Towards Resolving the Pro- and Anti-Tumor Effects of the Aryl Hydrocarbon Receptor

 and
and
Abstract
:1. Introduction
2. Results
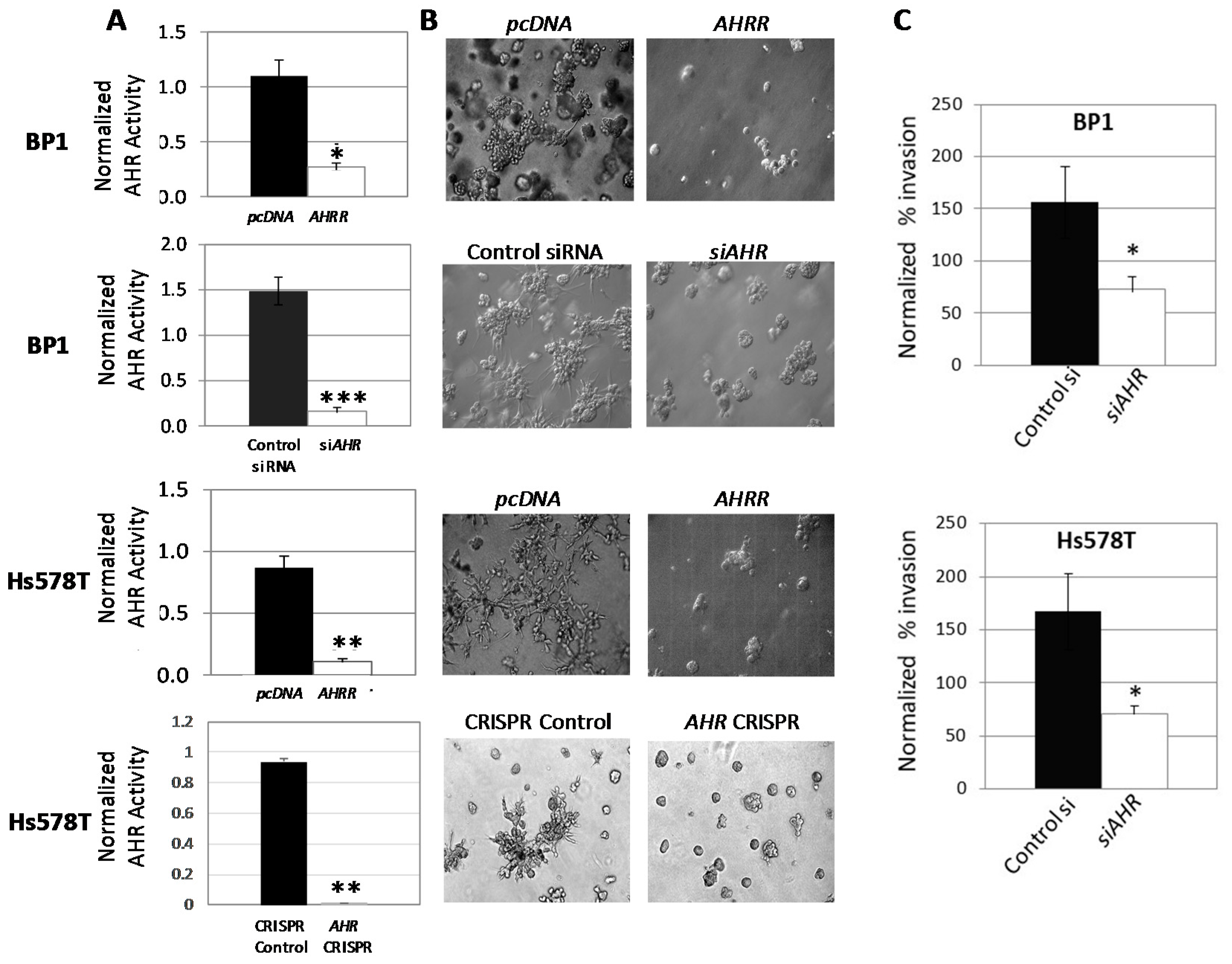
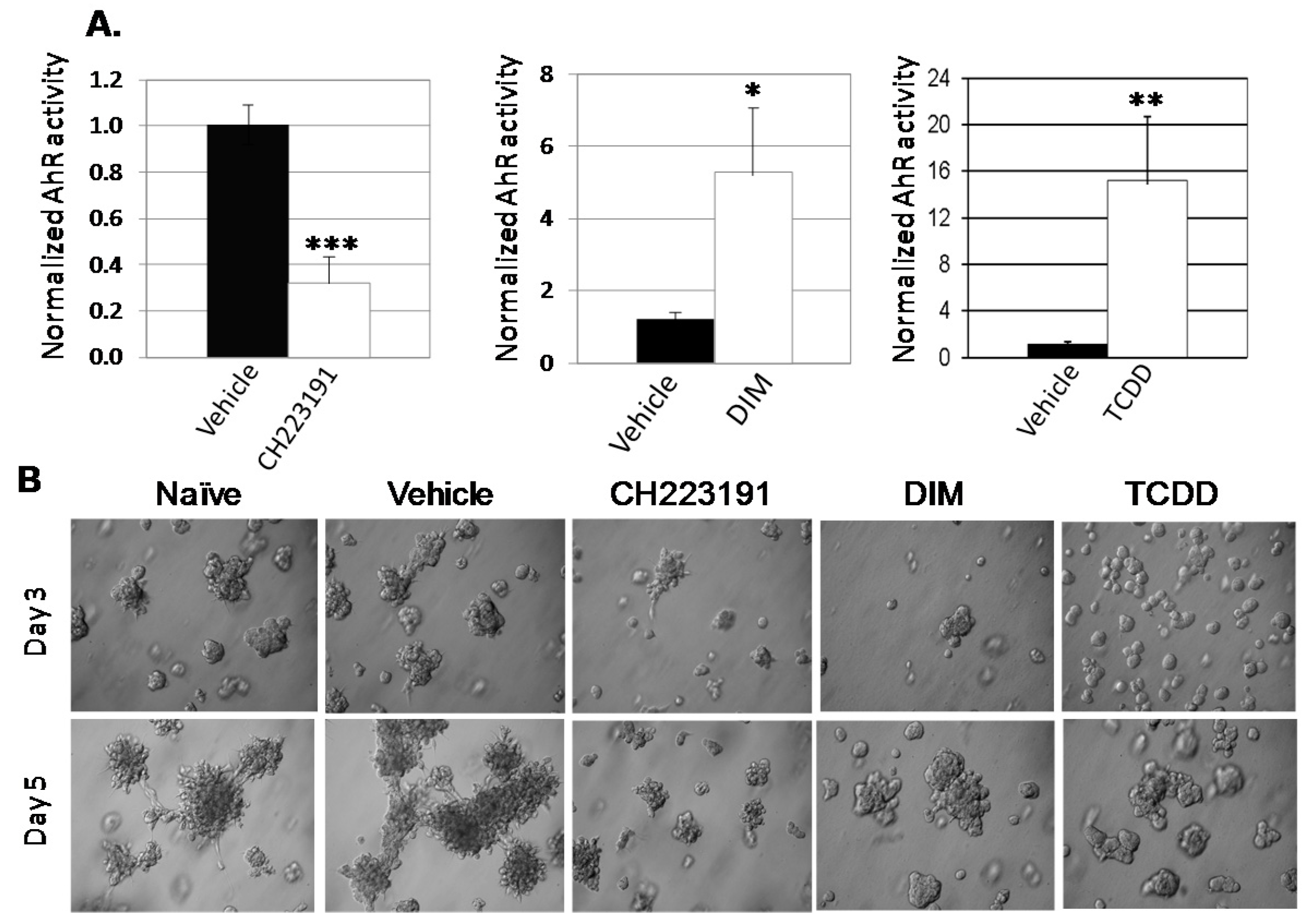
2.1. Inhibition of AHR Expression or Activity Significantly Reduces Irregular Colony Formation by Human Mammary Tumor Cells in 3D Cultures
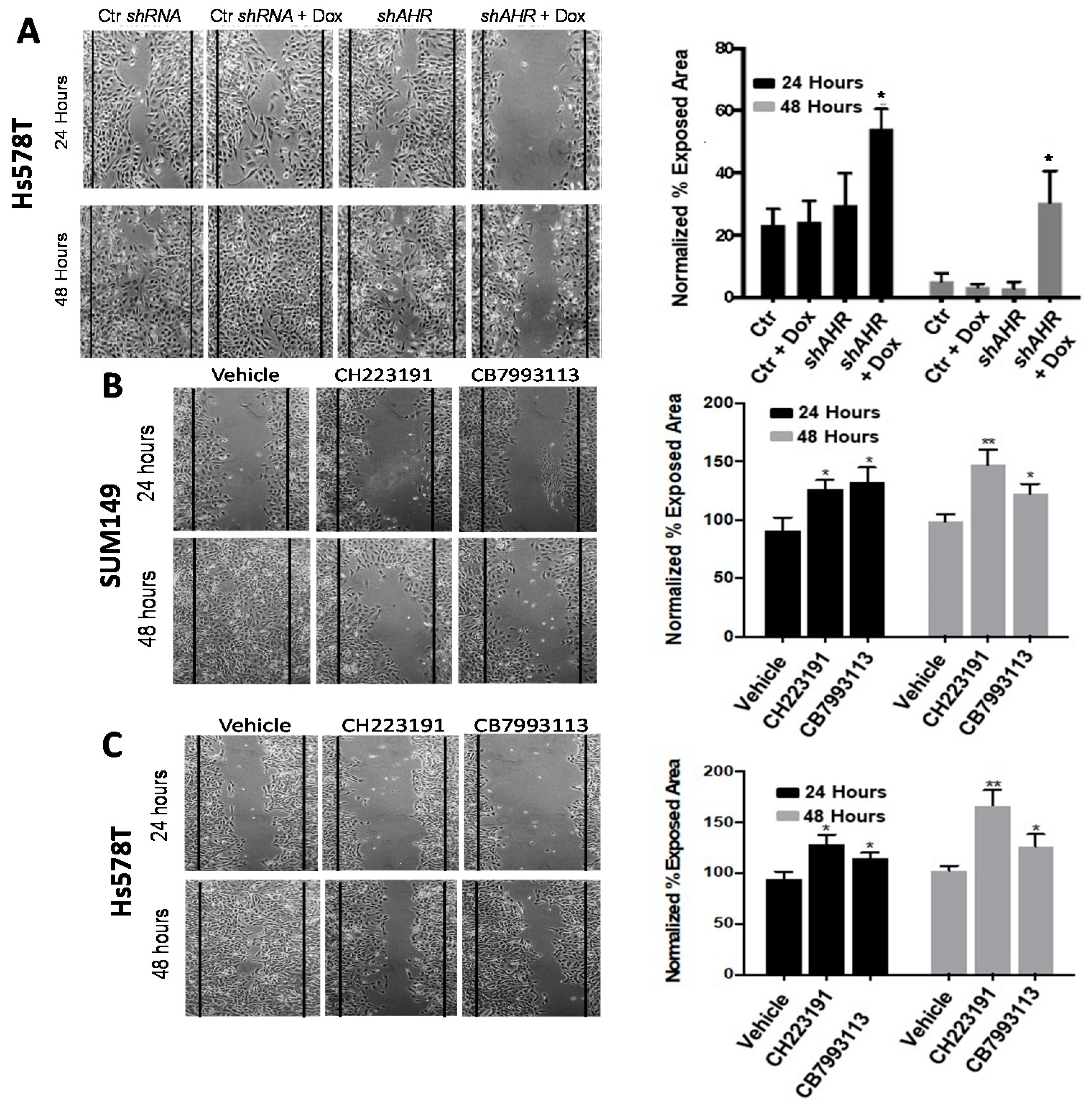
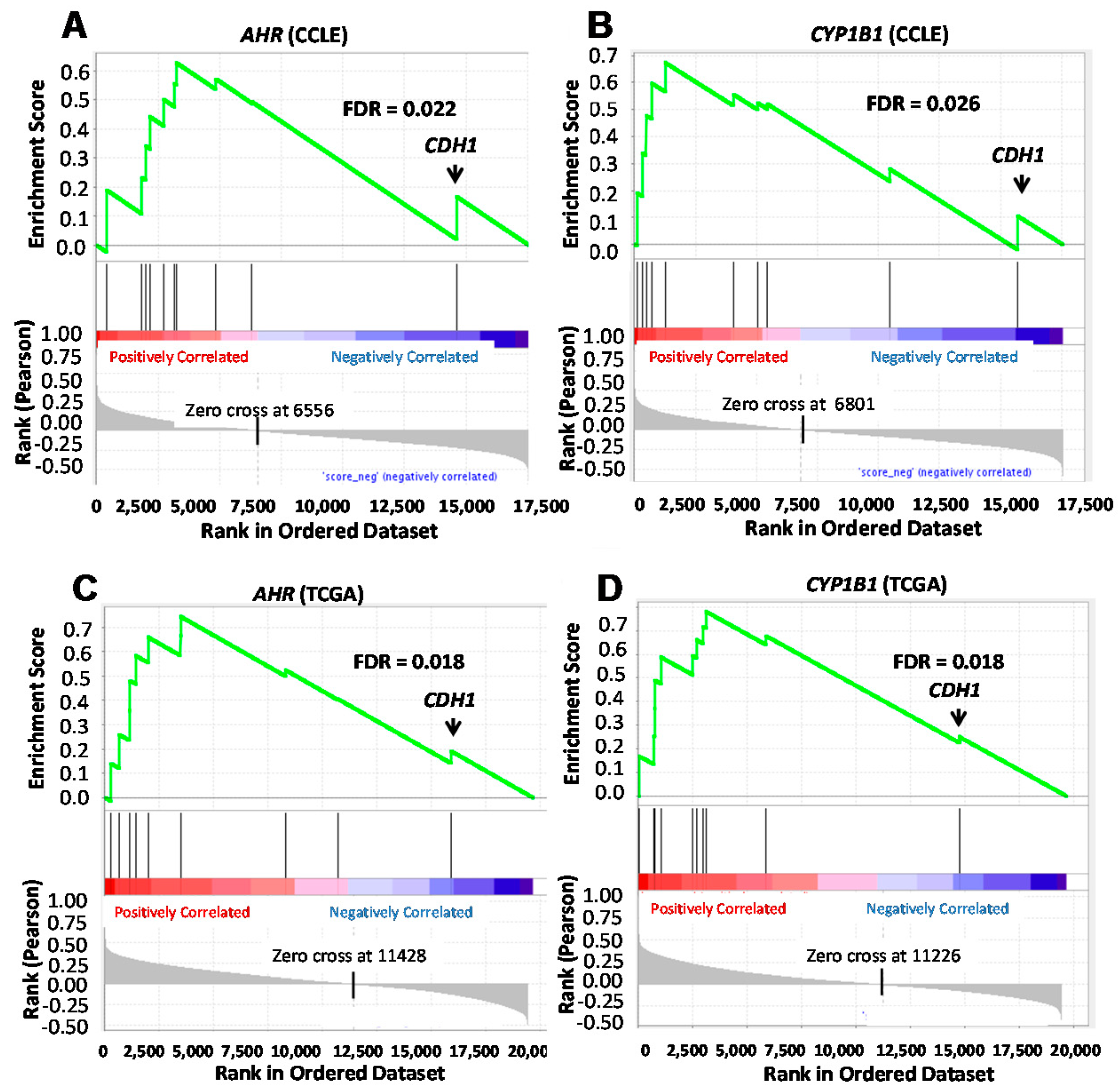
2.2. AHR Downregulation with Inducible shAHR Alters Expression of Multiple Tumor Invasion-Associated Genes
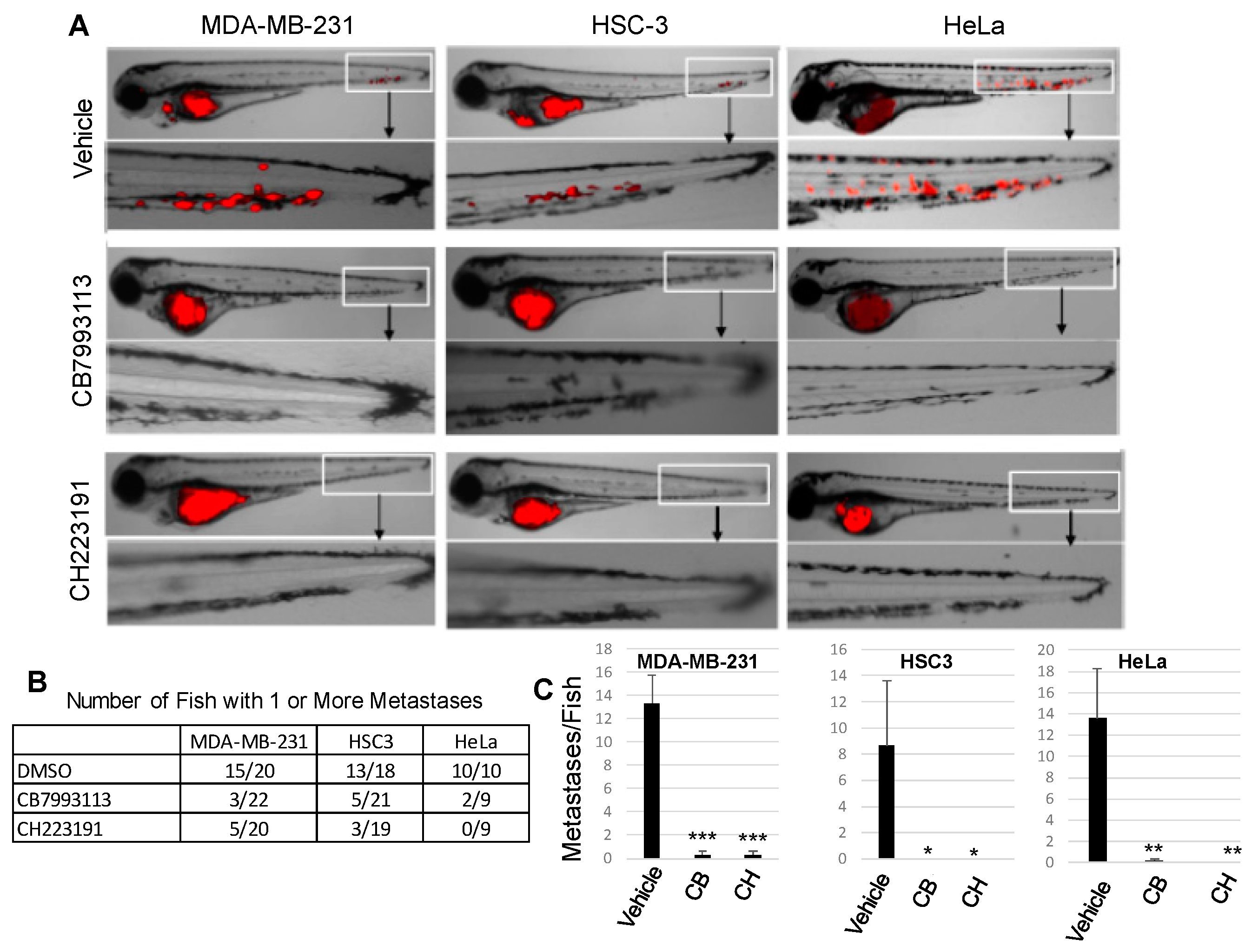
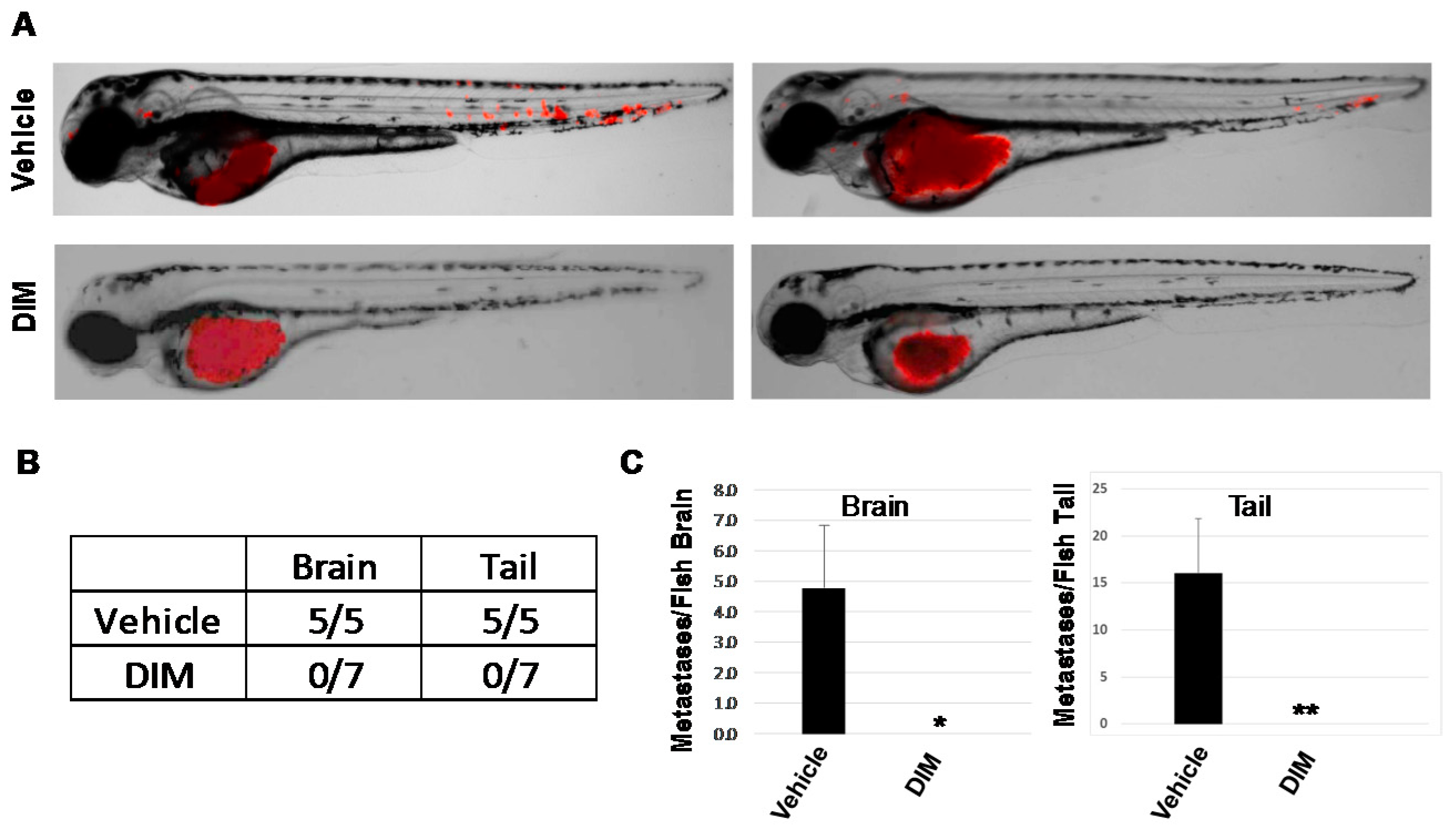
2.3. AHR Inhibition Blocks Human Cancer Cell Metastasis In Vivo
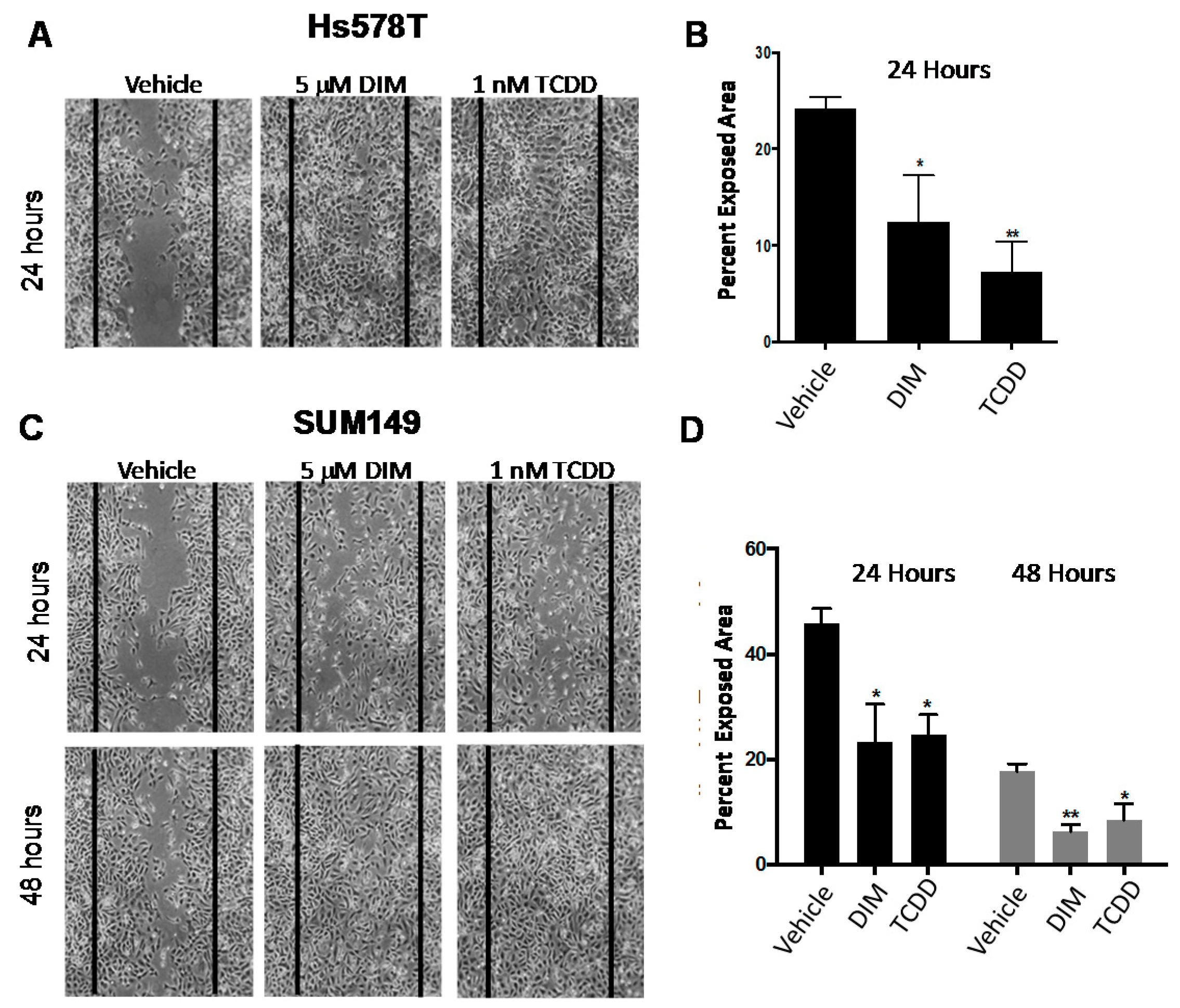
2.4. The Effects of AHR Agonists on Tumor Cell Invasion, Migration and Metastasis
3. Discussion
4. Materials and Methods
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| CCLE | Cancer cell line encyclopedia |
| DIM | Diindolylmethane |
| Dox | Doxycycline |
| FDR | False discovery rate |
| FICZ | 6-formyllindolo[3,2-b]carbazole |
| GSEA | Gene set enrichment analysis |
| IBC | Inflammatory breast cancer |
| TCDD | 2,3,7,8-tetrachlorodibenzo-p-dioxin |
| TNBC | Triple-negative breast cancer |
References
- IARC Monographs on the Evaluation of the Carcinogenic Risk of Chemicals to Humans; International Agency of Research on Cancer: Lyon, France, 2018; Volume 113.
- Hahn, M.E.; Karchner, S.I.; Shapiro, M.A.; Perera, S.A. Molecular evolution of two vertebrate aryl hydrocarbon (dioxin) receptors (AHR1 and AHR2) and the PAS family. Proc. Natl. Acad. Sci. USA 1997, 94, 13743–13748. [Google Scholar] [CrossRef] [PubMed]
- Knerr, S.; Schrenk, D. Carcinogenicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin in experimental models. Mol. Nutr. Food Res. 2006, 50, 897–907. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, C.; Kaina, B. The aryl hydrocarbon receptor (AhR) in the regulation of cell-cell contact and tumor growth. Carcinogenesis 2010, 31, 1319–1328. [Google Scholar] [CrossRef] [PubMed]
- DiNatale, B.C.; Smith, K.; John, K.; Krishnegowda, G.; Amin, S.G.; Perdew, G.H. Ah receptor antagonism represses head and neck tumor cell aggressive phenotype. Mol. Cancer Res. 2012, 10, 1369–1379. [Google Scholar] [CrossRef] [PubMed]
- Gramatzki, D.; Pantazis, G.; Schittenhelm, J.; Tabatabai, G.; Kohle, C.; Wick, W.; Schwarz, M.; Weller, M.; Tritschler, I. Aryl hydrocarbon receptor inhibition downregulates the TGF-beta/Smad pathway in human glioblastoma cells. Oncogene 2009, 28, 2593–2605. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.W.; Gazourian, L.; Quadri, S.A.; Romieu-Mourez, R.; Sherr, D.H.; Sonenshein, G.E. The RelA NF-kB subunit and the aryl hydrocarbon receptor (AhR) cooperate to transactivate the c-myc promoter in mammary cells Equal contributions. Oncogene 2000, 19, 5498–5506. [Google Scholar] [CrossRef] [PubMed]
- Litzenburger, U.M.; Opitz, C.A.; Sahm, F.; Rauschenbach, K.J.; Trump, S.; Winter, M.; Ott, M.; Ochs, K.; Lutz, C.; Liu, X.; et al. Constitutive IDO expression in human cancer is sustained by an autocrine signaling loop involving IL-6, STAT3 and the AHR. Oncotarget 2014, 5, 1038–1051. [Google Scholar] [CrossRef] [PubMed]
- Moennikes, O.; Loeppen, S.; Buchmann, A.; Andersson, P.; Ittrich, C.; Poellinger, L.; Schwarz, M. A constitutively active dioxin/aryl hydrocarbon receptor promotes hepatocarcinogenesis in mice. Cancer Res. 2004, 64, 4707–4710. [Google Scholar] [CrossRef] [PubMed]
- Murray, I.A.; Patterson, A.D.; Perdew, G.H. Aryl hydrocarbon receptor ligands in cancer: Friend and foe. Nat. Rev. Cancer 2014, 14, 801–814. [Google Scholar] [CrossRef] [PubMed]
- Safe, S.; Lee, S.O.; Jin, U.H. Role of the aryl hydrocarbon receptor in carcinogenesis and potential as a drug target. Toxicol. Sci. 2013, 135, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Schlezinger, J.J.; Liu, D.; Farago, M.; Seldin, D.C.; Belguise, K.; Sonenshein, G.E.; Sherr, D.H. A role for the aryl hydrocarbon receptor in mammary gland tumorigenesis. Biol. Chem. 2006, 387, 1175–1187. [Google Scholar] [CrossRef] [PubMed]
