of Cancer. Cancer Cell 2016, 29, 783–803. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, I.; Osada, H.; Fujii, M.; Fukatsu, A.; Hida, T.; Horio, Y.; Kondo, Y.; Sato, A.; Hasegawa, Y.; Tsujimura, T.; et al. LIM-domain protein AJUBA suppresses malignant mesothelioma cell proliferation via Hippo signaling cascade. Oncogene 2015, 34, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Murakami, H.; Mizuno, T.; Taniguchi, T.; Fujii, M.; Ishiguro, F.; Fukui, T.; Akatsuka, S.; Horio, Y.; Hida, T.; Kondo, Y.; et al. LATS2 is a tumor suppressor gene of malignant mesothelioma. Cancer Res. 2011, 71, 873–883. [Google Scholar] [CrossRef] [PubMed]
- Kakiuchi, T.; Takahara, T.; Kasugai, Y.; Arita, K.; Yoshida, N.; Karube, K.; Suguro, M.; Matsuo, K.; Nakanishi, H.; Kiyono, T.; et al. Modeling mesothelioma utilizing human mesothelial cells reveals involvement of phospholipase-C β4 in YAP-active mesothelioma cell proliferation. Carcinogenesis 2016, 37, 1098–1109. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, T.; Murakami, H.; Fujii, M.; Ishiguro, F.; Tanaka, I.; Kondo, Y.; Akatsuka, S.; Toyokuni, S.; Yokoi, K.; Osada, H.; et al. YAP induces malignant mesothelioma cell proliferation by upregulating transcription of cell cycle-promoting genes. Oncogene 2012, 31, 5117–5122. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; You, L.; Cooper, J.; Schiavon, G.; Pepe-Caprio, A.; Zhou, L.; Ishii, R.; Giovannini, M.; Hanemann, C.O.; Long, S.B.; et al. Merlin/NF2 suppresses tumorigenesis by inhibiting the E3 ubiquitin ligase CRL4(DCAF1) in the nucleus. Cell 2010, 140, 477–490. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Cooper, J.; Zhou, L.; Yang, C.; Erdjument-Bromage, H.; Zagzag, D.; Snuderl, M.; Ladanyi, M.; Hanemann, C.O.; Zhou, P.; et al. Merlin/NF2 loss-driven tumorigenesis linked to CRL4(DCAF1)-mediated inhibition of the Hippo pathway kinases LATS1 and 2 in the nucleus. Cancer Cell 2014, 26, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.; Xu, Q.; Zhou, L.; Pavlovic, M.; Ojeda, V.; Moulick, K.; de Stanchina, E.; Poirier, J.T.; Zauderer, M.; Rudin, C.M.; et al. Combined Inhibition of NEDD8-Activating Enzyme and mTOR Suppresses NF2 Loss-Driven Tumorigenesis. Mol. Cancer Ther. 2017, 16, 1693–1704. [Google Scholar] [CrossRef] [PubMed]
- Hikasa, H.; Sekido, Y.; Suzuki, A. Merlin/NF2-Lin28B-let-7 Is a Tumor-Suppressive Pathway that Is Cell-Density Dependent and Hippo Independent. Cell Rep. 2016, 14, 2950–2961. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed]
- Albert, V.; Hall, M.N. mTOR signaling in cellular and organismal energetics. Curr. Opin. Cell Biol. 2015, 33, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Rudge, D.G.; Koos, J.D.; Vaidialingam, B.; Yang, H.J.; Pavletich, N.P. mTOR kinase structure, mechanism and regulation. Nature 2013, 497, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Akasu, H.; Shimono, W.; Matsu, C.; Fujiwara, Y.; Shibagaki, Y.; Heard, J.J.; Tamanoi, F.; Hattori, S. Rheb protein binds CAD (carbamoyl-phosphate synthetase 2, aspartate transcarbamoylase, and dihydroorotase) protein in a GTP- and effector domain-dependent manner and influences its cellular localization and carbamoyl-phosphate synthetase (CPSase) activity. J. Biol. Chem. 2015, 290, 1096–1105. [Google Scholar] [CrossRef] [PubMed]
