Hemiptera Mitochondrial Control Region: New Sights into the Structural Organization, Phylogenetic Utility, and Roles of Tandem Repetitions of the Noncoding Segment


Abstract
:1. Introduction
2. Results
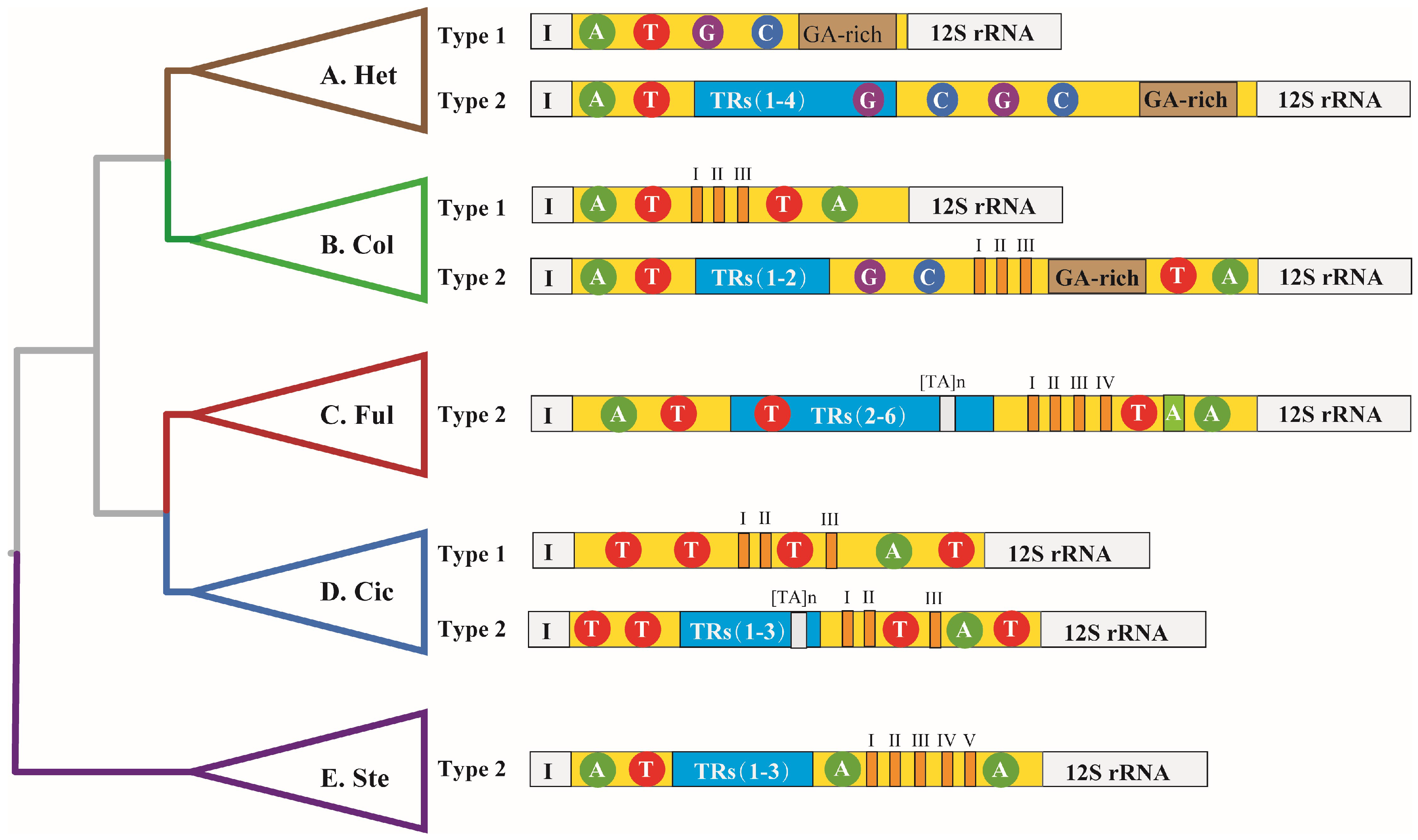
2.1. The Structural Organization
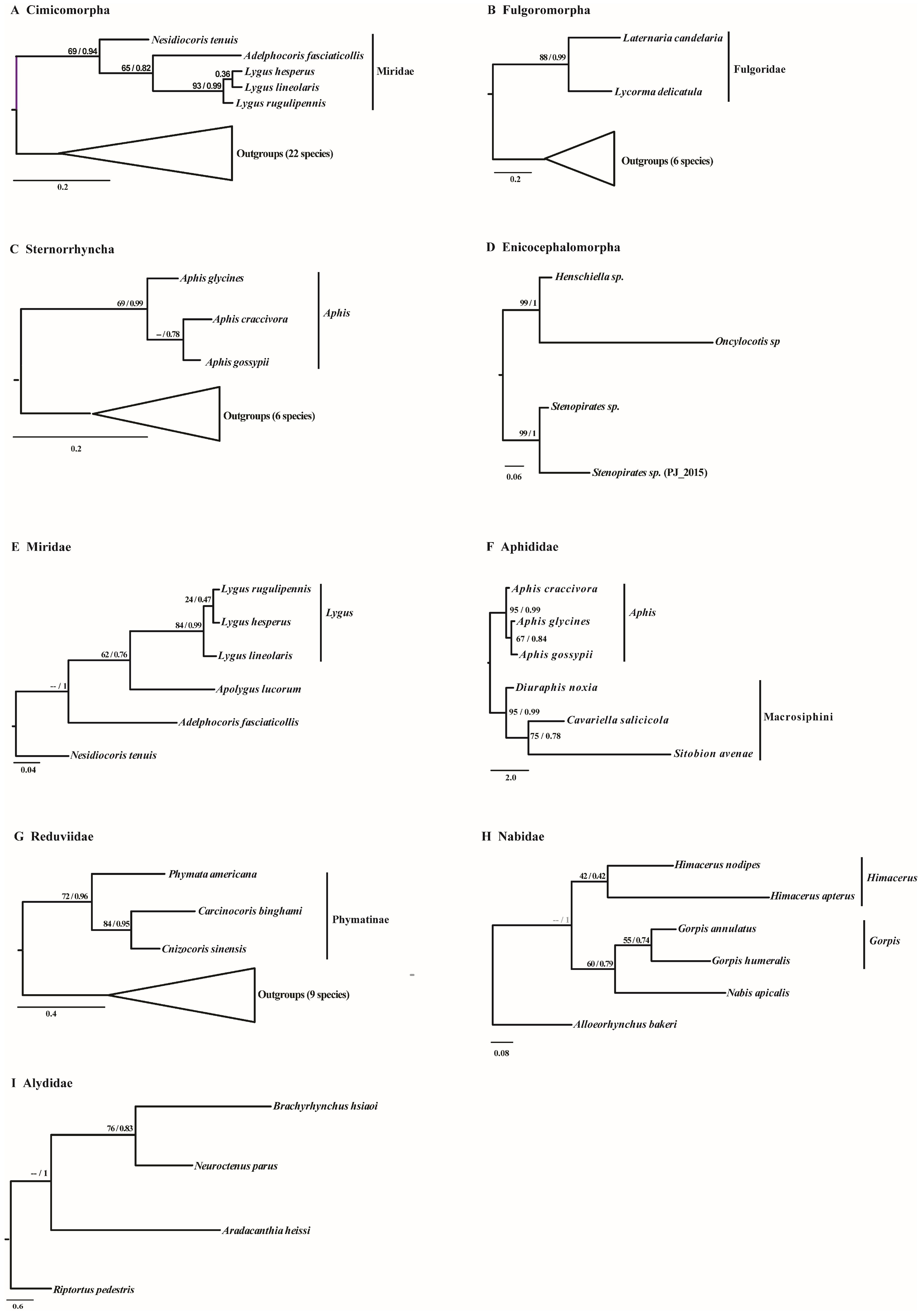
2.2. Phylogenetic Reconstruction
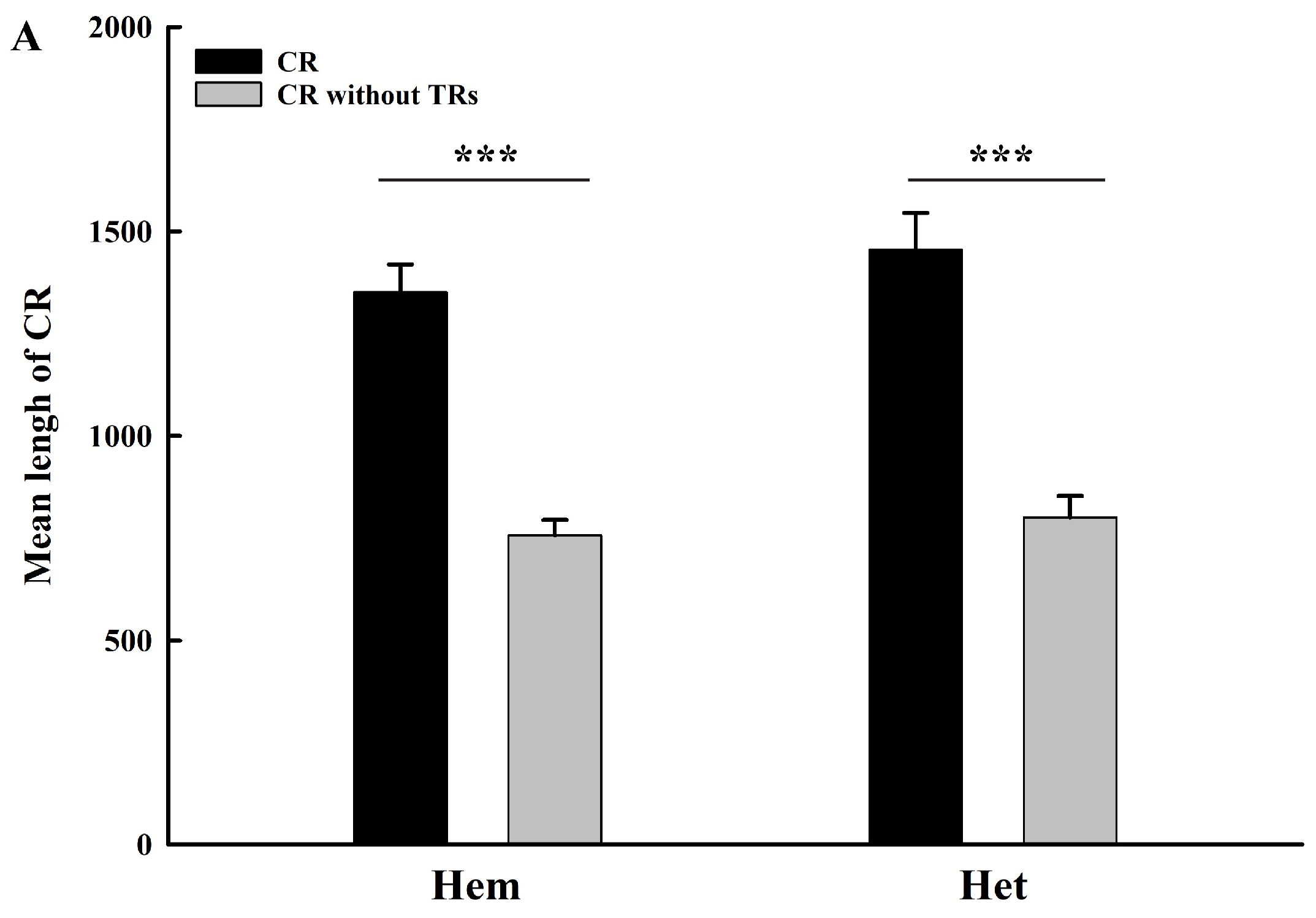
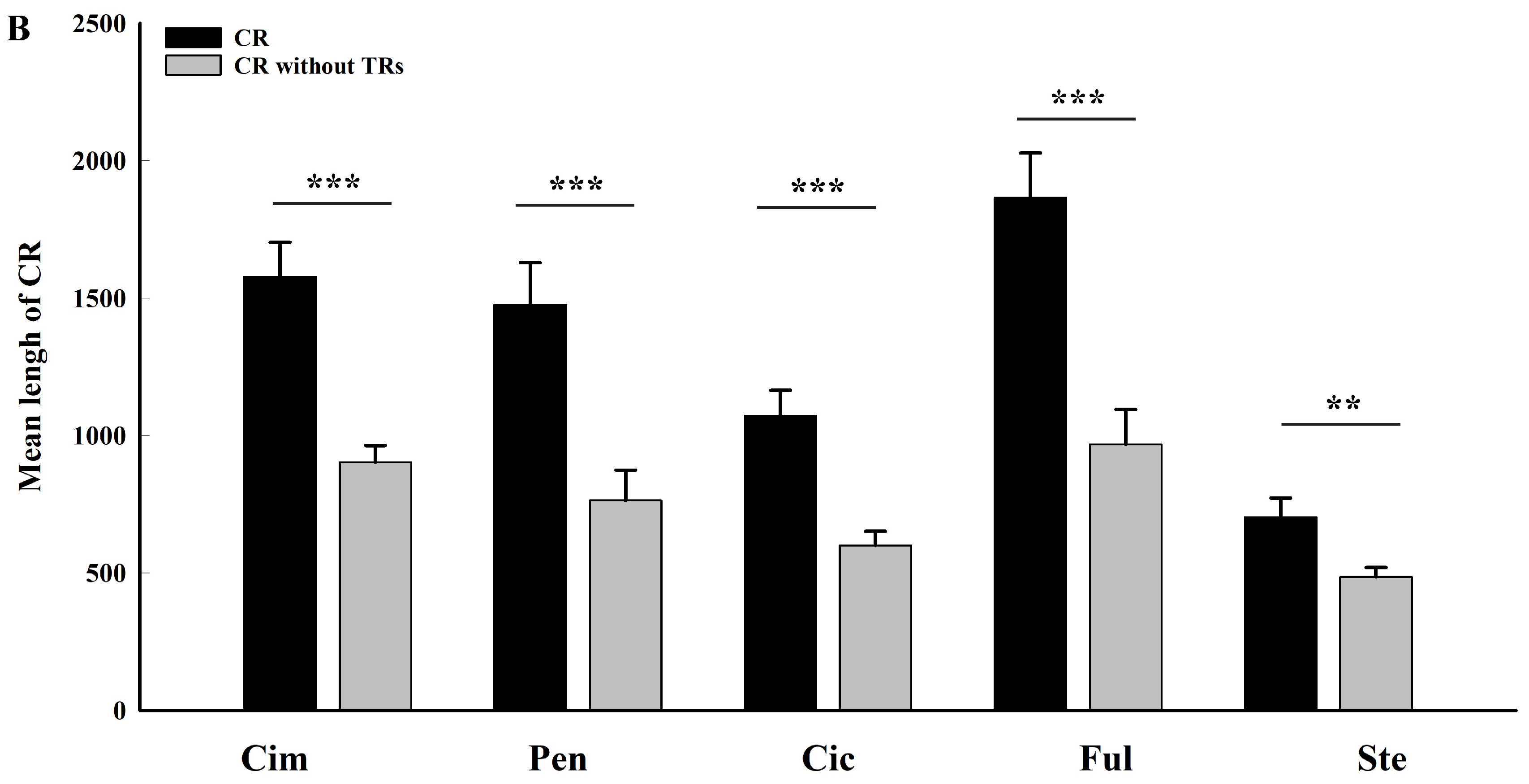
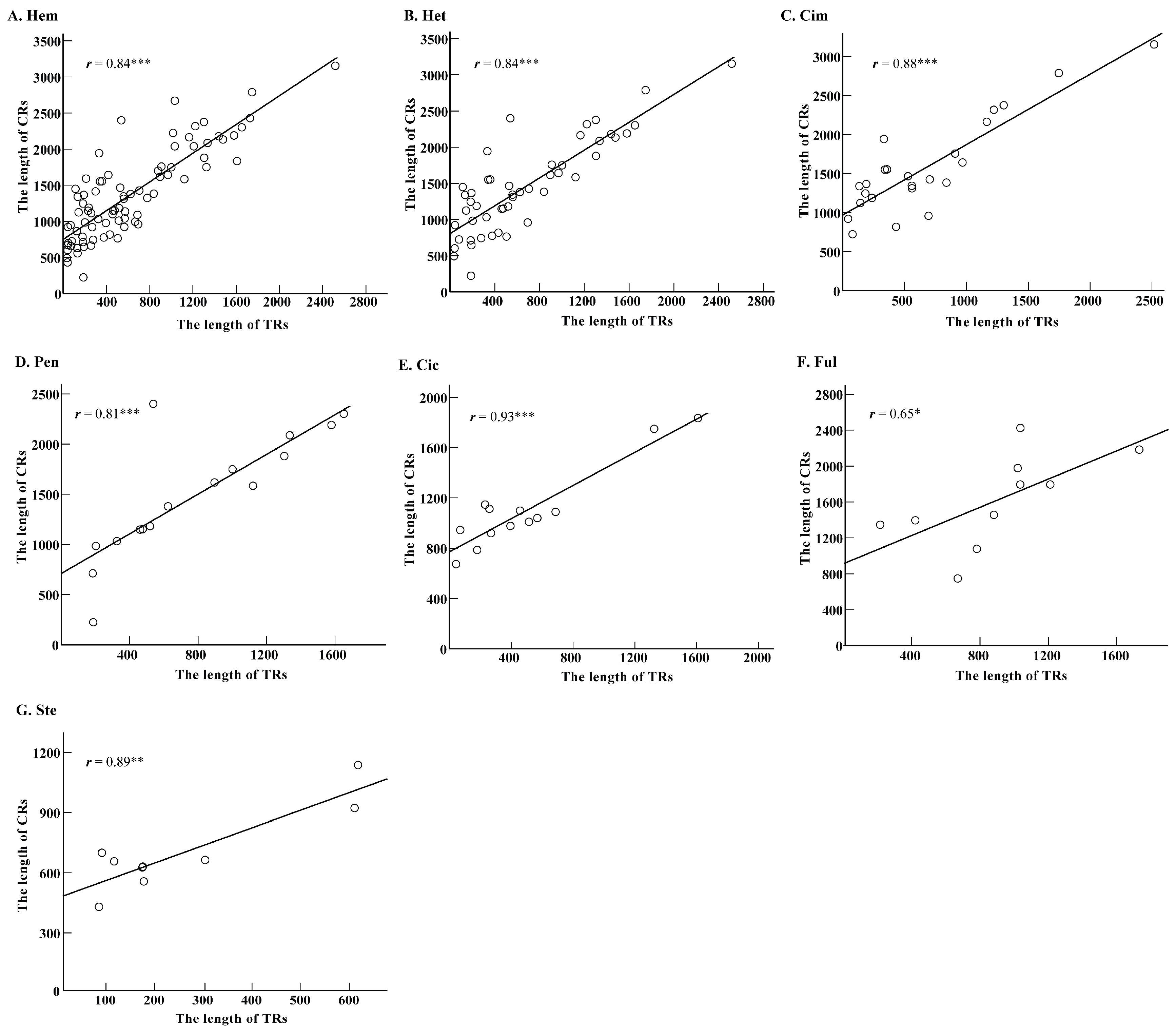
2.3. Roles of Tandem Replicationss on Length Variation of Control Region Sequences
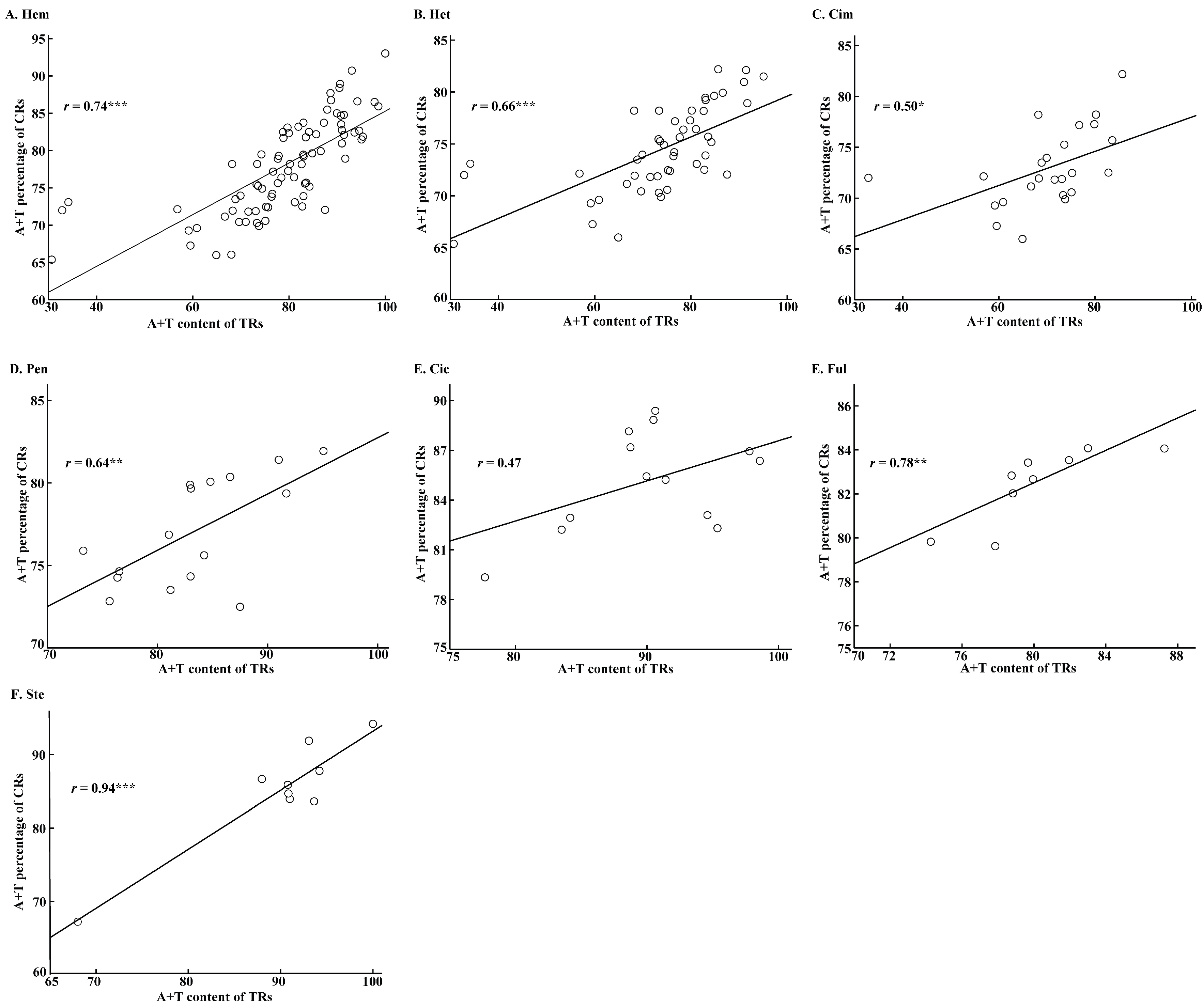
2.4. Roles of Tandem Replicationss on A+T Content and Base Skewing of Control Region Sequences
3. Discussion
3.1. The Conservation and Diversification of Control Regions in Hemiptera
3.2. The Potential Phylogenetic Utility of Control Regions in Hemiptera
3.3. Roles of Tandem Replications on Length Variation, AT Content, and Shift of Base Skewing in Hemiptera
4. Materials and Methods
4.1. Sequence Retrieval, Alignment, and Structural Analysis
4.2. Phylogenetic Reconstruction
4.3. Statistical Analyses
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| Cic | Cicadomorpha |
| Cim | Cimicomorpha |
| Col | Coleorrhyncha |
| CR | Control region |
| CSB | Conserved sequence block |
| Dip | Dipsocoromorpha |
| Eni | Enicocephalomorpha |
| Ful | Fulgoromorpha |
| Ger | Gerromorpha |
| Hem | Hemiptera |
| Het | Heteroptera |
| Lep | Leptopodomorpha |
| Pen | Pentatomomorpha |
| TR | Tandem repetition |
References
- Simon, C. Molecular Systematics at the Species Boundary: Exploiting Conserved and Variable Regions of the Mitochondrial Genome of Animals via Direct Sequencing from Amplified DNA; Springer: Berlin/Heidelberg, Germany, 1991; pp. 33–71. [Google Scholar]