- Duan, Z.; Li, Y.; Li, L. Promoting epithelial-to-mesenchymal transition by d-kynurenine via activating aryl hydrocarbon receptor. Mol. Cell. Biochem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Schiering, C.; Vonk, A.; Das, S.; Stockinger, B.; Wincent, E. Cytochrome P4501-inhibiting chemicals amplify aryl hydrocarbon receptor activation and IL-22 production in T helper 17 cells. Biochem. Pharmacol. 2018, 151, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Xue, P.; Fu, J.; Zhou, Y. The Aryl Hydrocarbon Receptor and Tumor Immunity. Front. Immunol. 2018, 9, 286. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, N.; Saito, N.; Zhao, S.; Terai, K.; Hiruta, N.; Park, Y.; Bujo, H.; Nemoto, K.; Kanno, Y. Heregulin-induced cell migration is promoted by aryl hydrocarbon receptor in HER2-overexpressing breast cancer cells. Exp. Cell Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Novikov, O.; Wang, Z.; Stanford, E.A.; Parks, A.J.; Ramirez-Cardenas, A.; Landesman, E.; Laklouk, I.; Sarita-Reyes, C.; Gusenleitner, D.; Li, A.; et al. An aryl hydrocarbon receptor-mediated amplification loop that enforces cell migration in ER-/PR-/Her2- human breast cancer cells. Mol. Pharmacol. 2016, 90, 674–688. [Google Scholar] [CrossRef] [PubMed]
- Opitz, C.A.; Litzenburger, U.M.; Sahm, F.; Ott, M.; Tritschler, I.; Trump, S.; Schumacher, T.; Jestaedt, L.; Schrenk, D.; Weller, M.; et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 2011, 478, 197–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.Y.; Puga, A. Constitutive activation of the aromatic hydrocarbon receptor. Mol. Cell. Biol. 1998, 18, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Barhoover, M.A.; Hall, J.M.; Greenlee, W.F.; Thomas, R.S. Aryl hydrocarbon receptor regulates cell cycle progression in human breast cancer cells via a functional interaction with cyclin-dependent kinase 4. Mol. Pharmacol. 2010, 77, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Stanford, E.A.; Wang, Z.; Novikov, O.; Mulas, F.; Landesman-Bollag, E.; Monti, S.; Smith, B.W.; Seldin, D.C.; Murphy, G.J.; Sherr, D.H. The role of the aryl hydrocarbon receptor in the development of cells with the molecular and functional characteristics of cancer stem-like cells. BMC Biol. 2016, 14, 20. [Google Scholar] [CrossRef] [PubMed]
- John, K.; Lahoti, T.S.; Wagner, K.; Hughes, J.M.; Perdew, G.H. The Ah receptor regulates growth factor expression in head and neck squamous cell carcinoma cell lines. Mol. Carcinog. 2014, 53, 765–776. [Google Scholar] [CrossRef] [PubMed]
- DiNatale, B.C.; Murray, I.A.; Schroeder, J.C.; Flaveny, C.A.; Lahoti, T.S.; Laurenzana, E.M.; Omiecinski, C.J.; Perdew, G.H. Kynurenic acid is a potent endogenous aryl hydrocarbon receptor ligand that synergistically induces interleukin-6 in the presence of inflammatory signaling. Toxicol. Sci. 2010, 115, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Prud'homme, G.J.; Glinka, Y.; Toulina, A.; Ace, O.; Subramaniam, V.; Jothy, S. Breast cancer stem-like cells are inhibited by a non-toxic aryl hydrocarbon receptor agonist. PLoS ONE 2010, 5, e13831. [Google Scholar] [CrossRef] [PubMed]
- Brooks, J.; Eltom, S.E. Malignant transformation of mammary epithelial cells by ectopic overexpression of the aryl hydrocarbon receptor. Curr. Cancer Drug Targets 2011, 11, 654–669. [Google Scholar] [CrossRef] [PubMed]
- Rothhammer, V.; Borucki, D.M.; Kenison, J.E.; Kaye, H.; Wang, Z.; Sherr, D.H.; Quintana, F.J. Detection of aryl hydrocarbon receptor agonists in human samples. Sci. Rep. 2018, in press. [Google Scholar] [CrossRef] [PubMed]