- Robitaille, A.M.; Christen, S.; Shimobayashi, M.; Cornu, M.; Fava, L.L.; Moes, S.; Prescianotto-Baschong, C.; Sauer, U.; Jenoe, P.; Hall, M.N. Quantitative phosphoproteomics reveal mTORC1 activates de novo pyrimidine synthesis. Science 2013, 339, 1320–1323. [Google Scholar] [CrossRef] [PubMed]
- Ben-Sahra, I.; Howell, J.J.; Asara, J.M.; Manning, B.D. Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science 2013, 339, 1323–1328. [Google Scholar] [CrossRef] [PubMed]
- López -Lago, M.A.; Okada, T.; Murillo, M.M.; Socci, N.; Giancotti, F.G. Loss of the tumor suppressor gene NF2, encoding merlin, constitutively activates integrin-dependent mTORC1 signaling. Mol. Cell. Biol. 2009, 29, 4235–4249. [Google Scholar] [CrossRef] [PubMed]
- James, M.F.; Han, S.; Polizzano, C.; Plotkin, S.R.; Manning, B.D.; Stemmer-Rachamimov, A.O.; Gusella, J.F.; Ramesh, V. NF2/merlin is a novel negative regulator of mTOR complex 1, and activation of mTORC1 is associated with meningioma and schwannoma growth. Mol. Cell. Biol. 2009, 29, 4250–4261. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Chirieac, L.R.; Bueno, R.; Pass, H.; Wu, W.; Malinowska, I.A.; Kwiatkowski, D.J. Tsc1-Tp53 loss induces mesothelioma in mice, and evidence for this mechanism in human mesothelioma. Oncogene 2014, 33, 3151–3160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altomare, D.A.; You, H.; Xiao, G.H.; Ramos-Nino, M.E.; Skele, K.L.; De Rienzo, A.; Jhanwar, S.C.; Mossman, B.T.; Kane, A.B.; Testa, J.R. Human and mouse mesotheliomas exhibit elevated AKT/PKB activity, which can be targeted pharmacologically to inhibit tumor cell growth. Oncogene 2005, 24, 6080–6089. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Murakami, H.; Kawaguchi, K.; Tanigushi, T.; Fujii, M.; Shinjo, K.; Kondo, Y.; Osada, H.; Shimokata, K.; Horio, Y.; et al. Activation of the PI3K-AKT pathway in human malignant mesothelioma cells. Mol. Med. Rep. 2009, 2, 181–188. [Google Scholar] [PubMed]
- Sato, T.; Nakashima, A.; Guo, L.; Coffman, K.; Tamanoi, F. Single amino-acid changes that confer constitutive activation of mTOR are discovered in human cancer. Oncogene 2010, 29, 2746–2752. [Google Scholar] [CrossRef] [PubMed]
- Grabiner, B.C.; Nardi, V.; Birsoy, K.; Possemato, R.; Shen, K.; Sinha, S.; Jordan, A.; Beck, A.H.; Sabatini, D.M. A diverse array of cancer-associated MTOR mutations are hyperactivating and can predict rapamycin sensitivity. Cancer Discov. 2014, 4, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Rehfeld, F.; Rohde, A.M.; Nguyen, D.T.; Wulczyn, F.G. Lin28 and let-7: Ancient milestones on the road from pluripotency to neurogenesis. Cell Tissue Res. 2015, 359, 145–160. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Ng, S.B.; Chng, W.J. LIN28/LIN28B: An emerging oncogenic driver in cancer stem cells. Int. J. Biochem. Cell Biol. 2013, 45, 973–978. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.M.; Grosshans, H.; Shingara, J.; Byrom, M.; Jarvis, R.; Cheng, A.; Labourier, E.; Reinert, K.L.; Brown, D.; Slack, F.J. RAS is regulated by the let-7 microRNA family. Cell 2005, 120, 635–647. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.S.; Lu, J.; Mercer, K.L.; Golub, T.R.; Jacks, T. Impaired microRNA processing enhances cellular transformation and tumorigenesis. Nat. Genet. 2007, 39, 673–677. [Google Scholar] [CrossRef] [PubMed]