- Clayton, D.A. Replication and transcription of vertebrate mitochondrial DNA. Annu. Rev. Cell Biol. 1991, 7, 453–478. [Google Scholar] [CrossRef] [PubMed]
- Carrodeguas, J.A.; Vallejo, C.G. Mitochondrial Transcription Initiation in the Crustacean Artemia franciscana. Eur. J. Biochem. 1997, 250, 514–523. [Google Scholar] [CrossRef] [PubMed]
- Shadel, G.S.; Clayton, D.A. Mitochondrial DNA maintenance in vertebrates. Annu. Rev. Biochem. 1997, 66, 409–435. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.X.; Hewitt, G.M. Insect mitochondrial control region: A review of its structure, evolution and usefulness in evolutionary studies. Biochem. Syst. Ecol. 1997, 25, 99–120. [Google Scholar] [CrossRef]
- Sbisà, E.; Tanzariello, F.; Reyes, A.; Pesole, G.; Saccone, C. Mammalian mitochondrial D-loop region structural analysis: Identification of new conserved sequences and their functional and evolutionary implications. Gene 1997, 205, 125–140. [Google Scholar] [CrossRef]
- Randi, E.; Lucchini, V. Organization and evolution of the mitochondrial DNA control region in the avian genus Alectoris. J. Mol. Evol. 1998, 47, 449–462. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, H.; Gao, T.; Miao, Z. Structure of mitochondrial DNA control region and molecular phylogenetic relationship among three flounders of genus Pleuronectes. Biochem. Syst. Ecol. 2011, 39, 627–634. [Google Scholar] [CrossRef]
- Goddard, J.M.; Wolstenholme, D.R. Origin and direction of replication in mitochondrial DNA molecules from the genus Drosophila. Nucleic Acids Res. 1980, 8, 741–757. [Google Scholar] [PubMed]