- Haarmann-Stemmann, T.; Esser, C.; Krutmann, J. The Janus-Faced Role of Aryl Hydrocarbon Receptor Signaling in the Skin: Consequences for Prevention and Treatment of Skin Disorders. J. Investig. Dermatol. 2015, 135, 2572–2576. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.T.; Chang, H.; Chen, P.H.; Lin, S.L.; Lin, P. Requirement of aryl hydrocarbon receptor overexpression for CYP1B1 up-regulation and cell growth in human lung adenocarcinomas. Clin. Cancer Res. 2007, 13, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Zudaire, E.; Cuesta, N.; Murty, V.; Woodson, K.; Adams, L.; Gonzalez, N.; Martinez, A.; Narayan, G.; Kirsch, I.; Franklin, W.; et al. The aryl hydrocarbon receptor repressor is a putative tumor suppressor gene in multiple human cancers. J. Clin. Investig. 2008, 118, 640–650. [Google Scholar] [CrossRef] [PubMed]
- Fritz, W.A.; Lin, T.M.; Cardiff, R.D.; Peterson, R.E. The aryl hydrocarbon receptor inhibits prostate carcinogenesis in TRAMP mice. Carcinogenesis 2007, 28, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Andersson, P.; McGuire, J.; Rubio, C.; Gradin, K.; Whitelaw, M.L.; Pettersson, S.; Hanberg, A.; Poellinger, L. A constitutively active dioxin/aryl hydrocarbon receptor induces stomach tumors. Proc. Natl. Acad. Sci. USA 2002, 99, 9990–9995. [Google Scholar] [CrossRef] [PubMed]
- Jin, U.H.; Kim, S.B.; Safe, S. Omeprazole Inhibits Pancreatic Cancer Cell Invasion through a Nongenomic Aryl Hydrocarbon Receptor Pathway. Chem. Res. Toxicol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Abdelrahim, M.; Smith, R., 3rd; Safe, S. Aryl hydrocarbon receptor gene silencing with small inhibitory RNA differentially modulates Ah-responsiveness in MCF-7 and HepG2 cancer cells. Mol. Pharmacol. 2003, 63, 1373–1381. [Google Scholar] [CrossRef] [PubMed]
- DiNatale, B.C.; Schroeder, J.C.; Perdew, G.H. Ah receptor antagonism inhibits constitutive and cytokine inducible IL6 production in head and neck tumor cell lines. Mol. Carcinog. 2011, 50, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Safe, S.; McDougal, A. Mechanism of action and development of selective aryl hydrocarbon receptor modulators for treatment of hormone-dependent cancers (Review). Int. J. Oncol. 2002, 20, 1123–1128. [Google Scholar] [CrossRef] [PubMed]
- Jin, U.H.; Lee, S.O.; Pfent, C.; Safe, S. The aryl hydrocarbon receptor ligand omeprazole inhibits breast cancer cell invasion and metastasis. BMC Cancer 2014, 14, 498. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Kim, K.; Jin, U.H.; Pfent, C.; Cao, H.; Amendt, B.; Liu, X.; Wilson-Robles, H.; Safe, S. Aryl hydrocarbon receptor agonists induce microRNA-335 expression and inhibit lung metastasis of estrogen receptor negative breast cancer cells. Mol. Cancer Ther. 2012, 11, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Roman, A.C.; Carvajal-Gonzalez, J.M.; Merino, J.M.; Mulero-Navarro, S.; Fernandez-Salguero, P.M. The aryl hydrocarbon receptor in the crossroad of signalling networks with therapeutic value. Pharmacol. Ther. 2017. [Google Scholar] [CrossRef] [PubMed]
- Safe, S.; Cheng, Y.; Jin, U.H. The Aryl hydrocarbon receptor (AhR) as a drug target for cancer chemotherapy. Curr. Opin. Toxicol. 2017, 2, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Powell, J.B.; Goode, G.D.; Eltom, S.E. The Aryl Hydrocarbon Receptor: A Target for Breast Cancer Therapy. J. Cancer Ther. 2013, 4, 1177–1186. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wang, W.; Safe, S. Regulation of constitutive gene expression through interactions of Sp1 protein with the nuclear aryl hydrocarbon receptor complex. Biochemistry 1999, 38, 11490–11500. [Google Scholar] [CrossRef] [PubMed]
- Larsen, M.C.; Brake, P.B.; Pollenz, R.S.; Jefcoate, C.R. Linked expression of Ah receptor, ARNT, CYP1A1, and CYP1B1 in rat mammary epithelia, in vitro, is each substantially elevated by specific extracellular matrix interactions that precede branching morphogenesis. Toxicol. Sci. 2004, 82, 46–61. [Google Scholar] [CrossRef] [PubMed]
