The Horizon of a Therapy for Rare Genetic Diseases: A “Druggable” Future for Fibrodysplasia Ossificans Progressiva


Abstract
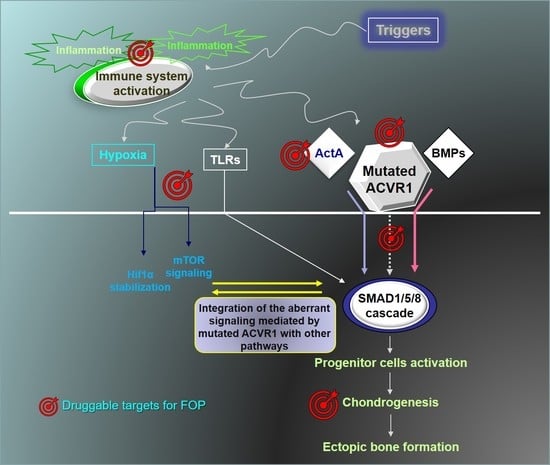
:
1. Fibrodysplasia Ossificans Progressiva (FOP) and the ACVR1 Gene
2. Translating the Results of Basic Research into the Development of New Therapeutic Strategies
2.1. Targeting the Altered Signaling
2.1.1. FOP Mutations and BMP Signaling
2.1.2. FOP and Neofunction of the Mutated Receptor
3. Targeting Cell Progenitors and Differentiation Processes
4. Targeting the Expression of the Receptor
5. Targeting the Immune System
6. Targeting the Microenvironment of FOP Local Lesions
7. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kaplan, F.S.; Xu, M.; Seemann, P.; Connor, J.M.; Glaser, D.L.; Carroll, L.; Delai, P.; Fastnacht-Urban, E.; Forman, S.J.; Gillessen-Kaesbach, G.; et al. Classic and atypical fibrodysplasia ossificans progressiva (FOP) phenotypes are caused by mutations in the bone morphogenetic protein (BMP) type I receptor ACVR1. Hum Mutat. 2009, 30, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Hüning, I.; Gillessen-Kaesbach, G. Fibrodysplasia ossificans progressiva: Clinical course, genetic mutations and genotype-phenotype correlation. Mol. Syndromol. 2014, 5, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Shore, E.M.; Xu, M.; Feldman, G.J.; Fenstermacher, D.A.; Cho, T.J.; Choi, I.H.; Connor, J.M.; Delai, P.; Glaser, D.L.; LeMerrer, M.; et al. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat. Genet. 2006, 38, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, M.; Horbelt, D.; Marom, B.; Knaus, P.; Henis, Y.I. Homomeric and heteromeric complexes among TGF-β and BMP receptors and their roles in signaling. Cell. Signal. 2011, 23, 1424–1432. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Li, B.Y.; Danielson, P.D.; Shah, P.C.; Rockwell, S.; Lechleider, R.J.; Martin, J.; Manganaro, T.; Donahoe, P.K. The immunophilin FKBP12 functions as a common inhibitor of the TGF beta family type I receptors. Cell 1996, 86, 435–444. [Google Scholar] [CrossRef]
- Bocciardi, R.; Bordo, D.; Di Duca, M.; Di Rocco, M.; Ravazzolo, R. Mutational analysis of the ACVR1 gene in Italian patients affected with fibrodysplasia ossificans progressiva: Confirmations and advancements. Eur. J. Hum. Genet. 2009, 17, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Ratbi, I.; Borcciadi, R.; Regragui, A.; Ravazzolo, R.; Sefiani, A. Rarely occurring mutation of ACVR1 gene in Moroccan patient with fibrodysplasia ossificans progressiva. Clin. Rheumatol. 2010, 29, 119–121. [Google Scholar] [CrossRef] [PubMed]
- Hatsell, S.J.; Idone, V.; Wolken, D.M.; Huang, L.; Kim, H.J.; Wang, L.; Wen, X.; Nannuru, K.C.; Jimenez, J.; Xie, L.; et al. ACVR1R206H receptor mutation causes fibrodysplasia ossificans progressiva by imparting responsiveness to activin A. Sci. Transl. Med. 2015, 7, 303ra137. [Google Scholar] [CrossRef] [PubMed]
- Hino, K.; Ikeya, M.; Horigome, K.; Matsumoto, Y.; Ebise, H.; Nishio, M.; Sekiguchi, K.; Shibata, M.; Nagata, S.; Matsuda, S.; et al. Neofunction of ACVR1 in fibrodysplasia ossificans progressiva. Proc. Natl. Acad. Sci. USA 2015, 112, 15438–15443. [Google Scholar] [CrossRef] [PubMed]
- Di Rocco, M.; Baujat, G.; Bertamino, M.; Brown, M.; De Cunto, C.L.; Delai, P.L.R.; Eekhoff, E.M.W.; Haga, N.; Hsiao, E.; Keen, R.; et al. International physician survey on management of FOP: A modified Delphi study. Orphanet J. Rare Dis. 2017, 12, 110. [Google Scholar] [CrossRef] [PubMed]