- Saito, S.; Tamura, K.; Aotsuka, T. Replication origin of mitochondrial DNA in insects. Genetics 2005, 171, 1695–1705. [Google Scholar] [CrossRef] [PubMed]
- Fenn, J.D.; Cameron, S.L.; Whiting, M.F. The complete mitochondrial genome sequence of the Mormon cricket (Anabrus simplex: Tettigoniidae: Orthoptera) and an analysis of control region variability. Insect Mol. Biol. 2007, 16, 239. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, J.; Jiang, L.; Qiao, G. Hemipteran Mitochondrial Genomes: Features, Structures and Implications for Phylogeny. Int. J. Mol. Sci. 2015, 16, 12382–12404. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.F.; Mckechnie, S.W.; Pierce, N.; Kreitman, M. The lepidopteran mitochondrial control region: Structure and evolution. Mol. Biol. Evol. 1993, 10, 1259–1272. [Google Scholar] [PubMed]
- Schultheis, A.S.; Weigt, L.A.; Hendricks, A.C. Arrangement and structural conservation of the mitochondrial control region of two species of Plecoptera: Utility of tandem repeat-containing regions in studies of population genetics and evolutionary history. Insect Mol. Biol. 2002, 11, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Dueñas, J.C.; Gardenal, C.N.; Llinás, G.A.; Panzetta-Dutari, G.M. Structural organization of the mitochondrial DNA control region in Aedes aegypti. Genome 2006, 49, 931–937. [Google Scholar] [CrossRef] [PubMed]
- Amaral, D.T.; Mitani, Y.; Oliveira, G.; Ohmiya, Y.; Viviani, V.R. Revisiting Coleoptera a + T-rich region: Structural conservation, phylogenetic and phylogeographic approaches in mitochondrial control region of bioluminescent Elateridae species (Coleoptera). Mitochondrial DNA Part A DNA Mapp. Seq. Anal. 2017, 28, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Solignac, M.; Monerot, M.; Mounolou, J.C. Concerted evolution of sequence repeats in Drosophila mitochondrial DNA. J. Mol. Evol. 1986, 24, 53–60. [Google Scholar] [CrossRef]
