Cancer Immunotherapy: A Focus on the Regulation of Immune Checkpoints

 ,
,
Abstract
:
1. Introduction
2. Regulation of Immune Checkpoints
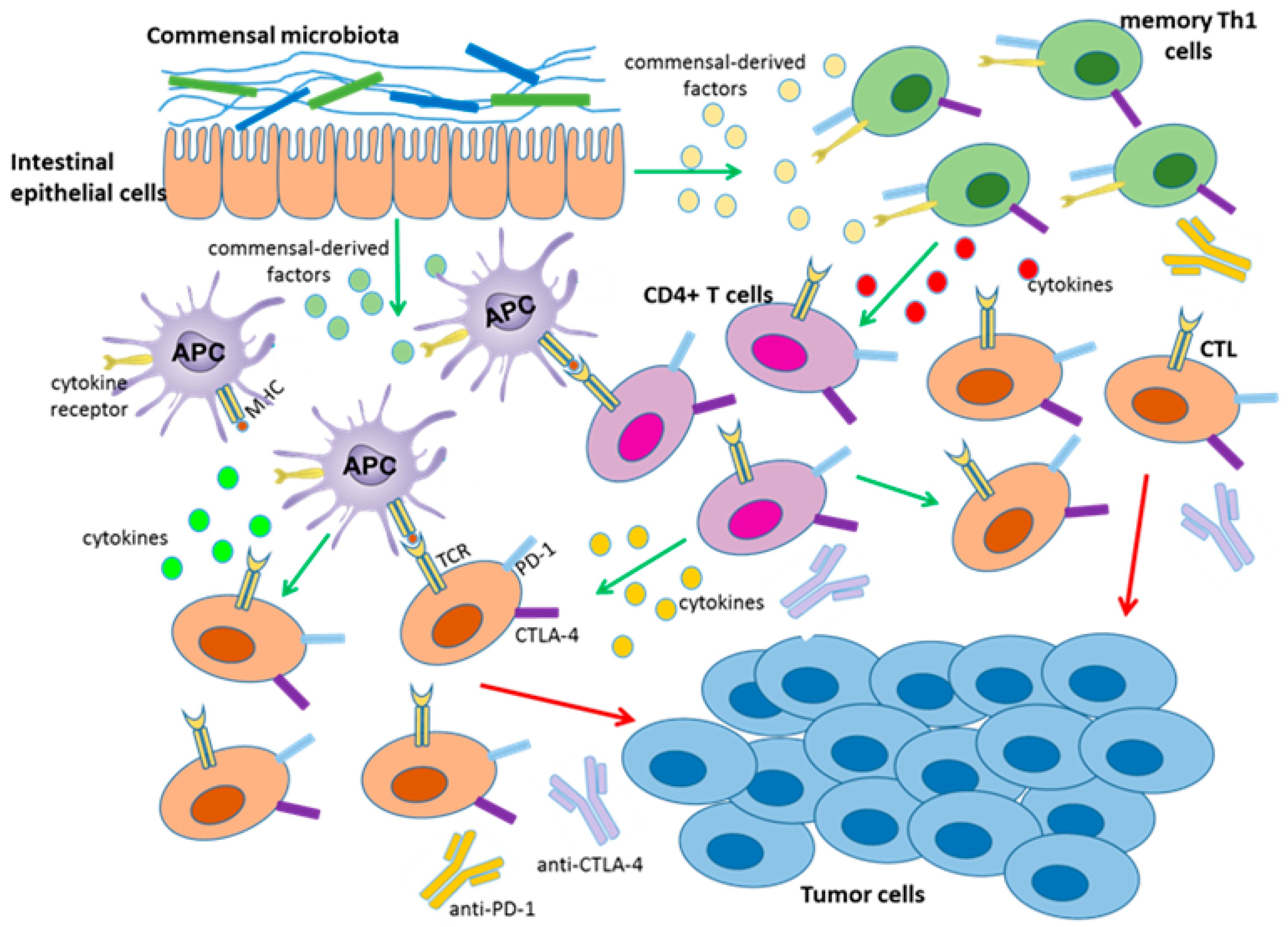
2.1. Regulation Mediated by Myeloid and Lymphoid Cells in the Tumor Microenvironment
2.2. Regulation Mediated by Oncolytic Viruses
2.3. Regulation Mediated by Epigenetics
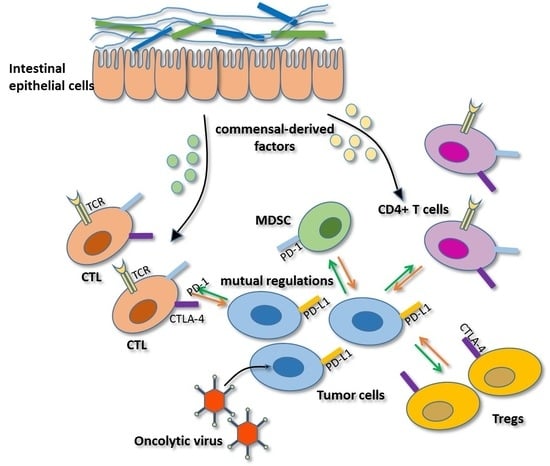
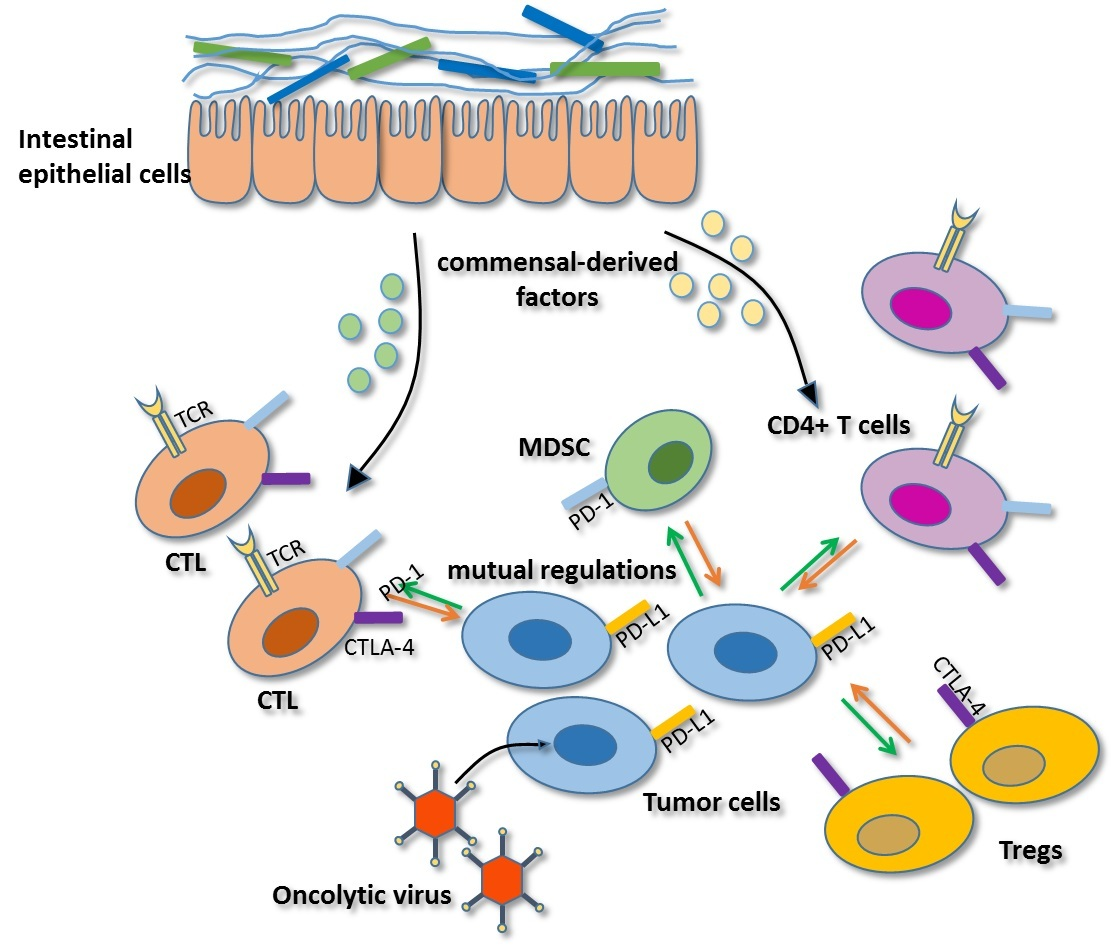
2.4. Regulation Mediated by Gut Microbiota
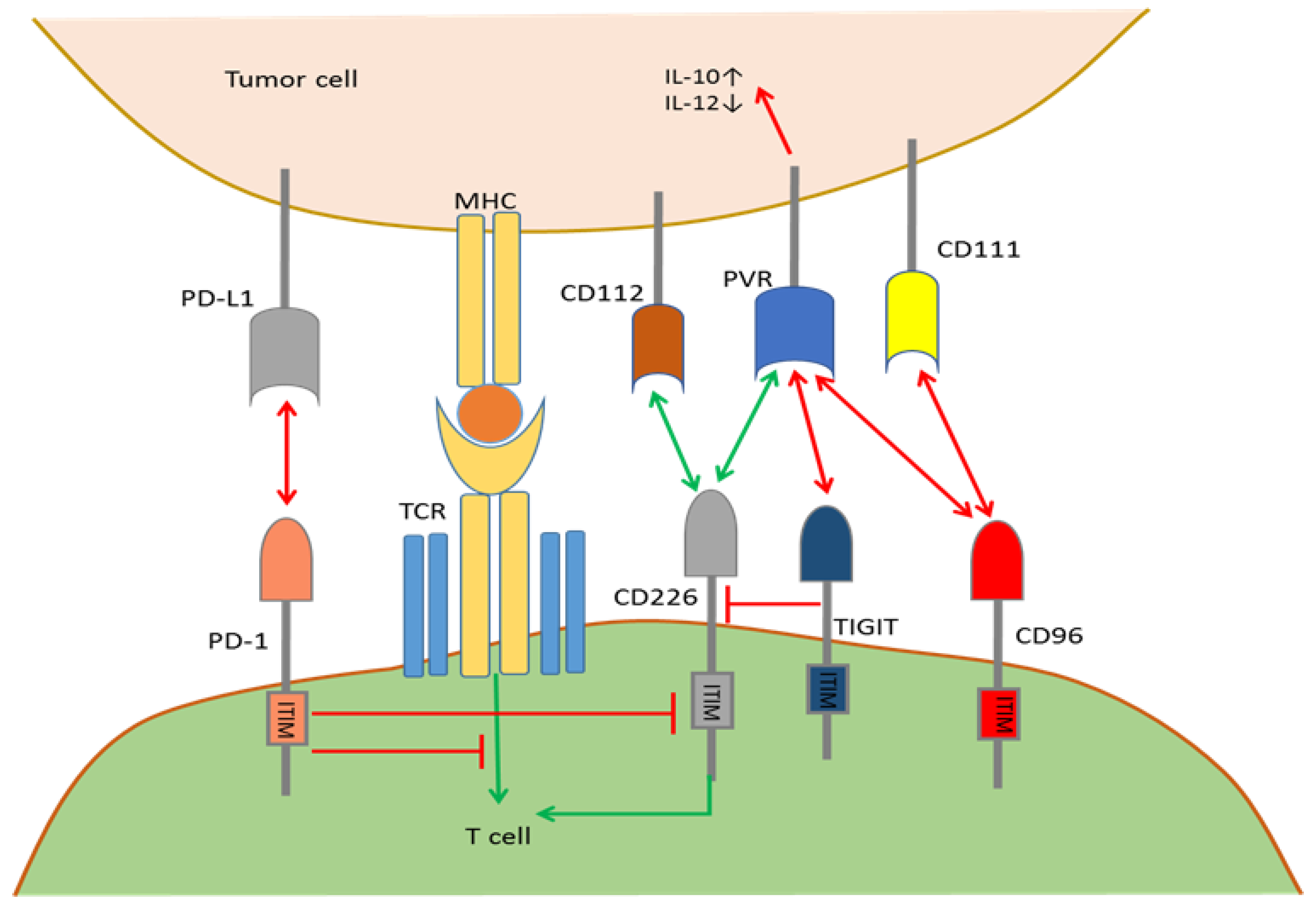
2.5. Mutual Regulation among Immune Checkpoints
3. Future Directions for Personalized Cancer Immunotherapy
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Couzin-Frankel, J. Cancer immunotherapy. Science 2013, 342, 1432–1433. [Google Scholar] [CrossRef] [PubMed]
- Mahoney, K.M.; Freeman, G.J.; McDermott, D.F. The next immune-checkpoint inhibitors: PD-1/PD-L1 blockade in melanoma. Clin. Ther. 2015, 37, 764–782. [Google Scholar] [CrossRef] [PubMed]
- Ott, P.A.; Hodi, F.S.; Robert, C. CTLA-4 and PD-1/PD-L1 blockade: New immunotherapeutic modalities with durable clinical benefit in melanoma patients. Clin. Cancer Res. 2013, 19, 5300–5309. [Google Scholar] [CrossRef] [PubMed]
- Lozano, E.; Dominguez-Villar, M.; Kuchroo, V.; Hafler, D.A. The TIGIT/CD226 axis regulates human T cell function. J. Immunol. 2012, 188, 3869–3875. [Google Scholar] [CrossRef] [PubMed]
- Durham, N.M.; Nirschl, C.J.; Jackson, C.M.; Elias, J.; Kochel, C.M.; Anders, R.A.; Drake, C.G. Lymphocyte activation Gene 3 (LAG-3) modulates the ability of CD4 T-cells to be suppressed in vivo. PLoS ONE 2014, 9, e109080. [Google Scholar] [CrossRef] [PubMed]
- Thaker, Y.R.; Schneider, H.; Rudd, C.E. TCR and CD28 activate the transcription factor NF-κB in T-cells via distinct adaptor signaling complexes. Immunol. Lett. 2015, 163, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Nurieva, R.; Thomas, S.; Nguyen, T.; Martin-Orozco, N.; Wang, Y.; Kaja, M.K.; Yu, X.Z.; Dong, C. T-cell tolerance or function is determined by combinatorial costimulatory signals. EMBO J. 2006, 25, 2623–2633. [Google Scholar] [CrossRef] [PubMed]
- Lipson, E.J.; Drake, C.G. Ipilimumab: An anti-CTLA-4 antibody for metastatic melanoma. Clin. Cancer Res. 2011, 17, 6958–6962. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Sznol, M.; McDermott, D.F.; Kluger, H.M.; Carvajal, R.D.; Sharfman, W.H.; Brahmer, J.R.; Lawrence, D.P.; Atkins, M.B.; Powderly, J.D.; et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J. Clin. Oncol. 2014, 32, 1020–1030. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.S.; D’Angelo, S.P.; Minor, D.; Hodi, F.S.; Gutzmer, R.; Neyns, B.; Hoeller, C.; Khushalani, N.I.; Miller, W.H., Jr.; Lao, C.D.; et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): A randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015, 16, 375–384. [Google Scholar] [CrossRef]