- Volek-Smith, H.; Urist, M.R. Recombinant human bone morphogenetic protein (rhBMP) induced heterotopic bone development in vivo and in vitro. Proc. Soc. Exp. Biol. Med. 1996, 211, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Lounev, V.Y.; Ramachandran, R.; Wosczyna, M.N.; Yamamoto, M.; Maidment, A.D.; Shore, E.M.; Glaser, D.L.; Goldhamer, D.J.; Kaplan, F.S. Identification of progenitor cells that contribute to heterotopic skeletogenesis. J. Bone Jt. Surg. Am. 2009, 91, 652–663. [Google Scholar] [CrossRef] [PubMed]
- Kan, L.; Hu, M.; Gomes, W.A.; Kessler, J.A. Transgenic mice overexpressing BMP4 develop a fibrodysplasia ossificans progressiva (FOP)-like phenotype. Am. J. Pathol. 2004, 165, 1107–1115. [Google Scholar] [CrossRef]
- De la Peña, L.S.; Billings, P.C.; Fiori, J.L.; Ahn, J.; Kaplan, F.S.; Shore, E.M. Fibrodysplasia ossificans progressiva (FOP), a disorder of ectopic osteogenesis, misregulates cell surface expression and trafficking of BMPRIA. J. Bone Miner. Res. 2005, 20, 1168–1176. [Google Scholar] [CrossRef] [PubMed]
- Fiori, J.L.; Billings, P.C.; de la Peña, L.S.; Kaplan, F.S.; Shore, E.M. Dysregulation of the BMP-p38 MAPK signaling pathway in cells from patients with fibrodysplasia ossificans progressiva (FOP). J. Bone Miner. Res. 2006, 21, 902–909. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, T.; Kohda, M.; Kanomata, K.; Nojima, J.; Nakamura, A.; Kamizono, J.; Noguchi, Y.; Iwakiri, K.; Kondo, T.; Kurose, J.; et al. Constitutively activated ALK2 and increased SMAD1/5 cooperatively induce bone morphogenetic protein signaling in fibrodysplasia ossificans progressiva. J. Biol. Chem. 2009, 284, 7149–7156. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.; Little, S.C.; Xu, M.; Haupt, J.; Ast, C.; Katagiri, T.; Mundlos, S.; Seemann, P.; Kaplan, F.S.; Mullins, M.C.; et al. The fibrodysplasia ossificans progressiva R206H ACVR1 mutation activates BMP-independent chondrogenesis and zebrafish embryo ventralization. J. Clin. Investig. 2009, 119, 3462–3472. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, T.; Kanomata, K.; Nojima, J.; Kokabu, S.; Akita, M.; Ikebuchi, K.; Jimi, E.; Komori, T.; Maruki, Y.; Matsuoka, M.; et al. A unique mutation of ALK2, G356D, found in a patient with fibrodysplasia ossificans progressiva is a moderately activated BMP type I receptor. Biochem. Biophys. Res. Commun. 2008, 377, 905–909. [Google Scholar] [CrossRef] [PubMed]
- Song, G.A.; Kim, H.J.; Woo, K.M.; Baek, J.H.; Kim, G.S.; Choi, J.Y.; Ryoo, H.M. Molecular consequences of the ACVR1R206H mutation of fibrodysplasia ossificans progressiva. J. Biol. Chem. 2010, 285, 22542–22553. [Google Scholar] [CrossRef] [PubMed]
- Ohte, S.; Shin, M.; Sasanuma, H.; Yoneyama, K.; Akita, M.; Ikebuchi, K.; Jimi, E.; Maruki, Y.; Matsuoka, M.; Namba, A.; et al. A novel mutation of ALK2, L196P, found in the most benign case of fibrodysplasia ossificans progressiva activates BMP-specific intracellular signaling equivalent to a typical mutation, R206H. Biochem. Biophys. Res. Commun. 2011, 407, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Haupt, J.; Deichsel, A.; Stange, K.; Ast, C.; Bocciardi, R.; Ravazzolo, R.; Di Rocco, M.; Ferrari, P.; Landi, A.; Kaplan, F.S.; et al. ACVR1 p.Q207E causes classic fibrodysplasia ossificans progressiva and is functionally distinct from the engineered constitutively active ACVR1 p.Q207D variant. Hum. Mol. Genet. 2014, 23, 5364–5377. [Google Scholar] [CrossRef] [PubMed]