- Monforte, A.; Barrio, E.; Latorre, A. Characterization of the length polymorphism in the A+T-rich region of the Drosophila obscura group species. J. Mol. Evol. 1993, 36, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, M.T.; Azeredo-Espin, A.M.L.; Lessinger, A.C. The mitochondrial DNA control region of muscidae flies: Evolution and structural conservation in a dipteran context. J. Mol. Evol. 2007, 64, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Forero, D. The systematics of the Hemiptera. Rev. Colomb. Entomol. 2008, 34, 1–21. [Google Scholar]
- Carver, M.; Gross, G.F.; Woodward, T.E. Hemiptera (Bugs, leafhoppers, cicadas, aphids, scale insects etc.). In Systematic and Applied Entomology; Melbourne University Press: Carlton, Australia, 1994; pp. 429–509. [Google Scholar]
- Cryan, J.R.; Urban, J.M. Higher-level phylogeny of the insect order Hemiptera: Is Auchenorrhyncha really paraphyletic? Syst. Entomol. 2012, 37, 7–21. [Google Scholar] [CrossRef]
- Min, L.; Ying, T.; Ying, Z.; Bu, W.; Min, L.; Ying, T.; Ying, Z.; Bu, W. Higher Level Phylogeny and the First Divergence Time Estimation of Heteroptera (Insecta: Hemiptera) Based on Multiple Genes. PLoS ONE 2012, 7, e32152. [Google Scholar] [CrossRef]
- Ball, E.D. 3. The life cycle in hemiptera (exc1. aphids and coccids.). Ann. Entomol. Soc. Am. 1920, 13, 142–151. [Google Scholar] [CrossRef]
- Sanborn, A. Cicada Thermoregulation (Hemiptera, Cicadoidea); Biologiezentrum: Linz, Austria, 2002; pp. 455–470. [Google Scholar]
- Weirauch, C.; Schuh, R.T. Systematics and evolution of Heteroptera: 25 years of progress. Annu. Rev. Entomol. 2011, 56, 487–510. [Google Scholar] [CrossRef] [PubMed]