- Bouwmeester, T.; Bauch, A.; Ruffner, H.; Angrand, P.O.; Bergamini, G.; Croughton, K.; Cruciat, C.; Eberhard, D.; Gagneur, J.; Ghidelli, S.; et al. A physical and functional map of the human TNF-α/NF-κB signal transduction pathway. Nat. Cell Biol. 2004, 6, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Scudiero, I.; Zotti, T.; Ferravante, A.; Vessichelli, M.; Reale, C.; Masone, M.C.; Leonardi, A.; Vito, P.; Stilo, R. Tumor necrosis factor (TNF) receptor-associated factor 7 is required for TNFα-induced Jun NH2-terminal kinase activation and promotes cell death by regulating polyubiquitination and lysosomal degradation of c-FLIP protein. J. Biol. Chem. 2012, 287, 6053–6061. [Google Scholar] [CrossRef] [PubMed]
- Rippo, M.R.; Moretti, S.; Vescovi, S.; Tomasetti, M.; Orecchia, S.; Amici, G.; Catalano, A.; Procopio, A. FLIP overexpression inhibits death receptor-induced apoptosis in malignant mesothelial cells. Oncogene 2004, 23, 7753–7760. [Google Scholar] [CrossRef] [PubMed]
- Clark, V.E.; Erson-Omay, E.Z.; Serin, A.; Yin, J.; Cotney, J.; Ozduman, K.; Avsar, T.; Li, J.; Murray, P.B.; Henegariu, O.; et al. Genomic analysis of non-NF2 meningiomas reveals mutations in TRAF7, KLF4, AKT1, and SMO. Science 2013, 339, 1077–1080. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, I.M.; Kolev, V.N.; Vidal, C.M.; Kadariya, Y.; Ring, J.E.; Wright, Q.; Weaver, D.T.; Menges, C.; Padval, M.; McClatchey, A.I.; et al. Merlin deficiency predicts FAK inhibitor sensitivity: A synthetic lethal relationship. Sci. Transl. Med. 2014, 6, 237ra68. [Google Scholar] [CrossRef] [PubMed]
- Liu-Chittenden, Y.; Huang, B.; Shim, J.S.; Chen, Q.; Lee, S.J.; Anders, R.A.; Liu, J.O.; Pan, D. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 2012, 26, 1300–1305. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.Q.; Dai, Y.Y.; Hsu, P.C.; Wang, H.; Cheng, L.; Yang, Y.L.; Wang, Y.C.; Xu, Z.D.; Liu, S.; Chan, G.; et al. Targeting YAP in malignant pleural mesothelioma. J. Cell. Mol. Med. 2017, 21, 2663–2676. [Google Scholar] [CrossRef] [PubMed]
- Tranchant, R.; Quetel, L.; Tallet, A.; Meiller, C.; Renier, A.; de Koning, L.; de Reynies, A.; Le Pimpec-Barthes, F.; Zucman-Rossi, J.; Jaurand, M.C.; et al. Co-occurring Mutations of Tumor Suppressor Genes, LATS2 and NF2, in Malignant Pleural Mesothelioma. Clin. Cancer Res. 2017, 23, 3191–3202. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Skouta, R.; Kaplan, A.; Yang, W.S.; Hayano, M.; Dixon, S.J.; Brown, L.M.; Valenzuela, C.A.; Wolpaw, A.J.; Stockwell, B.R. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat. Chem. Biol. 2016, 12, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Xie, M.; Scott, A.W.; Jin, J.; Ma, L.; Dong, X.; Skinner, H.D.; Johnson, R.L.; Ding, S.; Ajani, J.A. A Novel YAP1 Inhibitor Targets CSC-Enriched Radiation-Resistant Cells and Exerts Strong Antitumor Activity in Esophageal Adenocarcinoma. Mol. Cancer Ther. 2018, 17, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.C.; Moroishi, T.; Meng, Z.; Jeong, H.S.; Plouffe, S.W.; Sekido, Y.; Han, J.; Park, H.W.; Guan, K.L. Regulation of Hippo pathway transcription factor TEAD by p38 MAPK-induced cytoplasmic translocation. Nat. Cell Biol. 2017, 19, 996–1002. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.H.; Moon, J.; Garland, L.L.; Mack, P.C.; Testa, J.R.; Tsao, A.S.; Wozniak, A.J.; Gandara, D.R. SWOG S0722: Phase II study of mTOR inhibitor everolimus (RAD001) in advanced malignant pleural mesothelioma (MPM). J. Thorac. Oncol. 2015, 10, 387–391. [Google Scholar] [CrossRef] [PubMed]