- Mucha, B.E.; Hashiguchi, M.; Zinski, J.; Shore, E.M.; Mullins, M.C. Variant BMP receptor mutations causing fibrodysplasia ossificans progressiva (FOP) in humans show BMP ligand-independent receptor activation in zebrafish. Bone 2018, 109, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.B.; Deng, D.Y.; Lai, C.S.; Hong, C.C.; Cuny, G.D.; Bouxsein, M.L.; Hong, D.W.; McManus, P.M.; Katagiri, T.; Sachidanandan, C.; et al. BMP type I receptor inhibition reduces heterotopic ossification. Nat. Med. 2008, 14, 1363–1369. [Google Scholar] [CrossRef] [PubMed]
- Cuny, G.D.; Yu, P.B.; Laha, J.K.; Xing, X.; Liu, J.F.; Lai, C.S.; Deng, D.Y.; Sachidanandan, C.; Bloch, K.D.; Peterson, R.T. Structure-activity relationship study of bone orphogenetic protein (BMP) signaling inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 4388–4392. [Google Scholar] [CrossRef] [PubMed]
- Sanvitale, C.E.; Kerr, G.; Chaikuad, A.; Ramel, M.C.; Mohedas, A.H.; Reichert, S.; Wang, Y.; Triffitt, J.T.; Cuny, G.D.; Yu, P.B.; et al. A new class of small molecule inhibitor of BMP signaling. PLoS ONE 2013, 8, e62721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohedas, A.H.; Xing, X.; Armstrong, K.A.; Bullock, A.N.; Cuny, G.D.; Yu, P.B. Development of n ALK2-biased BMP type I receptor kinase inhibitor. ACS Chem. Biol. 2013, 8, 1291–1302. [Google Scholar] [CrossRef] [PubMed]
- Mohedas, A.H.; Wang, Y.; Sanvitale, C.E.; Canning, P.; Choi, S.; Xing, X.; Bullock, A.N.; Cuny, G.D.; Yu, P.B. Structure-activity relationship of 3,5-diaryl-2-aminopyridine ALK2 inhibitors reveals unaltered binding affinity for fibrodysplasia ossificans progressiva causing mutants. J. Med. Chem. 2014, 57, 7900–7915. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, T.; Scott, G.; Komatsu, Y.; Araya, R.; Kawano, M.; Ray, M.K.; Yamada, M.; Mishina, Y. Generation of a mouse with conditionally activated signaling through the BMP eceptor, ALK2. Genesis 2006, 44, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Wieser, R.; Wrana, J.L.; Massague, J. GS domain mutations that constitutively activate T beta R-I, the downstream signaling component in the TGF-beta receptor complex. EMBO J. 1995, 14, 2199–2208. [Google Scholar] [PubMed]
- Williams, E.; Bullock, A.N. Structural basis for the potent and selective binding of LDN-212854 to the BMP receptor kinase ALK2. Bone 2017. [Google Scholar] [CrossRef] [PubMed]
- Mall, M.A.; Galietta, L.J. Targeting ion channels in cystic fibrosis. J. Cyst. Fibros. 2015, 14, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Yadin, D.; Knaus, P.; Mueller, T.D. Structural insights into BMP receptors: Specificity, activation and inhibition. Cytokine Growth Factor Rev. 2016, 27, 13–34. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Zheng, W.; Simeonov, A. Drug discovery and development for rare genetic disorders. Am. J. Med. Genet A 2017, 173, 2307–2322. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, R.; Matsushita, M.; Kitoh, H.; Masuda, A.; Ito, M.; Katagiri, T.; Kawai, T.; Ishiguro, N.; Ohno, K. Clinically applicable antianginal agents suppress osteoblastic transformation of myogenic cells and heterotopic ossifications in mice. J. Bone Miner. Metab. 2013, 31, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Kitoh, H.; Achiwa, M.; Kaneko, H.; Mishima, K.; Matsushita, M.; Kadono, I.; Horowitz, J.D.; Sallustio, B.C.; Ohno, K.; Ishiguro, N. Perhexiline maleate in the treatment of fibrodysplasia ossificans progressiva: An open-labeled clinical trial. Orphanet J. Rare Dis. 2013, 8, 163. [Google Scholar] [CrossRef] [PubMed]
- Lees-Shepard, J.B.; Yamamoto, M.; Biswas, A.A.; Stoessel, S.J.; Nicholas, S.E.; Cogswell, C.A.; Devarakonda, P.M.; Schneider, M.J., Jr.; Cummins, S.M.; Legendre, N.P.; et al. Activin-dependent signaling in fibro/adipogenic progenitors causes fibrodysplasia ossificans progressiva. Nat. Commun. 2018, 9, 471. [Google Scholar] [CrossRef] [PubMed]