- Burrows, M. Jumping mechanisms in jumping plant lice (Hemiptera, Sternorrhyncha, Psyllidae). J. Exp. Biol. 2012, 215, 3612–3621. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Leavengood, J.M., Jr.; Chapman, E.G.; Burkhardt, D.; Song, F.; Jiang, P.; Liu, J.; Zhou, X.; Cai, W. Mitochondrial phylogenomics of Hemiptera reveals adaptive innovations driving the diversification of true bugs. Proc. Biol. Sci. 2017, 284, 20171223. [Google Scholar] [CrossRef] [PubMed]
- Cameron, S.L. Insect mitochondrial genomics: Implications for evolution and phylogeny. Annu. Rev. Entomol. 2014, 59, 95–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.X.; Szymura, J.M.; Hewitt, G.M. Evolution and structural conservation of the control region of insect mitochondrial DNA. J. Mol. Evol. 1995, 40, 382–391. [Google Scholar] [CrossRef] [PubMed]
- Gullan, P.J.; Cranston, P.S. The Insects: An Outline of Entomology, 4th ed.; Wiley-Blackwell: Oxford, UK, 2010. [Google Scholar]
- Weirauch, C.; Munro, J.B. Molecular phylogeny of the assassin bugs (Hemiptera: Reduviidae), based on mitochondrial and nuclear ribosomal genes. Mol. Phylogenet. Evol. 2009, 53, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Ortizrivas, B.; Martíneztorres, D. Combination of molecular data support the existence of three main lineages in the phylogeny of aphids (Hemiptera: Aphididae) and the basal position of the subfamily Lachninae. Mol. Phylogenet. Evol. 2010, 55, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, H.; Song, F.; Shi, A.; Zhou, X.; Cai, W. Comparative mitogenomic analysis of damsel bugs representing three tribes in the family nabidae (Insecta: Hemiptera). PLoS ONE 2012, 7, e45925. [Google Scholar] [CrossRef] [PubMed]