- Belguise, K.; Guo, S.; Yang, S.; Rogers, A.E.; Seldin, D.C.; Sherr, D.H.; Sonenshein, G.E. Green tea polyphenols reverse cooperation between c-Rel and CK2 that induces the aryl hydrocarbon receptor, slug, and an invasive phenotype. Cancer Res. 2007, 67, 11742–11750. [Google Scholar] [CrossRef] [PubMed]
- Currier, N.; Solomon, S.E.; Demicco, E.G.; Chang, D.L.; Farago, M.; Ying, H.; Dominguez, I.; Sonenshein, G.E.; Cardiff, R.D.; Xiao, Z.X.; et al. Oncogenic signaling pathways activated in DMBA-induced mouse mammary tumors. Toxicol. Pathol. 2005, 33, 726–737. [Google Scholar] [CrossRef] [PubMed]
- Trombino, A.F.; Near, R.I.; Matulka, R.A.; Yang, S.; Hafer, L.J.; Toselli, P.A.; Kim, D.W.; Rogers, A.E.; Sonenshein, G.E.; Sherr, D.H. Expression of the aryl hydrocarbon receptor/transcription factor (AhR) and AhR-regulated CYP1 gene transcripts in a rat model of mammary tumorigenesis. Breast Cancer Res. Treat. 2000, 63, 117–131. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Solomon, S.; Fraser, L.R.; Trombino, A.F.; Liu, D.; Sonenshein, G.E.; Hestermann, E.V.; Sherr, D.H. Constitutive regulation of CYP1B1 by the aryl hydrocarbon receptor (AhR) in pre-malignant and malignant mammary tissue. J. Cell. Biochem. 2008, 104, 402–417. [Google Scholar] [CrossRef] [PubMed]
- Safe, S.; Wang, F.; Porter, W.; Duan, R.; McDougal, A. Ah receptor agonists as endocrine disruptors: Antiestrogenic activity and mechanisms. Toxicol. Lett. 1998, 102–103, 343–347. [Google Scholar] [CrossRef]
- Wormke, M.; Stoner, M.; Saville, B.; Safe, S. Crosstalk between estrogen receptor alpha and the aryl hydrocarbon receptor in breast cancer cells involves unidirectional activation of proteasomes. FEBS Lett. 2000, 478, 109–112. [Google Scholar] [CrossRef]
- Spink, D.C.; Katz, B.H.; Hussain, M.M.; Pentecost, B.T.; Cao, Z.; Spink, B.C. Estrogen regulates Ah responsiveness in MCF-7 breast cancer cells. Carcinogenesis 2003, 24, 1941–1950. [Google Scholar] [CrossRef] [PubMed]
- Beischlag, T.V.; Perdew, G.H. ER α-AHR-ARNT protein-protein interactions mediate estradiol-dependent transrepression of dioxin-inducible gene transcription. J. Biol. Chem. 2005, 280, 21607–21611. [Google Scholar] [CrossRef] [PubMed]
- Reen, R.K.; Cadwallader, A.; Perdew, G.H. The subdomains of the transactivation domain of the aryl hydrocarbon receptor (AhR) inhibit AhR and estrogen receptor transcriptional activity. Arch. Biochem. Biophys. 2002, 408, 93–102. [Google Scholar] [CrossRef]
- Hockings, J.K.; Thorne, P.A.; Kemp, M.Q.; Morgan, S.S.; Selmin, O.; Romagnolo, D.F. The ligand status of the aromatic hydrocarbon receptor modulates transcriptional activation of BRCA-1 promoter by estrogen. Cancer Res. 2006, 66, 2224–2232. [Google Scholar] [CrossRef] [PubMed]
- Wulf, G.; Garg, P.; Liou, Y.C.; Iglehart, D.; Lu, K.P. Modeling breast cancer in vivo and ex vivo reveals an essential role of Pin1 in tumorigenesis. EMBO J. 2004, 23, 3397–3407. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Henry, E.C.; Kim, D.K.; Kim, Y.H.; Shin, K.J.; Han, M.S.; Lee, T.G.; Kang, J.K.; Gasiewicz, T.A.; Ryu, S.H.; et al. Novel compound 2-methyl-2H-pyrazole-3-carboxylic acid (2-methyl-4-o-tolylazo-phenyl)-amide (CH-223191) prevents 2,3,7,8-TCDD-induced toxicity by antagonizing the aryl hydrocarbon receptor. Mol. Pharmacol. 2006, 69, 1871–1878. [Google Scholar] [CrossRef] [PubMed]
- Parks, A.J.; Pollastri, M.P.; Hahn, M.E.; Stanford, E.A.; Novikov, O.; Franks, D.G.; Haigh, S.E.; Narasimhan, S.; Ashton, T.D.; Hopper, T.G.; et al. In silico identification of an aryl hydrocarbon receptor antagonist with biological activity in vitro and in vivo. Mol. Pharmacol. 2014, 86, 593–608. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Guo, H.; Li, Y.; Xu, X.; Yang, K.; Bai, Y. Association between polymorphisms in the promoter regions of matrix metalloproteinases (MMPs) and risk of cancer metastasis: A meta-analysis. PLoS ONE 2012, 7, e31251. [Google Scholar] [CrossRef] [PubMed]