- Hino, K.; Horigome, K.; Nishio, M.; Komura, S.; Nagata, S.; Zhao, C.; Jin, Y.; Kawakami, K.; Yamada, Y.; Ohta, A.; et al. Activin-A enhances mTOR signaling to promote aberrant chondrogenesis in fibrodysplasia ossificans progressiva. J. Clin. Investig. 2017, 127, 3339–3352. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, F.S.; Zeitlin, L.; Dunn, S.P.; Benor, S.; Hagin, D.; Al Mukaddam, M.; Pignolo, R.J. Acute and chronic rapamycin use in patients with Fibrodysplasia Ossificans Progressiva: A report of two cases. Bone 2017. [Google Scholar] [CrossRef] [PubMed]
- Medici, D.; Shore, E.M.; Lounev, V.Y.; Kaplan, F.S.; Kalluri, R.; Olsen, B.R. Conversion of vascular endothelial cells into multipotent stem-like cells. Nat. Med. 2010, 16, 1400–1406. [Google Scholar] [CrossRef] [PubMed]
- Medici, D.; Olsen, B.R. The role of endothelial-mesenchymal transition in heterotopic ossification. J. Bone Miner. Res. 2012, 27, 1619–1622. [Google Scholar] [CrossRef] [PubMed]
- Dey, D.; Bagarova, J.; Hatsell, S.J.; Armstrong, K.A.; Huang, L.; Ermann, J.; Vonner, A.J.; Shen, Y.; Mohedas, A.H.; Lee, A.; et al. Two tissue-resident progenitor lineages drive distinct phenotypes of heterotopic ossification. Sci. Transl. Med. 2016, 8, 366ra163. [Google Scholar] [CrossRef] [PubMed]
- Wosczyna, M.N.; Biswas, A.A.; Cogswell, C.A.; Goldhamer, D.J. Multipotent progenitors resident in the skeletal muscle interstitium exhibit robust BMP-dependent osteogenic activity and mediate heterotopic ossification. J. Bone Miner. Res. 2012, 27, 1004–1017. [Google Scholar] [CrossRef] [PubMed]
- Biswas, A.A.; Goldhamer, D.J. FACS Fractionation and Differentiation of Skeletal-Muscle Resident Multipotent Tie2+ Progenitors. Methods Mol. Biol. 2016, 1460, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Shore, E.M.; Kaplan, F.S. Inherited human diseases of heterotopic bone formation. Nat. Rev. Rheumatol. 2010, 6, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Mackie, E.J.; Tatarczuch, L.; Mirams, M. The skeleton: A multi-functional complex organ. The growth plate chondrocyte and endochondral ossification. J. Endocrinol. 2011, 211, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Van Dinther, M.; Visser, N.; de Gorter, D.J.; Doorn, J.; Goumans, M.J.; de Boer, J.; ten Dijke, P. ALK2 R206H mutation linked to fibrodysplasia ossificans progressiva confers constitutive activity to the BMP type I receptor and sensitizes mesenchymal cells to BMP-induced osteoblast differentiation and bone formation. J. Bone Miner. Res. 2010, 25, 1208–1215. [Google Scholar] [CrossRef] [PubMed]
- Culbert, A.L.; Chakkalakal, S.A.; Theosmy, E.G.; Brennan, T.A.; Kaplan, F.S.; Shore, E.M. Alk2 regulates early chondrogenic fate in fibrodysplasia ossificans progressiva heterotopic endochondral ossification. Stem Cells 2014, 32, 1289–1300. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, L.M.; Weston, A.D.; Underhill, T.M. Molecular mechanisms regulating chondroblast differentiation. J. Bone Jt. Surg. Am. 2003, 85 (Suppl. 2), 124–132. [Google Scholar] [CrossRef]
- Pacifici, M. Retinoid roles and action in skeletal development and growth provide the rationale for an ongoing heterotopic ossification prevention trial. Bone 2017. [Google Scholar] [CrossRef] [PubMed]