- Catakovic, K.; Gassner, F.J.; Ratswohl, C.; Zaborsky, N.; Rebhandl, S.; Schubert, M.; Steiner, M.; Gutjahr, J.C.; Pleyer, L.; Egle, A.; et al. TIGIT expressing CD4+ T cells represent a tumor-supportive T cell subset in chronic lymphocytic leukemia. Oncoimmunology 2017, 7, e1371399. [Google Scholar] [CrossRef] [PubMed]
- Fleury, M.; Belkina, A.C.; Proctor, E.A.; Zammitti, C.; Simms, R.W.; Lauffenburger, D.A.; Snyder-Cappione, J.E.; Lafyatis, R.; Dooms, H. Increased expression and modulated regulatory activity of co-inhibitory receptors PD-1, TIGIT and TIM-3 in lymphocytes of systemic sclerosis patients. Arthritis Rheumatol. 2018, 70, 566–577. [Google Scholar] [CrossRef] [PubMed]
- Lines, J.L.; Pantazi, E.; Mak, J.; Sempere, L.F.; Wang, L.; O’Connell, S.; Ceeraz, S.; Suriawinata, A.A.; Yan, S.; Ernstoff, M.S.; et al. VISTA is an immune checkpoint molecule for human T cells. Cancer Res. 2014, 74, 1924–1932. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; Yang, S.; Gu, A.; Zhan, F.; He, C.; Qin, C.; Zhang, X.; Feng, P. Murine b7-h3 is a co-stimulatory molecule for T cell activation. Monoclon. Antib. Immunodiagn. Immunother. 2013, 32, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Altan, M.; Pelekanou, V.; Schalper, K.A.; Toki, M.; Gaule, P.; Syrigos, K.; Herbst, R.S.; Rimm, D.L. B7-H3 expression in NSCLC and its association with B7-H4, PD-L1 and tumor-infiltrating lymphocytes. Clin. Cancer Res. 2017, 23, 5202–5209. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Martin-Orozco, N.; Zheng, P.; Li, J.; Zhang, P.; Tan, H.; Park, H.J.; Jeong, M.; Chang, S.H.; Kim, B.S.; et al. Inhibition of the B7-H3 immune checkpoint limits tumor growth by enhancing cytotoxic lymphocyte function. Cell Res. 2017, 27, 1034–1045. [Google Scholar] [CrossRef] [PubMed]
- Elpek, K.G.; Cremasco, V.; Shen, H.; Harvey, C.J.; Wucherpfennig, K.W.; Goldstein, D.R.; Monach, P.A.; Turley, S.J. The tumor microenvironment shapes lineage, transcriptional, and functional diversity of infiltrating myeloid cells. Cancer Immunol. Res. 2014, 2, 655–667. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, T.; Qian, B.Z.; Soong, D.; Cassetta, L.; Noy, R.; Sugano, G.; Kato, Y.; Li, J.; Pollard, J.W. CCL2-induced chemokine cascade promotes breast cancer metastasis by enhancing retention of metastasis-associated macrophages. J. Exp. Med. 2015, 212, 1043–1059. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Lei, Z.; Zhao, J.; Gong, W.; Liu, J.; Chen, Z.; Liu, Y.; Li, D.; Yuan, Y.; Zhang, G.M.; et al. CCL2/CCR2 pathway mediates recruitment of myeloid suppressor cells to cancers. Cancer Lett. 2007, 252, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Skora, A.D.; Li, Z.; Liu, Q.; Tam, A.J.; Blosser, R.L.; Diaz, L.A., Jr.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B.; et al. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc. Natl. Acad. Sci. USA 2014, 111, 11774–11779. [Google Scholar] [CrossRef] [PubMed]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Ridder, K.; Sevko, A.; Heide, J.; Dams, M.; Rupp, A.K.; Macas, J.; Starmann, J.; Tjwa, M.; Plate, K.H.; Sültmann, H.; et al. Extracellular vesicle-mediated transfer of functional RNA in the tumor microenvironment. Oncoimmunology 2015, 4, e1008371. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, M.; Ramirez, M.E.; Sierra, R.A.; Raber, P.; Thevenot, P.; Al-Khami, A.A.; Sanchez-Pino, D.; Hernandez, C.; Wyczechowska, D.D.; Ochoa, A.C.; et al. l-Arginine depletion blunts antitumor T-cell responses by inducing myeloid-derived suppressor cells. Cancer Res. 2015, 75, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Redd, P.S.; Ibrahim, M.L.; Klement, J.D.; Sharman, S.K.; Paschall, A.V.; Yang, D.; Nayak-Kapoor, A.; Liu, K. SETD1B activates iNOS expression in myeloid-derived suppressor cells. Cancer Res. 2017, 77, 2834–2843. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F.R. The chemokine system and cancer. J. Pathol. 2012, 226, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.R.; Maute, R.L.; Dulken, B.W.; Hutter, G.; George, B.M.; McCracken, M.N.; Gupta, R.; Tsai, J.M.; Sinha, R.; Corey, D.; et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 2017, 545, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Arlauckas, S.P.; Garris, C.S. In vivo imaging reveals a tumor-associated macrophage-mediated resistance pathway in anti-PD-1 therapy. Sci. Transl. Med. 2017, 9, e3604. [Google Scholar] [CrossRef] [PubMed]
- Akbay, E.A.; Koyama, S.; Liu, Y.; Dries, R.; Bufe, L.E.; Silkes, M.; Alam, M.M.; Magee, D.M.; Jones, R.; Jinushi, M.; et al. Interleukin-17A Promotes Lung Tumor Progression through Neutrophil Attraction to Tumor Sites and Mediating Resistance to PD-1 Blockade. J. Thorac. Oncol. 2017, 12, 1268–1279. [Google Scholar] [CrossRef] [PubMed]
- Luo, Q.; Huang, Z.; Ye, J.; Deng, Y.; Fang, L.; Li, X.; Guo, Y.; Jiang, H.; Ju, B.; Huang, Q.; et al. PD-L1-expressing neutrophils as a novel indicator to assess disease activity and severity of systemic lupus erythematosus. Arthritis Res. Ther. 2016, 18, 47. [Google Scholar] [CrossRef] [PubMed]
- Hori, S.; Nomura, T.; Sakaguchi, S. Control of regulatory T cell development by the transcription factor Foxp3. Science 2003, 299, 1057–1061. [Google Scholar] [CrossRef] [PubMed]
- Tai, X.; Van Laethem, F.; Pobezinsky, L. Basis of CTLA-4 function in regulatory and conventional CD4+ T cells. Blood 2012, 119, 5155–5163. [Google Scholar] [CrossRef] [PubMed]
- Wing, J.B.; Ise, W.; Kurosaki, T.; Sakaguchi, S. Regulatory T cells control antigen-specific expansion of Tfh cell number and humoral immune responses via the coreceptor CTLA-4. Immunity 2014, 41, 1013–1025. [Google Scholar] [CrossRef] [PubMed]