- Song, N.; Liang, A.P. A preliminary molecular phylogeny of planthoppers (Hemiptera: Fulgoroidea) based on nuclear and mitochondrial DNA sequences. PLoS ONE 2013, 8, e58400. [Google Scholar] [CrossRef] [PubMed]
- Song, N.; Cai, W.; Li, H. Deep-level phylogeny of Cicadomorpha inferred from mitochondrial genomes sequenced by NGS. Sci Rep. 2017, 7, 10429. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Hall, T. BioEdit Sequence Alignment Editor for Windows 95/98/NT/XP. In Ibis Therapeutics; A Division of Isis Pharmaceuticals; Isis Pharmaceuticals, Inc.: Carlsbad, CA, USA, 2005. [Google Scholar]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and high-performance computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Gascuel, O.; Rannala, B. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Bryman, A.; Cramer, D. Quantitative Data Analysis with IBM SPSS 17, 18 & 19: A Guide for Social Scientists; Routledge: New York, NY, USA, 2011; p. 497. [Google Scholar]
- Massey, F.J. The kolmogorov-smirnov test for goodness of fit. J. Am. Stat. Assoc. 1951, 46, 68–78. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | Best-Fit Model | −lnL | BIC | |
|---|---|---|---|---|
| Infraorder level | Cicadomorpha | HKY+G | 840.293 | 1883.474 |
| Cimicomorpha | HKY+G | 745.3419 | 1700.95 | |
| Coleorrhyncha | HKY | 227.3687 | 490.2786 | |
| Dipsocoromorpha | HKY | 160.6696 | 352.9492 | |
| Enicocephalomorpha | F81 | 251.9445 | 543.0995 | |
| Fulgoromorpha | HKY | 330.6916 | 745.6133 | |
| Nepomorpha | HKY | 111.504 | 264.2301 | |
| Sternorrhyncha | HKY+G | 811.118 | 1721.88 | |
| Family level | Alydidae | HKY | 197.9267 | 434.1398 |