- Shimono, K.; Morrison, T.N.; Tung, W.E.; Chandraratna, R.A.; Williams, J.A.; Iwamoto, M.; Pacifici, M. Inhibition of ectopic bone formation by a selective retinoic acid receptor alpha-agonist: A new therapy for heterotopic ossification? J. Orthop. Res. 2010, 28, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Shimono, K.; Tung, W.E.; Macolino, C.; Chi, A.H.; Didizian, J.H.; Mundy, C.; Chandraratna, R.A.; Mishina, Y.; Enomoto-Iwamoto, M.; Pacifici, M.; et al. Potent inhibition of heterotopic ossification by nuclear retinoic acid receptor-γ agonists. Nat. Med. 2011, 17, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Hind, M.; Stinchcombe, S. Palovarotene, a novel retinoic acid receptor gamma agonist for the treatment of emphysema. Curr. Opin. Investig. Drugs 2009, 10, 1243–1250. [Google Scholar] [PubMed]
- Sinha, S.; Uchibe, K.; Usami, Y.; Pacifici, M.; Iwamoto, M. Effectiveness and mode of action of a combination therapy for heterotopic ossification with a retinoid agonist and an anti-inflammatory agent. Bone 2016, 90, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Chakkalakal, S.A.; Uchibe, K.; Convente, M.R.; Zhang, D.; Economides, A.N.; Kaplan, F.S.; Pacifici, M.; Iwamoto, M.; Shore, E.M. Palovarotene Inhibits Heterotopic Ossification and Maintains Limb Mobility and Growth in Mice With the Human ACVR1R206H Fibrodysplasia Ossificans Progressiva (FOP) Mutation. J. Bone Miner. Res. 2016, 31, 1666–1675. [Google Scholar] [CrossRef] [PubMed]
- Giacopelli, F.; Cappato, S.; Tonachini, L.; Mura, M.; Di Lascio, S.; Fornasari, D.; Ravazzolo, R.; Bocciardi, R. Identification and characterization of regulatory elements in the promoter of ACVR1, the gene mutated in Fibrodysplasia Ossificans Progressiva. Orphanet J. Rare Dis. 2013, 8, 145. [Google Scholar] [CrossRef] [PubMed]
- Cappato, S.; Tonachini, L.; Giacopelli, F.; Tirone, M.; Galietta, L.J.; Sormani, M.; Giovenzana, A.; Spinelli, A.E.; Canciani, B.; Brunelli, S.; et al. High-throughput screening for modulators of ACVR1 transcription: Discovery of potential therapeutics for fibrodysplasia ossificans progressiva. Dis. Model Mech. 2016, 9, 685–696. [Google Scholar] [CrossRef] [PubMed]
- Karbiener, M.; Neuhold, C.; Opriessnig, P.; Prokesch, A.; Bogner-Strauss, J.G.; Scheideler, M. MicroRNA-30c promotes human adipocyte differentiation and co-represses PAI-1 and ALK2. RNA Biol. 2011, 8, 850–860. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Wang, Q.; Wen, J.; Liu, S.; Gao, X.; Cheng, J.; Zhang, D. ACVR1, a therapeutic target of fibrodysplasia ossificans progressiva, is negatively regulated by miR-148a. Int. J. Mol. Sci. 2012, 13, 2063–2077. [Google Scholar] [CrossRef] [PubMed]
- Mura, M.; Cappato, S.; Giacopelli, F.; Ravazzolo, R.; Bocciardi, R. The role of the 3′UTR region in the regulation of the ACVR1/Alk-2 gene expression. PLoS ONE 2012, 7, e50958. [Google Scholar] [CrossRef] [PubMed]
- Zumbrennen-Bullough, K.B.; Wu, Q.; Core, A.B.; Canali, S.; Chen, W.; Theurl, I.; Meynard, D.; Babitt, J.L. MicroRNA-130a is up-regulated in mouse liver by iron deficiency and targets the bone morphogenetic protein (BMP) receptor ALK2 to attenuate BMP signaling and hepcidin transcription. J. Biol. Chem. 2014, 289, 23796–23808. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Liu, Y.; Guo, Y.; Liu, B.; Zhao, Y.; Li, P.; Song, F.; Zheng, H.; Yu, J.; Song, T.; et al. Regulatory MiR-148a-ACVR1/BMP circuit defines a cancer stem cell-like aggressive subtype of hepatocellular carcinoma. Hepatology 2015, 61, 574–584. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A.; Fokkema, I.; Verschuuren, J.; Ginjaar, I.; van Deutekom, J.; van Ommen, G.J.; den Dunnen, J.T. Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations. Hum. Mutat. 