- Radisky, E.S.; Radisky, D.C. Matrix metalloproteinase-induced epithelial-mesenchymal transition in breast cancer. J. Mammary Gland Biol. Neoplasia 2010, 15, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Li, D.M.; Feng, Y.M. Signaling mechanism of cell adhesion molecules in breast cancer metastasis: Potential therapeutic targets. Breast Cancer Res. Treat. 2011, 128, 7–21. [Google Scholar] [CrossRef] [PubMed]
- Byrne, G.J.; Hayden, K.E.; McDowell, G.; Lang, H.; Kirwan, C.C.; Tetlow, L.; Kumar, S.; Bundred, N.J. Angiogenic characteristics of circulating and tumoural thrombospondin-1 in breast cancer. Int. J. Oncol. 2007, 31, 1127–1132. [Google Scholar] [PubMed]
- Peng, T.L.; Chen, J.; Mao, W.; Song, X.; Chen, M.H. Aryl hydrocarbon receptor pathway activation enhances gastric cancer cell invasiveness likely through a c-Jun-dependent induction of matrix metalloproteinase-9. BMC Cell Biol. 2009, 10, 27. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.A.; Villano, C.M.; Dorn, R.; White, L.A. Interaction between the aryl hydrocarbon receptor and retinoic acid pathways increases matrix metalloproteinase-1 expression in keratinocytes. J. Biol. Chem. 2004, 279, 25284–25293. [Google Scholar] [CrossRef] [PubMed]
- Villano, C.M.; Murphy, K.A.; Akintobi, A.; White, L.A. 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) induces matrix metalloproteinase (MMP) expression and invasion in A2058 melanoma cells. Toxicol. Appl. Pharmacol. 2006, 210, 212–224. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.F.; Chen, J.; Mao, W.; Wang, Y.H.; Chen, M.H. Downregulation of aryl hydrocarbon receptor expression decreases gastric cancer cell growth and invasion. Oncol. Rep. 2013, 30, 364–370. [Google Scholar] [CrossRef] [PubMed]
- Pickl, M.; Ries, C.H. Comparison of 3D and 2D tumor models reveals enhanced HER2 activation in 3D associated with an increased response to trastuzumab. Oncogene 2009, 28, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Nelson, C.M.; Bissell, M.J. Of extracellular matrix, scaffolds, and signaling: Tissue architecture regulates development, homeostasis, and cancer. Annu. Rev. Cell Dev. Biol. 2006, 22, 287–309. [Google Scholar] [CrossRef] [PubMed]
- Al Saleh, S.; Sharaf, L.H.; Luqmani, Y.A. Signalling pathways involved in endocrine resistance in breast cancer and associations with epithelial to mesenchymal transition (Review). Int. J. Oncol. 2011. [Google Scholar] [CrossRef]
- Bambang, I.F.; Xu, S.; Zhou, J.; Salto-Tellez, M.; Sethi, S.K.; Zhang, D. Overexpression of endoplasmic reticulum protein 29 regulates mesenchymal-epithelial transition and suppresses xenograft tumor growth of invasive breast cancer cells. Lab. Investig. 2009, 89, 1229–1242. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.Z.; Chan, J.; Wang, Q.; Zhang, W.; Sun, C.D.; Wang, L.H. Twist transcriptionally up-regulates AKT2 in breast cancer cells leading to increased migration, invasion, and resistance to paclitaxel. Cancer Res. 2007, 67, 1979–1987. [Google Scholar] [CrossRef] [PubMed]
- Foty, R.A.; Steinberg, M.S. Cadherin-mediated cell-cell adhesion and tissue segregation in relation to malignancy. Int. J. Dev. Biol. 2004, 48, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.; Qi, M.; Chen, D.Z.; Yuan, R.; Goldberg, I.D.; Rosen, E.M.; Auborn, K.; Fan, S. Suppression of breast cancer invasion and migration by indole-3-carbinol: Associated with up-regulation of BRCA1 and E-cadherin/catenin complexes. J. Mol. Med. 2000, 78, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.R.; Sanchez-Velar, N.; Sherr, D.H.; Sonenshein, G.E. 7,12-dimethylbenz(a)anthracene treatment of a c-rel mouse mammary tumor cell line induces epithelial to mesenchymal transition via activation of nuclear factor-kappaB. Cancer Res. 2006, 66, 2570–2575. [Google Scholar] [CrossRef] [PubMed]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–307. [Google Scholar] [CrossRef] [PubMed]