- Hannani, D.; Vétizou, M.; Enot, D. Anticancer immunotherapy by CTLA-4 blockade: Obligatory contribution of IL-2 receptors and negative prognostic impact of soluble CD25. Cell Res. 2015, 25, 208–224. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Park, J.S.; Jeong, Y.H.; Son, J.; Ban, Y.H.; Lee, B.H.; Chen, L.; Chang, J.; Chung, D.H.; Choi, I. PD-1 upregulated on regulatory T cells during chronic virus infection enhances the suppression of CD8+ T cell immune response via the interaction with PD-L1 expressed on CD8+ T cells. J. Immunol. 2015, 194, 5801–5811. [Google Scholar] [CrossRef] [PubMed]
- Joller, N.; Lozano, E.; Burkett, P.R.; Patel, B.; Xiao, S.; Zhu, C.; Xia, J.R.; Tan, T.G.; Sefik, E.; Yajnik, V. Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity 2014, 40, 569–5681. [Google Scholar] [CrossRef] [PubMed]
- Andtbacka, R.H.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef] [PubMed]
- Garber, K. China approves world’s first oncolytic virus therapy for cancer treatment. J. Natl. Cancer Inst. 2006, 98, 298–300. [Google Scholar] [CrossRef] [PubMed]
- Engeland, C.E.; Grossardt, C.; Veinaide, R.; Bossow, S.; Lutz, D.; Kaufmann, J.K.; Shevchenko, I.; Umansky, V.; Nettelbeck, D.M.; Weichert, W. CTLA-4 and PD-L1 checkpoint blockade enhances oncolytic measles virus therapy. Mol. Ther. 2014, 22, 1949–1959. [Google Scholar] [CrossRef] [PubMed]
- Bartee, M.Y.; Dunlap, K.M.; Bartee, E. Tumor-localized secretion of soluble PD1 enhances oncolytic virotherapy. Cancer Res. 2017, 77, 2952–2963. [Google Scholar] [CrossRef] [PubMed]
- Rojas, J.J.; Sampath, P.; Hou, W.Z.; Thorne, S.H. Defining Effective Combinations of Immune Checkpoint Blockade and Oncolytic Virotherapy. Clin. Cancer Res. 2015, 21, 5543–5551. [Google Scholar] [CrossRef] [PubMed]
- Quetglas, J.I.; Labiano, S.; Aznar, M.A.; Bolanos, E.; Azpilikueta, A.; Rodriguez, I.; Casales, E.; Sanchez-Paulete, A.R.; Segura, V.; Smerdou, C.; et al. Virotherapy with a semliki forest virus-based vector encoding IL12 synergizes with PD-1/PD-L1 blockade. Cancer Immunol. Res. 2015, 3, 449–454. [Google Scholar] [CrossRef] [PubMed]
- Puzanov, I.; Milhem, M.M.; Minor, D.; Hamid, O.; Li, A.; Chen, L.S.; Chastain, M.; Gorski, K.S.; Anderson, A.; Chou, J. Talimogene laherparepvec in combination with ipilimumab in previously untreated, unresectable stage IIIB-IV melanoma. J. Clin. Oncol. 2016, 34, 2619–2626. [Google Scholar] [CrossRef] [PubMed]
- Woller, N.; Gurlevik, E.; Fleischmann-Mundt, B.; Schumacher, A.; Knocke, S.; Kloos, A.M.; Saborowski, M.; Geffers, R.; Manns, M.P.; Wirth, T.C. Viral infection of tumors overcomes resistance to PD-1-immunotherapy by broadening neoantigenome-directed T-cell responses. Mol. Ther. 2015, 23, 1630–1640. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.A.; Kouzarides, T. Cancer epigenetics: from mechanism to therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Woods, D.M.; Sodre, A.L.; Villagra, A.; Sarnaik, A.; Sotomayor, E.M.; Weber, J. HDAC inhibition upregulates PD-1 ligands in melanoma and augments immunotherapy with PD-1 blockade. Cancer Immunol. Res. 2015, 3, 1375–1385. [Google Scholar] [CrossRef] [PubMed]