- Thoreen, C.C.; Kang, S.A.; Chang, J.W.; Liu, Q.; Zhang, J.; Gao, Y.; Reichling, L.J.; Sim, T.; Sabatini, D.M.; Gray, N.S. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J. Biol. Chem. 2009, 284, 8023–8032. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, V.; Campbell, A.; Beaumont, K.L.; Cawkwell, L.; Lind, M.J. PTEN protein expression in malignant pleural mesothelioma. Tumour Biol. 2013, 34, 847–851. [Google Scholar] [CrossRef] [PubMed]
- Goel, S.; Wang, Q.; Watt, A.C.; Tolaney, S.M.; Dillon, D.A.; Li, W.; Ramm, S.; Palmer, A.C.; Yuzugullu, H.; Varadan, V.; et al. Overcoming Therapeutic Resistance in HER2-Positive Breast Cancers with CDK4/6 Inhibitors. Cancer Cell 2016, 29, 255–269. [Google Scholar] [CrossRef] [PubMed]
- Franco, J.; Balaji, U.; Freinkman, E.; Witkiewicz, A.K.; Knudsen, E.S. Metabolic Reprogramming of Pancreatic Cancer Mediated by CDK4/6 Inhibition Elicits Unique Vulnerabilities. Cell Rep. 2016, 14, 979–990. [Google Scholar] [CrossRef] [PubMed]
- Bonelli, M.A.; Digiacomo, G.; Fumarola, C.; Alfieri, R.; Quaini, F.; Falco, A.; Madeddu, D.; La Monica, S.; Cretella, D.; Ravelli, A.; et al. Combined Inhibition of CDK4/6 and PI3K/AKT/mTOR Pathways Induces a Synergistic Anti-Tumor Effect in Malignant Pleural Mesothelioma Cells. Neoplasia 2017, 19, 637–648. [Google Scholar] [CrossRef] [PubMed]
- Rodrik-Outmezguine, V.S.; Okaniwa, M.; Yao, Z.; Novotny, C.J.; McWhirter, C.; Banaji, A.; Won, H.; Wong, W.; Berger, M.; de Stanchina, E.; et al. Overcoming mTOR resistance mutations with a new-generation mTOR inhibitor. Nature 2016, 534, 272–276. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.; Aksoy, O.; Wong, R.A.; Ilkhanizadeh, S.; Novotny, C.J.; Gustafson, W.C.; Truong, A.Y.; Cayanan, G.; Simonds, E.F.; Haas-Kogan, D.; et al. A Kinase Inhibitor Targeted to mTORC1 Drives Regression in Glioblastoma. Cancer Cell 2017, 31, 424–435. [Google Scholar] [CrossRef] [PubMed]
- Demierre, M.F.; Higgins, P.D.; Gruber, S.B.; Hawk, E.; Lippman, S.M. Statins and cancer prevention. Nat. Rev. Cancer 2005, 5, 930–942. [Google Scholar] [CrossRef] [PubMed]
- Rubins, J.B.; Greatens, T.; Kratzke, R.A.; Tan, A.T.; Polunovsky, V.A.; Bitterman, P. Lovastatin induces apoptosis in malignant mesothelioma cells. Am. J. Respir. Crit. Care Med. 1998, 157, 1616–1622. [Google Scholar] [CrossRef] [PubMed]
- Asakura, K.; Izumi, Y.; Yamamoto, M.; Yamauchi, Y.; Kawai, K.; Serizawa, A.; Mizushima, T.; Ohmura, M.; Kawamura, M.; Wakui, M.; et al. The cytostatic effects of lovastatin on ACC-MESO-1 cells. J. Surg. Res. 2011, 170, e197–e209. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, Y.; Izumi, Y.; Asakura, K.; Fukutomi, T.; Serizawa, A.; Kawai, K.; Wakui, M.; Suematsu, M.; Nomori, H. Lovastatin and valproic acid additively attenuate cell invasion in ACC-MESO-1 cells. Biochem. Biophys. Res. Commun. 2011, 410, 328–332. [Google Scholar] [CrossRef] [PubMed]
- Tuerdi, G.; Ichinomiya, S.; Sato, H.; Siddig, S.; Suwa, E.; Iwata, H.; Yano, T.; Ueno, K. Synergistic effect of combined treatment with gamma-tocotrienol and statin on human malignant mesothelioma cells. Cancer Lett. 2013, 339, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Hwang, K.E.; Kim, Y.S.; Hwang, Y.R.; Kwon, S.J.; Park, D.S.; Cha, B.K.; Kim, B.R.; Yoon, K.H.; Jeong, E.T.; Kim, H.R. Enhanced apoptosis by pemetrexed and simvastatin in malignant mesothelioma and lung cancer cells by reactive oxygen species-dependent mitochondrial dysfunction and Bim induction. Int. J. Oncol. 2014, 45, 1769–1777. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Osada, H.; Murakami-Tonami, Y.; Horio, Y.; Hida, T.; Sekido, Y. Statin suppresses Hippo pathway-inactivated malignant mesothelioma cells and blocks the YAP/CD44 growth stimulatory axis. Cancer Lett. 2017, 385, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wu, Y.; Wang, H.; Zhang, Y.; Mei, L.; Fang, X.; Zhang, X.; Zhang, F.; Chen, H.; Liu, Y.; et al. Interplay of mevalonate and Hippo pathways regulates RHAMM transcription via YAP to modulate breast cancer cell motility. Proc. Natl. Acad. Sci. USA 2014, 111, E89–E98. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, G.; Ruggeri, N.; Specchia, V.; Cordenonsi, M.; Mano, M.; Dupont, S.; Manfrin, A.; Ingallina, E.; Sommaggio, R.; Piazza, S.; et al. Metabolic control of YAP and TAZ by the mevalonate pathway. Nat. Cell Biol. 2014, 16, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Guerrant, W.; Kota, S.; Troutman, S.; Mandati, V.; Fallahi, M.; Stemmer-Rachamimov, A.; Kissil, J.L. YAP Mediates Tumorigenesis in Neurofibromatosis Type 2 by Promoting Cell Survival and Proliferation through a COX-2-EGFR Signaling Axis. Cancer Res. 2016, 76, 3507–3519. [Google Scholar] [CrossRef] [PubMed]
- Wahle, B.M.; Hawley, E.T.; He, Y.; Smith, A.E.; Yuan, J.; Masters, A.R.; Jones, D.R.; Gehlhausen, J.R.; Park, S.J.; Conway, S.J.; et al. Chemopreventative celecoxib fails to prevent schwannoma formation or sensorineural hearing loss in genetically engineered murine model of neurofibromatosis type 2. Oncotarget 2018, 9, 718–725. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Aerts, J.G.; Popat, S.; Fennell, D.A. Novel insights into mesothelioma biology and implications for therapy. Nat. Rev. Cancer 2017, 17, 475–488. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.E.; Ngo, B.; Modrek, A.S.; Lee, W.H. Targeting tumor suppressor networks for cancer therapeutics. Curr. Drug Targets 2014, 15, 2–16. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.; Giancotti, F.G. Molecular insights into NF2/Merlin tumor suppressor function. FEBS Lett. 2014, 588, 2743–2752. [Google Scholar] [CrossRef] [PubMed]
- McCambridge, A.J.; Napolitano, A.; Mansfield, A.S.; Fennell, D.A.; Sekido, Y.; Nowak, A.K.; Reungwetwattana, T.; Mao, W.; Pass, H.I.; Carbone, M.; et al. State of the art: Advances in Malignant Pleural Mesothelioma in 2017. J. Thorac. Oncol. 2018, in press. [Google Scholar] [CrossRef] [PubMed]
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Mikkelson, T.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.G.; et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 2014, 343, 84–87. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sato, T.; Sekido, Y. NF2/Merlin Inactivation and Potential Therapeutic Targets in Mesothelioma. Int. J. Mol. Sci. 2018, 19, 988. https://doi.org/10.3390/ijms19040988
Sato T, Sekido Y. NF2/Merlin Inactivation and Potential Therapeutic Targets in Mesothelioma. International Journal of Molecular Sciences. 2018; 19(4):988. https://doi.org/10.3390/ijms19040988
Chicago/Turabian StyleSato, Tatsuhiro, and Yoshitaka Sekido. 2018. "NF2/Merlin Inactivation and Potential Therapeutic Targets in Mesothelioma" International Journal of Molecular Sciences 19, no. 4: 988. https://doi.org/10.3390/ijms19040988
APA StyleSato, T., & Sekido, Y. (2018). NF2/Merlin Inactivation and Potential Therapeutic Targets in Mesothelioma. International Journal of Molecular Sciences, 19(4), 988. https://doi.org/10.3390/ijms19040988