- Tokizane, T.; Shiina, H.; Igawa, M.; Enokida, H.; Urakami, S.; Kawakami, T.; Ogishima, T.; Okino, S.T.; Li, L.C.; Tanaka, Y.; et al. Cytochrome P450 1B1 is overexpressed and regulated by hypomethylation in prostate cancer. Clin. Cancer Res. 2005, 11, 5793–5801. [Google Scholar] [CrossRef] [PubMed]
- Cirillo, F.; Pellegrino, M.; Malivindi, R.; Rago, V.; Avino, S.; Muto, L.; Dolce, V.; Vivacqua, A.; Rigiracciolo, D.C.; De Marco, P.; et al. GPER is involved in the regulation of the estrogen-metabolizing CYP1B1 enzyme in breast cancer. Oncotarget 2017, 8, 106608–106624. [Google Scholar] [CrossRef] [PubMed]
- Mao, M.; Wu, Z.; Chen, J. MicroRNA-187-5p suppresses cancer cell progression in non-small cell lung cancer (NSCLC) through down-regulation of CYP1B1. Biochem. Biophys. Res. Commun. 2016, 478, 649–655. [Google Scholar] [CrossRef] [PubMed]
- Shehin, S.E.; Stephenson, R.O.; Greenlee, W.F. Transcriptional regulation of the human CYP1B1 gene. Evidence for involvement of an aryl hydrocarbon receptor response element in constitutive expression. J. Biol. Chem. 2000, 275, 6770–6776. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.X.; Ellis, M.J. The Cancer Genome Atlas: Clinical applications for breast cancer. Oncology 2013, 27, 1263–1269. [Google Scholar] [PubMed]
- Teng, Y.; Xie, X.; Walker, S.; White, D.T.; Mumm, J.S.; Cowell, J.K. Evaluating human cancer cell metastasis in zebrafish. BMC Cancer 2013, 13, 453. [Google Scholar] [CrossRef] [PubMed]
- Drabsch, Y.; He, S.; Zhang, L.; Snaar-Jagalska, B.E.; ten Dijke, P. Transforming growth factor-beta signalling controls human breast cancer metastasis in a zebrafish xenograft model. Breast Cancer Res. 2013, 15, R106. [Google Scholar] [CrossRef] [PubMed]
- Brown, H.K.; Schiavone, K.; Tazzyman, S.; Heymann, D.; Chico, T.J. Zebrafish xenograft models of cancer and metastasis for drug discovery. Expert Opin. Drug. Discov. 2017, 12, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Groenewoud, A.; Tulotta, C.; Zoni, E.; Kruithof-de Julio, M.; van der Horst, G.; van der Pluijm, G.; Ewa Snaar-Jagalska, B. A zebrafish xenograft model for studying human cancer stem cells in distant metastasis and therapy response. Methods Cell Biol. 2017, 138, 471–496. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Ordonez, A.; Seoane, S.; Cabezas, P.; Eiro, N.; Sendon-Lago, J.; Macia, M.; Garcia-Caballero, T.; Gonzalez, L.O.; Sanchez, L.; Vizoso, F.; et al. Breast cancer metastasis to liver and lung is facilitated by Pit-1-CXCL12-CXCR4 axis. Oncogene 2018, 37, 1430–1444. [Google Scholar] [CrossRef] [PubMed]
- Stuelten, C.H.; Parent, C.A.; Montell, D.J. Cell motility in cancer invasion and metastasis: Insights from simple model organisms. Nat. Rev. Cancer 2018. [Google Scholar] [CrossRef] [PubMed]
- Stanford, E.A.; Ramirez-Cardenas, A.; Wang, Z.; Novikov, O.; Alamoud, K.; Koutrakis, P.; Mizgerd, J.P.; Genco, C.A.; Kukuruzinska, M.; Monti, S.; et al. Role for the aryl hydrocarbon receptor and diverse ligands in oral squamous cell carcinoma migration and tumorigenesis. Mol. Cancer Res. 2016, 14, 696–706. [Google Scholar] [CrossRef] [PubMed]
- Goode, G.D.; Ballard, B.R.; Manning, H.C.; Freeman, M.L.; Kang, Y.; Eltom, S.E. Knockdown of aberrantly upregulated aryl hydrocarbon receptor reduces tumor growth and metastasis of MDA-MB-231 human breast cancer cell line. Int. J. Cancer 2013, 133, 2769–2780. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.S.; Hord, N.G.; Perdew, G.H. Characterization of the activated form of the aryl hydrocarbon receptor in the nucleus of HeLa cells in the absence of exogenous ligand. Arch. Biochem. Biophys. 1996, 329, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Holcomb, M.; Safe, S. Inhibition of 7,12-dimethylbenzanthracene-induced rat mammary tumor growth by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Cancer Lett. 1994, 82, 43–47. [Google Scholar] [CrossRef]