- Goswami, S.; Chen, J.F.; Zhao, H.; Zhang, X.J.; Sharma, P. Abstract 4026: Epigenetic changes in T cells in response to immune checkpoint blockade. Cancer Res. 2016, 76, 4026. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, L.Q.; Predina, J.; Han, R.X.; Beier, U.H.; Wang, L.C.S.; Kapoor, V.; Bhatti, T.R.; Akimova, T.; Singhal, S. Inhibition of p300 impairs Foxp3+ T regulatory cell function and promotes antitumor immunity. Nat. Med. 2013, 19, 1173–1177. [Google Scholar] [CrossRef] [PubMed]
- Wrangle, J.; Wang, W.; Koch, A.; Easwaran, H.; Mohammad, H.P.; Vendetti, F.; VanCriekinge, W.; De Meyer, T.; Du, Z.Z.; Parsana, P. Alterations of immune response of non-small cell lung cancer with azacytidine. Oncotarget 2013, 4, 2067–2079. [Google Scholar] [CrossRef] [PubMed]
- Chiappinelli, K.B.; Strissel, P.L.; Desrichard, A.; Li, H.L.; Henke, C.; Akman, B.; Hein, A.; Rote, N.S.; Cope, L.M.; Snyder, A. Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell 2015, 162, 974–986. [Google Scholar] [CrossRef] [PubMed]
- Roulois, D.; Yau, H.L.; Singhania, R.; Wang, Y.D.; Danesh, A.; Shen, S.Y.; Han, H.; Liang, G.N.; Jones, P.A.; Pugh, T.J. DNA-demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell 2015, 162, 961–973. [Google Scholar] [CrossRef] [PubMed]
- Peng, D.; Kryczek, I.; Nagarsheth, N.; Zhao, L.L.; Wei, S.; Wang, W.M.; Sun, Y.Q.; Zhao, E.D.; Vatan, L.; Szeliga, W. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 2015, 527, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Nduom, E.K.; Kong, L.Y.; Hashimoto, Y.; Xu, S.; Gabrusiewicz, K.; Ling, X.Y.; Huang, N.; Qiao, W.; Zhou, S.H. MiR-138 exerts anti-glioma efficacy by targeting immune checkpoints. Neuro. Oncol. 2016, 18, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Gibbons, D.L.; Goswami, S.; Cortez, M.A.; Ahn, Y.H.; Byers, L.A.; Zhang, X.J.; Yi, X.H.; Dwyer, D.; Lin, W. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat. Commun. 2014, 5, 5241. [Google Scholar] [CrossRef] [PubMed]
- Iwamura, K.; Kato, T.; Miyahara, Y.; Naota, H.; Mineno, J.; Ikeda, H.; Shiku, H. siRNA-mediated silencing of PD-1 ligands enhances tumor-specific human T-cell effector functions. Gene Ther. 2012, 19, 959–966. [Google Scholar] [CrossRef] [PubMed]
- Belkaid, Y.; Hand, T.W. Role of the microbiota in immunity and inflammation. Cell 2014, 157, 121–141. [Google Scholar] [CrossRef] [PubMed]
- Iida, N.; Dzutsev, A.; Stewart, C.A.; Smith, L.; Bouladoux, N.; Weingarten, R.A.; Molina, D.A.; Salcedo, R.; Back, T.; Cramer, S. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science 2013, 342, 967–970. [Google Scholar] [CrossRef] [PubMed]
- Sivan, A.; Corrales, L.; Hubert, N.; Williams, J.B.; Aquino-Michaels, K.; Earley, Z.M.; Benyamin, F.W.; Lei, Y.M.; Jabri, B.; Alegre, M.L. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 2015, 350, 1084–1089. [Google Scholar] [CrossRef] [PubMed]