| Aphididae | HKY+G | 792.3208 | 1664.487 | |
| Cercopidae | HKY | 258.9371 | 586.3942 | |
| Cicadellidae | HKY | 437.4311 | 964.5663 | |
| Cicadidae | F81 | 79.49639 | 182.3182 | |
| Delphacidae | F81 | 440.9577 | 937.0325 | |
| Lygaeoidea | F81 | 216.8583 | 464.3028 | |
| Miridae | F81 | 188.896 | 428.9057 | |
| Nabidae | HKY+G | 807.0794 | 1689.418 | |
| Pentatomiodea | HKY | 294.1281 | 669.885 | |
| Reduviidae | GTR+G | 1281.578 | 2712.585 | |
| Infraorder | A-Skew | T-Skew | G-Skew | C-Skew |
|---|---|---|---|---|
| Cim | -- | -- | √ | -- |
| Dip | -- | √ | -- | √ |
| Eni | -- | √ | -- | -- |
| Ger | √ | -- | √ | -- |
| Nep | √ | -- | √ | -- |
| Pen | -- | √ | √ | -- |
| Col | -- | √ | √ | -- |
| Cic | -- | √ | √ | -- |
| Ful | -- | √ | -- | -- |
| Ste | -- | -- | √ | -- |
| Infraorder | Species | AT-Skew | AT-Skew a | GC-Skew | GC-Skew b |
|---|---|---|---|---|---|
| Cim | Cimex lectularius | 0.01 | −0.08 | −0.14 | 0.40 |
| Cim | Corythucha ciliata | -- | -- | −0.1 | 0.01 |
| Cim | Gorpis annulatus | -- | -- | −0.02 | 0.10 |
| Cim | Gorpis humeralis | 0.01 | 0.00 | 0 | 0.09 |
| Cim | Himacerus apterus | -- | -- | −0.04 | 0.12 |
| Cim | Himacerus nodipes | -- | -- | −0.04 | 0.00 |
| Cim | Lygus hesperus | −0.07 | 0.04 | -- | -- |
| Cim | Lygus lineolaris | −0.01 | 0.04 | -- | -- |
| Cim | Nesidiocoris tenuis | −0.01 | 0.09 | -- | -- |
| Cim | Scotomedes sp. | 0.04 | −0.08 | -- | -- |
| Pen | Aradacanthia heissi | −0.03 | 0.06 | -- | -- |
| Pen | Dalcantha dilatata | −0.01 | 0.00 | -- | -- |
| Pen | Dolycoris baccarum | −0.06 | 0.00 | -- | -- |
| Pen | Dysdercus cingulatus | −0.18 | 0.04 | -- | -- |
| Pen | Nezara viridula | −0.09 | 0.03 | -- | -- |
| Pen | Sastragala edessoides | 0.02 | −0.01 | -- | -- |