- Hall, J.M.; Barhoover, M.A.; Kazmin, D.; McDonnell, D.P.; Greenlee, W.F.; Thomas, R.S. Activation of the aryl-hydrocarbon receptor inhibits invasive and metastatic features of human breast cancer cells and promotes breast cancer cell differentiation. Mol. Endocrinol. 2010, 24, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.; McDougal, A.; Wang, F.; Safe, S. Aryl hydrocarbon receptor-mediated antiestrogenic and antitumorigenic activity of diindolylmethane. Carcinogenesis 1998, 19, 1631–1639. [Google Scholar] [CrossRef] [PubMed]
- Hoivik, D.; Willett, K.; Wilson, C.; Safe, S. Estrogen does not inhibit 2,3,7, 8-tetrachlorodibenzo-p-dioxin-mediated effects in MCF-7 and Hepa 1c1c7 cells. J. Biol. Chem. 1997, 272, 30270–30274. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.; Hsieh, T.; Thomas, T.; Safe, S. Identification of estrogen-induced genes downregulated by AhR agonists in MCF-7 breast cancer cells using suppression subtractive hybridization. Gene 2001, 262, 207–214. [Google Scholar] [CrossRef]
- Su, M.; Qian, C.; Hu, Y.; Lu, W.; Huang, R.; Chen, M.; Chen, J. Inhibitory effect of the low-toxic exogenous aryl hydrocarbon receptor modulator 3′3-diindolylmethane on gastric cancer in mice. Oncol. Lett. 2017, 14, 8100–8105. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.F.; Chen, J.; Mao, W.; Wang, Y.H.; Chen, M.H. A selective aryl hydrocarbon receptor modulator 3,3′-Diindolylmethane inhibits gastric cancer cell growth. J. Exp. Clin. Cancer Res. 2012, 31, 46. [Google Scholar] [CrossRef] [PubMed]
- Esser, C. Biology and function of the aryl hydrocarbon receptor: Report of an international and interdisciplinary conference. Arch. Toxicol. 2012, 86, 1323–1329. [Google Scholar] [CrossRef] [PubMed]
- Esser, C. The Aryl Hydrocarbon Receptor in Immunity: Tools and Potential. Methods Mol. Biol. 2016, 1371, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Kadow, S.; Jux, B.; Zahner, S.P.; Wingerath, B.; Chmill, S.; Clausen, B.E.; Hengstler, J.; Esser, C. Aryl hydrocarbon receptor is critical for homeostasis of invariant gammadelta T cells in the murine epidermis. J. Immunol. 2011, 187, 3104–3110. [Google Scholar] [CrossRef] [PubMed]
- Berg, J.; Mahmoudjanlou, Y.; Duscha, A.; Massa, M.G.; Thone, J.; Esser, C.; Gold, R.; Haghikia, A. The immunomodulatory effect of laquinimod in CNS autoimmunity is mediated by the aryl hydrocarbon receptor. J. Neuroimmunol. 2016, 298, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Bekki, K.; Vogel, H.; Li, W.; Ito, T.; Sweeney, C.; Haarmann-Stemmann, T.; Matsumura, F.; Vogel, C.F. The aryl hydrocarbon receptor (AhR) mediates resistance to apoptosis induced in breast cancer cells. Pestic. Biochem. Physiol. 2015, 120, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Quintana, F.J.; Sherr, D.H. Aryl hydrocarbon receptor control of adaptive immunity. Pharmacol. Rev. 2013, 65, 1148–1161. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Monti, S.; Sherr, D.H. The diverse and important contributions of the AHR to cancer and cancer immunity. Curr. Opin. Toxicol. 2017, 2, 102–107. [Google Scholar] [CrossRef]
- Kong, L.; Guo, S.; Liu, C.; Zhao, Y.; Feng, C.; Liu, Y.; Wang, T.; Li, C. Overexpression of SDF-1 activates the NF-kappaB pathway to induce epithelial to mesenchymal transition and cancer stem cell-like phenotypes of breast cancer cells. Int. J. Oncol. 2016, 48, 1085–1094. [Google Scholar] [CrossRef] [PubMed]
- Na, Y.; Kaul, S.C.; Ryu, J.; Lee, J.S.; Ahn, H.M.; Kaul, Z.; Kalra, R.S.; Li, L.; Widodo, N.; Yun, C.O.; et al. Stress chaperone mortalin contributes to epithelial-mesenchymal transition and cancer metastasis. Cancer Res. 2016, 76, 2754–2765. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.B.; Onder, T.T.; Jiang, G.; Tao, K.; Kuperwasser, C.; Weinberg, R.A.; Lander, E.S. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell 2009, 138, 645–659. [Google Scholar] [CrossRef] [PubMed]
- Minn, A.J.; Gupta, G.P.; Siegel, P.M.; Bos, P.D.; Shu, W.; Giri, D.D.; Viale, A.; Olshen, A.B.; Gerald, W.L.; Massague, J. Genes that mediate breast cancer metastasis to lung. Nature 2005, 436, 518–524. [Google Scholar] [CrossRef] [PubMed]