- Vetizou, M.; Pitt, J.M.; Daillere, R.; Lepage, P.; Waldschmitt, N.; Flament, C.; Rusakiewicz, S.; Routy, B.; Roberti, M.P.; Duong, C.P.M. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 2015, 350, 1079–1084. [Google Scholar] [CrossRef] [PubMed]
- Belkaid, Y.; Bouladoux, N.; Hand, T.W. Effector and memory T cell responses to commensal bacteria. Trends Immunol. 2013, 34, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, S.; Erturk-Hasdemir, D.; Ochoa-Reparaz, J.; Reinecker, H.C.; Kasper, D.L. Plasmacytoid dendritic cells mediate anti-inflammatory responses to a gut commensal molecule via both innate and adaptive mechanisms. Cell Host Microbe 2014, 15, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Kwon, E.D.; Drake, C.G.; Scher, H.I.; Fizazi, K.; Bossi, A.; van den Eertwegh, A.J.M.; Krainer, M.; Houede, N.; Santos, R.; Mahammedi, H. Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184-043): A multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol. 2014, 15, 700–712. [Google Scholar] [CrossRef]
- Johnston, R.J.; Comps-Agrar, L.; Hackney, J.; Yu, X.; Huseni, M.; Yang, Y.; Park, S.; Javina, V.; Chiu, H.; Irving, B. The immunoreceptor TIGIT regulates antitumor and antiviral CD8+ T cell effector function. Cancer Cell 2014, 26, 923–937. [Google Scholar] [CrossRef] [PubMed]
- Stanietsky, N.; Simic, H.; Arapovic, J.; Toporik, A.; Levy, O.; Novik, A.; Levine, Z.; Beiman, M.; Dassa, L.; Achdout, H. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 17858–17863. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Harden, K.; Gonzalez, L.C.; Francesco, M.; Chiang, E.; Irving, B.; Tom, I.; Ivelja, S.; Refino, C.J.; Clark, H. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat. Immunol. 2009, 10, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Meyer, D.; Seth, S.; Albrecht, J.; Maier, M.K.; du Pasquier, L.; Ravens, I.; Dreyer, L.; Burger, R.; Gramatzki, M.; Schwinzer, R. CD96 interaction with CD155 via its first Ig-like domain is modulated by alternative splicing or mutations in distal Ig-like domains. J. Biol. Chem. 2009, 284, 2235–2244. [Google Scholar] [CrossRef] [PubMed]
- Foks, A.C.; Ran, I.A.; Frodermann, V.; Bot, I.; van Santbrink, P.J.; Kuiper, J.; van Puijvelde, G.H.M. Agonistic anti-TIGIT treatment inhibits T cell responses in LDLr deficient mice without affecting atherosclerotic lesion development. PLoS ONE 2013, 8, e83134. [Google Scholar] [CrossRef] [PubMed]
- Stanietsky, N.; Rovis, T.L.; Glasner, A.; Seidel, E.; Tsukerman, P.; Yamin, R.; Enk, J.; Jonjic, S.; Mandelboim, O. Mouse TIGIT inhibits NK-cell cytotoxicity upon interaction with PVR. Eur. J. Immunol. 2013, 43, 2138–2150. [Google Scholar] [CrossRef] [PubMed]
- Chauvin, J.M.; Pagliano, O.; Fourcade, J.; Sun, Z.J.; Wang, H.; Sander, C.; Kirkwood, J.M.; Chen, T.H.T.; Maurer, M.; Korman, A.J. TIGIT and PD-1 impair tumor antigen-specific CD8+ T cells in melanoma patients. J. Clin. Investig. 2015, 125, 2046–2058. [Google Scholar] [CrossRef] [PubMed]