2009, 30, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Cai, J.; de Gorter, D.J.; Sanchez-Duffhues, G.; Kemaladewi, D.U.; Hoogaars, W.M.; Aartsma-Rus, A.; ‘t Hoen, P.A.C.; ten Dijke, P. Antisense-oligonucleotide mediated exon skipping in activin-receptor-like kinase 2: Inhibiting the receptor that is overactive in fibrodysplasia ossificans progressiva. PLoS ONE 2013, 8, e69096. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, J.; Kaplan, F.S.; Shore, E.M. Restoration of normal BMP signaling levels and osteogenic differentiation in FOP mesenchymal progenitor cells by mutant allele-specific targeting. Gene Ther. 2012, 19, 786–790. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Katagiri, T.; Furuya, H.; Hohjoh, H. Disease-causing allele-specific silencing against the ALK2 mutants, R206H and G356D, in fibrodysplasia ossificans progressiva. Gene Ther. 2012, 19, 781–785. [Google Scholar] [CrossRef] [PubMed]
- Gannon, F.H.; Valentine, B.A.; Shore, E.M.; Zasloff, M.A.; Kaplan, F.S. Acute lymphocytic infiltration in an extremely early lesion of fibrodysplasia ossificans progressiva. Clin. Orthop. Relat. Res. 1998, 346, 9–25. [Google Scholar] [CrossRef]
- Gannon, F.H.; Glaser, D.; Caron, R.; Thompson, L.D.; Shore, E.M.; Kaplan, F.S. Mast cell involvement in fibrodysplasia ossificans progressiva. Hum. Pathol. 2001, 32, 842–848. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, F.S.; Pignolo, R.J.; Shore, E.M. Granting immunity to FOP and catching heterotopic ossification in the Act. Semin. Cell Dev. Biol. 2016, 49, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Phillips, D.J.; de Kretser, D.M.; Hedger, M.P. Activin and related proteins in inflammation: Not just interested bystanders. Cytokine Growth Factor Rev. 2009, 20, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Aleman-Muench, G.R.; Soldevila, G. When versatility matters: Activins/inhibins as key regulators of immunity. Immunol. Cell Biol. 2012, 90, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Behrens, E.M.; Pignolo, R.J.; Kaplan, F.S. ECSIT links TLR and BMP signaling in FOP connective tissue progenitor cells. Bone 2017. [Google Scholar] [CrossRef] [PubMed]
- Kan, L.; Liu, Y.; McGuire, T.L.; Berger, D.M.; Awatramani, R.B.; Dymecki, S.M.; Kessler, J.A. Dysregulation of local stem/progenitor cells as a common cellular mechanism for heterotopic ossification. Stem Cells 2009, 27, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Brennan, T.A.; Lindborg, C.M.; Bergbauer, C.R.; Wang, H.; Kaplan, F.S.; Pignolo, R.J. Mast cell inhibition as a therapeutic approach in fibrodysplasia ossificans progressiva (FOP). Bone 2017. [Google Scholar] [CrossRef] [PubMed]
- Kan, L.; Lounev, V.Y.; Pignolo, R.J.; Duan, L.; Liu, Y.; Stock, S.R.; McGuire, T.L.; Lu, B.; Gerard, N.P.; Shore, E.M.; et al. Substance P signaling mediates BMP-dependent heterotopic ossification. J. Cell. Biochem. 2011, 112, 2759–2772. [Google Scholar] [CrossRef] [PubMed]
- Convente, M.R.; Chakkalakal, S.A.; Yang, E.; Caron, R.J.; Zhang, D.; Kambayashi, T.; Kaplan, F.S.; Shore, E.M. Depletion of Mast Cells and Macrophages Impairs Heterotopic Ossification in an ACVR1R206H Mouse Model of Fibrodysplasia Ossificans Progressiva. J. Bone Miner. Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, F.S.; Glaser, D.L.; Shore, E.M.; Pignolo, R.J.; Xu, M.; Zhang, Y.; Senitzer, D.; Forman, S.J.; Emerson, S.G. Hematopoietic stem-cell contribution to ectopic skeletogenesis. J. Bone Jt. Surg. Am. 2007, 89, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Del Zotto, G.; Antonini, F.; Azzari, I.; Ortolani, C.; Tripodi, G.; Giacopelli, F.; Cappato, S.; Moretta, L.; Ravazzolo, R.; Bocciardi, R. Peripheral blood mononuclear cell immunophenotyping in fibrodysplasia ossificans progressiva patients: Evidence for monocyte DNAM1 up-regulation. Cytom. B Clin. Cytom. 