| Pen | Urochela quadrinotata | 0.03 | −0.01 | -- | -- |
| Cic | Callitettix braconoides | -- | -- | −0.04 | 0.07 |
| Cic | Empoasca vitis | 0.01 | −0.01 | -- | -- |
| Cic | Homalodisca vitripennis | -- | -- | 0.01 | −0.02 |
| Cic | Leptobelus sp. | 0.01 | −0.02 | -- | -- |
| Cic | Yanocephalus yanonis | 0.01 | −0.05 | -- | -- |
| Ful | Nilaparvata lugens | -- | -- | −0.17 | 0.03 |
| Ful | Sogatella furcifera | 0.00 | 0.05 | −0.11 | 0.10 |
| Ste | Aleurocanthus spiniferus | 0.05 | −0.15 | −0.16 | 0.02 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, K.; Liang, A.-P. Hemiptera Mitochondrial Control Region: New Sights into the Structural Organization, Phylogenetic Utility, and Roles of Tandem Repetitions of the Noncoding Segment. Int. J. Mol. Sci. 2018, 19, 1292. https://doi.org/10.3390/ijms19051292
Li K, Liang A-P. Hemiptera Mitochondrial Control Region: New Sights into the Structural Organization, Phylogenetic Utility, and Roles of Tandem Repetitions of the Noncoding Segment. International Journal of Molecular Sciences. 2018; 19(5):1292. https://doi.org/10.3390/ijms19051292
Chicago/Turabian StyleLi, Kui, and Ai-Ping Liang. 2018. "Hemiptera Mitochondrial Control Region: New Sights into the Structural Organization, Phylogenetic Utility, and Roles of Tandem Repetitions of the Noncoding Segment" International Journal of Molecular Sciences 19, no. 5: 1292. https://doi.org/10.3390/ijms19051292
APA StyleLi, K., & Liang, A. -P. (2018). Hemiptera Mitochondrial Control Region: New Sights into the Structural Organization, Phylogenetic Utility, and Roles of Tandem Repetitions of the Noncoding Segment. International Journal of Molecular Sciences, 19(5), 1292. https://doi.org/10.3390/ijms19051292