- Seth, S.; Maier, M.K.; Qiu, Q.; Ravens, I.; Kremmer, E.; Forster, R.; Bernhardt, G. The murine pan T cell marker CD96 is an adhesion receptor for CD155 and nectin-1. Biochem. Biophys. Res. Commun. 2007, 364, 959–965. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Ward, J.F.; Pettaway, C.A.; Shi, L.Z.; Subudhi, S.K.; Vence, L.M.; Zhao, H.; Chen, J.F.; Chen, H.; Efstathiou, E. VISTA is an inhibitory immune checkpoint that is increased after ipilimumab therapy in patients with prostate cancer. Nat. Med. 2017, 23, 551–555. [Google Scholar] [CrossRef] [PubMed]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Herter-Sprie, G.S.; Buczkowski, K.A.; Richards, W.G.; Gandhi, L.; Redig, A.J.; Rodig, S.J.; Asahina, H. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 2016, 7, 10501. [Google Scholar] [CrossRef] [PubMed]
- Teng, M.W.; Ngiow, S.F.; Ribas, A.; Smyth, M.J. Classifying Cancers Based on T-cell Infiltration and PD-L1. Cancer Res. 2015, 75, 2139–2145. [Google Scholar] [CrossRef] [PubMed]
- Obeid, J.M.; Erdag, G.; Smolkin, M.E.; Deacon, D.H.; Patterson, J.W.; Chen, L.P.; Bullock, T.N.; Slingluff, C.L. PD-L1, PD-L2 and PD-1 expression in metastatic melanoma: Correlation with tumor-infiltrating immune cells and clinical outcome. Oncoimmunology 2016, 5, e1235107. [Google Scholar] [CrossRef] [PubMed]
- Taube, J.M.; Klein, A.; Brahmer, J.R.; Xu, H.Y.; Pan, X.Y.; Kim, J.H.; Chen, L.P.; Pardoll, D.M.; Topalian, S.L.; Anders, R.A. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin. Cancer Res. 2014, 20, 5064–5074. [Google Scholar] [CrossRef] [PubMed]
- Krieg, C.; Nowicka, M.; Guglietta, S.; Schindler, S.; Hartmann, F.J.; Weber, L.M.; Dummer, R.; Robinson, M.D.; Levesque, M.P.; Becher, B. High-dimensional single-cell analysis predicts response to anti-PD-1 immunotherapy. Nat. Med. 2018, 24, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, E.A.; Gore, S.D. DNA methyltransferase and histone deacetylase inhibitors in the treatment of myelodysplastic syndromes. Semin. Hematol. 2008, 45, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Bulliard, Y.; Jolicoeur, R.; Zhang, J.M.; Dranoff, G.; Wilson, N.S.; Brogdon, J.L. OX40 engagement depletes intratumoral Tregs via activating FcgammaRs, leading to antitumor efficacy. Immunol. Cell Biol. 2014, 92, 475–480. [Google Scholar] [CrossRef] [PubMed]
- Sukumar, S.; Wilson, D.C.; Yu, Y.; Wong, J.; Naravula, S.; Ermakov, G.; Riener, R.; Bhagwat, B.; Necheva, A.S.; Grein, J. Characterization of MK-4166, a clinical agonistic antibody that targets human GITR and inhibits the generation and suppressive effects of T regulatory cells. Cancer Res. 2017, 77, 4378–4388. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, D.L.; Liu, Z.Q.; Sathaiah, M.; Ravindranathan, R.; Guo, Z.B.; He, Y.K.; Guo, Z.S. Oncolytic viruses as therapeutic cancer vaccines. Mol. Cancer 2013, 12, 103. [Google Scholar] [CrossRef] [PubMed]