2017. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.T.; Doherty, G.; Fallon, P.G.; Cummins, E.P. Hypoxia-dependent regulation of inflammatory pathways in immune cells. J. Clin. Investig. 2016, 126, 3716–3724. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Liu, H.; Li, J.; Zhou, Y. Roles of hypoxia during the chondrogenic differentiation of mesenchymal stem cells. Curr. Stem Cell Res. Ther. 2014, 9, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Loder, S.; Brownley, C.; Cholok, D.; Mangiavini, L.; Li, J.; Breuler, C.; Sung, H.H.; Li, S.; Ranganathan, K.; et al. Inhibition of Hif1α prevents both trauma-induced and genetic heterotopic ossification. Proc. Natl. Acad. Sci. USA 2016, 113, E338–E347. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Lindborg, C.; Lounev, V.; Kim, J.H.; McCarrick-Walmsley, R.; Xu, M.; Mangiavini, L.; Groppe, J.C.; Shore, E.M.; Schipani, E.; et al. Cellular Hypoxia Promotes Heterotopic Ossification by Amplifying BMP Signaling. J. Bone Miner. Res. 2016, 31, 1652–1665. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Cholok, D.; Loder, S.; Li, J.; Breuler, C.; Chung, M.T.; Sung, H.H.; Ranganathan, K.; Habbouche, J.; Drake, J.; et al. mTOR inhibition and BMP signaling act synergistically to reduce muscle fibrosis and improve myofiber regeneration. JCI Insight 2016, 1, e89805. [Google Scholar] [CrossRef] [PubMed]
- Bower, H.; Björkholm, M.; Dickman, P.W.; Höglund, M.; Lambert, P.C.; Andersson, T.M. Life Expectancy of Patients With Chronic Myeloid Leukemia Approaches the Life Expectancy of the General Population. J. Clin. Oncol. 2016, 34, 2851–2857. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, F.S.; Andolina, J.R.; Adamson, P.C.; Teachey, D.T.; Finklestein, J.Z.; Ebb, D.H.; Whitehead, B.; Jacobs, B.; Siegel, D.M.; Keen, R.; et al. Early clinical observations on the use of imatinib mesylate in FOP: A report of seven cases. Bone 2017. [Google Scholar] [CrossRef] [PubMed]
- Stein, S.; Bogard, E.; Boice, N.; Fernandez, V.; Field, T.; Gilstrap, A.; Kahn, S.R.; Larkindale, J.; Mathieson, T. Principles for interactions with biopharmaceutical companies: The development of guidelines for patient advocacy organizations in the field of rare diseases. Orphanet J. Rare Dis. 2018, 13, 18. [Google Scholar] [CrossRef] [PubMed]


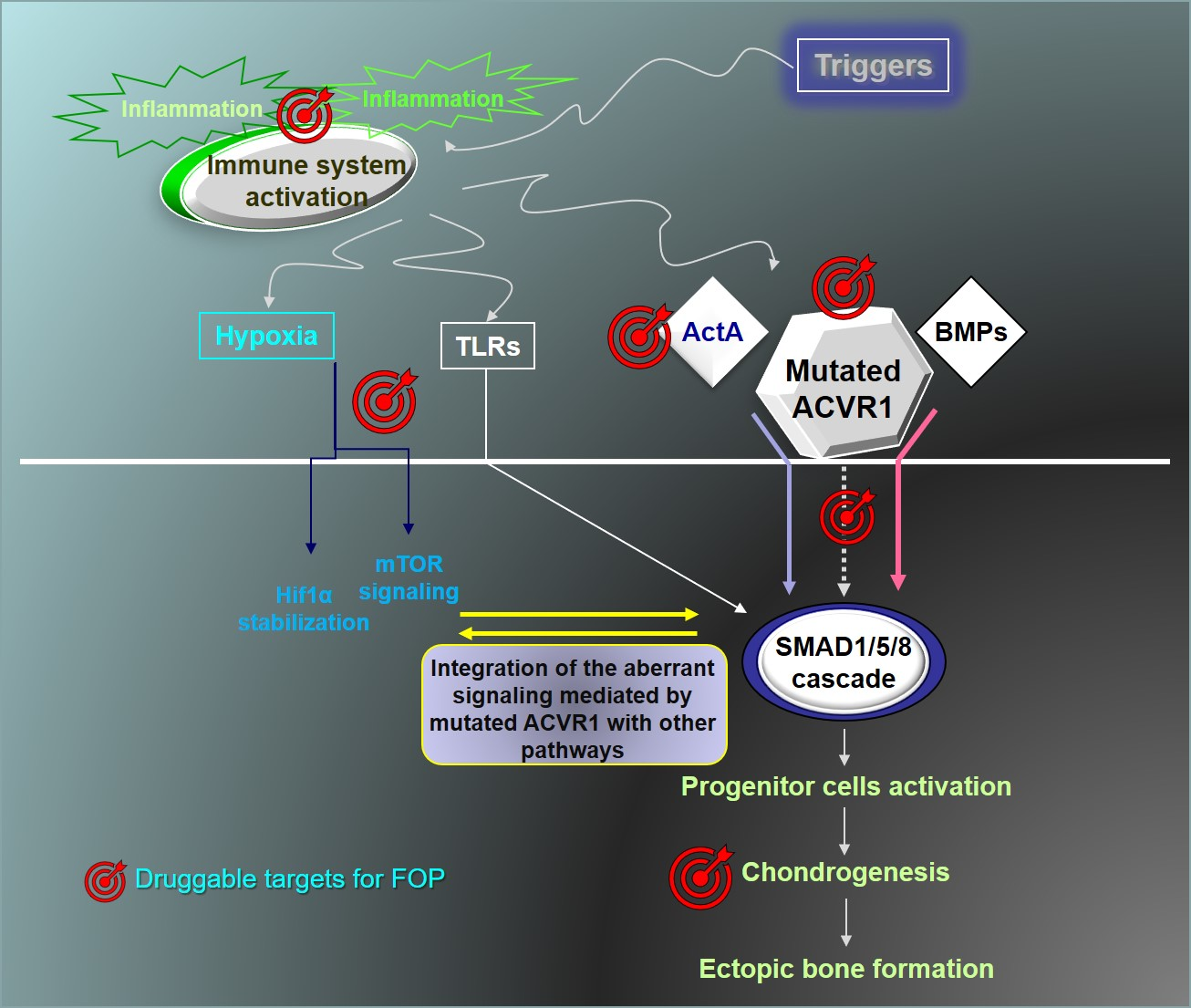{kind=link}
{kind=link}
| Exon | Nucleotide * | Residue ** | Domain |
|---|---|---|---|
| 6 | c.587T>C | p.Leu196Pro | GS |
| 6 | c.590-592delCTT | p.delPro197-Phe198insLeu | GS |
| 6 | c.605G>T | p.Arg202Ile | GS |
| 6 | c.617G>A | p.Arg206His | GS |
| 6 | c.619C>G | p.Gln207Glu | GS |
| 7 | c.774G>C/T | p.Arg258Ser | Kinase |
| 7 | c.774G>T | p.Arg258Ser | Kinase |
| 8 | c.974G>C | p.Gly325Ala | Kinase |
| 8 | c.982G>A/C/T | p.Gly328Arg/Trp | Kinase |
| 8 | c.983G>T/A | p.Gly328Val/Glu | Kinase |
| 9 | c.1067G>A | p.Gly356Asp | Kinase |
| 9 | c.1124G>C | p.Arg375Pro | Kinase |
| Drug | Company | Trial Phase | Title * | ClinicalTrials.gov Identifier * |
|---|---|---|---|---|
| Palovarotene | Clementia | Phase3 (pivotal) | An Efficacy and Safety Study of Palovarotene for the Treatment of FOP | NCT03312634 |
| REGN2477 | Regeneron | Phase 2 | A Study to Examine the Safety, Tolerability and Effects on Abnormal Bone Formation of REGN2477 in Patients With Fibrodysplasia Ossificans Progressiva (LUMINA-1) | NCT03188666 |
| Identification of Sensitive Targets | Strategy | Comments | |
|---|---|---|---|