- Pol, J.G.; Zhang, L.; Bridle, B.W.; Stephenson, K.B.; Resseguier, J.; Hanson, S.; Chen, L.; Kazdhan, N.; Bramson, J.L.; Stojdl, D.F. Maraba virus as a potent oncolytic vaccine vector. Mol. Ther. 2014, 22, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Viaud, S.; Saccheri, F.; Mignot, G.; Yamazaki, T.; Daillere, R.; Hannani, D.; Enot, D.P.; Pfirschke, C.; Engblom, C.; Pittet, M.J. The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science 2013, 342, 971–976. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Viruses | Target Checkpoints | Modifications | Cancers Selected for Clinical Trials |
|---|---|---|---|
| Measles virus (MV) | CTLA-4, PD-1 | α-PD-1 and α-CTLA-4 encoding | Melanoma, lymphoma, hepatocellular carcinoma, ovarian cancer, myeloma |
| Herpes virus (HSV) | CTLA-4, PD-1 | GM-CSF encoding; Ipilimumab combination; ICP34.5 deletion | Melanoma, head and neck cancer, pancreatic cancer, breast cancer, glioblastoma |
| Adenoviruses (Ad) | CTLA-4, PD-1, 4-1BB, PVR | GM-CSF, IL-2, IL-12, TNF-a encoding; E1B deletion; a-PD-1, a-CTLA-4,4-1BBL encoding | Head and neck cancer, pancreatic cancer, ovarian cancer, colorectal cancer, melanoma, glioblastoma |
| Reovirus | PD-1 | None | Glioma, sarcomas, colorectal cancer, non-small cell lung cancer (NSCLC), ovarian cancer, melanoma, pancreatic cancer, head and neck cancer |
| Coxsackievirus | CTLA-4, PD-1 | None | Melanoma, breast cancer, and prostate cancer |
| Newcastle disease virus (NDV) | CTLA-4, PD-1, ICOS | GM-CSF, IL-2, EGFP encoding | Colorectal cancer, hepatoma, lung cancer, prostate cancer |
| Vaccinia virus (VV) | CTLA-4, PD-1 | GM-CSF, IL-10, VEGF encoding | Melanoma, liver cancer, colorectal cancer, breast cancer, and hepatocellular carcinoma, pancreatic cancer |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, T.; Ma, Y.; Yu, L.; Jiang, J.; Shen, S.; Hou, Y.; Wang, T. Cancer Immunotherapy: A Focus on the Regulation of Immune Checkpoints. Int. J. Mol. Sci. 2018, 19, 1389. https://doi.org/10.3390/ijms19051389
Shi T, Ma Y, Yu L, Jiang J, Shen S, Hou Y, Wang T. Cancer Immunotherapy: A Focus on the Regulation of Immune Checkpoints. International Journal of Molecular Sciences. 2018; 19(5):1389. https://doi.org/10.3390/ijms19051389
Chicago/Turabian StyleShi, Tao, Yanyu Ma, Lingfeng Yu, Jiaxuan Jiang, Sunan Shen, Yayi Hou, and Tingting Wang. 2018. "Cancer Immunotherapy: A Focus on the Regulation of Immune Checkpoints" International Journal of Molecular Sciences 19, no. 5: 1389. https://doi.org/10.3390/ijms19051389
APA StyleShi, T., Ma, Y., Yu, L., Jiang, J., Shen, S., Hou, Y., & Wang, T. (2018). Cancer Immunotherapy: A Focus on the Regulation of Immune Checkpoints. International Journal of Molecular Sciences, 19(5), 1389. https://doi.org/10.3390/ijms19051389