| Targeting the altered signaling | Dysregulated BMP signaling | Development of pharmacological inhibitors of the kinase function | In vitro evidences; preclinical mouse models (Dorsomorphin, LDN-189193; LDN LDN-212854 other derivatives, …) Possible application of kinase inhibitors developed for other conditions? |
| Screening of FDA-approved compounds able to interfere with the dysregulated BMP signaling | In vitro evidences; preclinical mouse models. Perhexiline tested in patients on a off-label basis in a non-trial setting | ||
| Neofunction of the mutated receptor (responsiveness to ActA) | Development of blocking anti-ActA antibodies | In vitro evidences; preclinical FOP mouse models; REGN2477 recruiting Phase 2 trial | |
| Screening of FDA-approved compounds able to interfere with the ActA/mutated ACVR1 signaling | In vitro evidences; preclinical mouse models (mTOR inhibitors, Rapamycin) | ||
| Targeting differentiation processes | Chondrogenesis as a critical differentiation step in HO formation | Maintenance of the retinoid signaling active to block chondrogenic differentiation by using RARγ agonists | In vitro evidences; preclinical FOP mouse models. Palovarotene on Phase 3 trial |
| Targeting the expression of the ACVR1/Alk-2 receptor | Transcriptional level | Screening of molecules potentially able to down-modulate expression of the ACVR1 gene at transcriptional level | In vitro evidences; preclinical HO mouse models (Dipyridamole) |
| Post-transcriptional level | ACVR1 targeting by microRNA | Proof of principle | |
| Development of Anti-sense oligonucleotide (AON) to promote exon skipping and ACVR1 mRNA targeting to the non-sense mediated degradation pathway | |||
| Development of allele-specific RNAi molecules (ASP-RNAi) able to target the expression of mutant ACVR1 alleles | |||
| Targeting the immune system | Modulation of the immune response & inflammation | Targeted ablation of macrophages and mast cells | In vitro evidences; preclinical HO/FOP mouse models |
| Use of corticosteroids or immunosuppressant drugs | Corticosteroids are currently in use to manage FOP flare-ups; single case report of long term use of immunosuppressant is reported | ||
| Targeting the microenvironment of FOP local lesions | Modulation of hypoxia | Pharmacological inhibition of HIF-1α pathway (Apigenin, Imatinib, PX-478 and Rapamycin) | In vitro evidences; preclinical mouse models. Imatinib and Rapamycin tested in patients on a off-label basis in a non-trial setting |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cappato, S.; Giacopelli, F.; Ravazzolo, R.; Bocciardi, R. The Horizon of a Therapy for Rare Genetic Diseases: A “Druggable” Future for Fibrodysplasia Ossificans Progressiva. Int. J. Mol. Sci. 2018, 19, 989. https://doi.org/10.3390/ijms19040989
Cappato S, Giacopelli F, Ravazzolo R, Bocciardi R. The Horizon of a Therapy for Rare Genetic Diseases: A “Druggable” Future for Fibrodysplasia Ossificans Progressiva. International Journal of Molecular Sciences. 2018; 19(4):989. https://doi.org/10.3390/ijms19040989
Chicago/Turabian StyleCappato, Serena, Francesca Giacopelli, Roberto Ravazzolo, and Renata Bocciardi. 2018. "The Horizon of a Therapy for Rare Genetic Diseases: A “Druggable” Future for Fibrodysplasia Ossificans Progressiva" International Journal of Molecular Sciences 19, no. 4: 989. https://doi.org/10.3390/ijms19040989
APA StyleCappato, S., Giacopelli, F., Ravazzolo, R., & Bocciardi, R. (2018). The Horizon of a Therapy for Rare Genetic Diseases: A “Druggable” Future for Fibrodysplasia Ossificans Progressiva. International Journal of Molecular Sciences, 19(4), 989. https://doi.org/10.3390/ijms19040989