- Agiostratidou, G.; Li, M.; Suyama, K.; Badano, I.; Keren, R.; Chung, S.; Anzovino, A.; Hulit, J.; Qian, B.; Bouzahzah, B.; et al. Loss of retinal cadherin facilitates mammary tumor progression and metastasis. Cancer Res. 2009, 69, 5030–5038. [Google Scholar] [CrossRef] [PubMed]
- Ibaragi, S.; Shimo, T.; Hassan, N.M.; Isowa, S.; Kurio, N.; Mandai, H.; Kodama, S.; Sasaki, A. Induction of MMP-13 expression in bone-metastasizing cancer cells by type I collagen through integrin alpha1beta1 and alpha2beta1-p38 MAPK signaling. Anticancer Res. 2011, 31, 1307–1313. [Google Scholar] [PubMed]
- Shah, M.; Huang, D.; Blick, T.; Connor, A.; Reiter, L.A.; Hardink, J.R.; Lynch, C.C.; Waltham, M.; Thompson, E.W. An MMP13-selective inhibitor delays primary tumor growth and the onset of tumor-associated osteolytic lesions in experimental models of breast cancer. PLoS ONE 2012, 7, e29615. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Massague, J. Molecular pathways: VCAM-1 as a potential therapeutic target in metastasis. Clin. Cancer Res. 2012, 18, 5520–5525. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Mu, E.; Wei, Y.; Riethdorf, S.; Yang, Q.; Yuan, M.; Yan, J.; Hua, Y.; Tiede, B.J.; Haffty, B.G.; et al. VCAM-1 promotes osteolytic expansion of indolent bone micrometastasis of breast cancer by engaging alpha4beta1-positive osteoclast progenitors. Cancer Cell 2011, 20, 701–714. [Google Scholar] [CrossRef] [PubMed]
- Byrne, G.J.; Ghellal, A.; Iddon, J.; Blann, A.D.; Venizelos, V.; Kumar, S.; Howell, A.; Bundred, N.J. Serum soluble vascular cell adhesion molecule-1: Role as a surrogate marker of angiogenesis. J. Natl. Cancer Inst. 2000, 92, 1329–1336. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Zhang, X.H.; Massague, J. Macrophage binding to receptor VCAM-1 transmits survival signals in breast cancer cells that invade the lungs. Cancer Cell 2011, 20, 538–549. [Google Scholar] [CrossRef] [PubMed]
- Goto, R.; Nakamura, Y.; Takami, T.; Sanke, T.; Tozuka, Z. Quantitative LC-MS/MS Analysis of Proteins Involved in Metastasis of Breast Cancer. PLoS ONE 2015, 10, e0130760. [Google Scholar] [CrossRef] [PubMed]
- Chang, X.; Fan, Y.; Karyala, S.; Schwemberger, S.; Tomlinson, C.R.; Sartor, M.A.; Puga, A. Ligand-independent regulation of transforming growth factor beta1 expression and cell cycle progression by the aryl hydrocarbon receptor. Mol. Cell. Biol. 2007, 27, 6127–6139. [Google Scholar] [CrossRef] [PubMed]
- Elizondo, G.; Fernandez-Salguero, P.; Sheikh, M.S.; Kim, G.Y.; Fornace, A.J.; Lee, K.S.; Gonzalez, F.J. Altered cell cycle control at the G2/M phases in aryl hydrocarbon receptor-null embryo fibroblast. Mol. Pharmacol. 2000, 57, 1056–1063. [Google Scholar] [PubMed]
- Wiercinska, E.; Naber, H.P.; Pardali, E.; van der Pluijm, G.; van Dam, H.; ten Dijke, P. The TGF-β/Smad pathway induces breast cancer cell invasion through the up-regulation of matrix metalloproteinase 2 and 9 in a spheroid invasion model system. Breast Cancer Res. Treat. 2011, 128, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.D.; Kim, D.J.; Peters, J.M.; Perdew, G.H. The aryl hydrocarbon receptor directly regulates expression of the potent mitogen epiregulin. Toxicol. Sci. 2006, 89, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Carvajal-Gonzalez, J.M.; Mulero-Navarro, S.; Roman, A.C.; Sauzeau, V.; Merino, J.M.; Bustelo, X.R.; Fernandez-Salguero, P.M. The dioxin receptor regulates the constitutive expression of the Vav3 proto-oncogene and modulates cell shape and adhesion. Mol. Biol. Cell. 2009, 20, 1715–1727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Salguero, P.M. A remarkable new target gene for the dioxin receptor: The Vav3 proto-oncogene links AhR to adhesion and migration. Cell Adh. Migr. 2010, 4, 172–175. [Google Scholar] [CrossRef] [PubMed]
- Hayashida, T.; Takahashi, F.; Chiba, N.; Brachtel, E.; Takahashi, M.; Godin-Heymann, N.; Gross, K.W.; Vivanco, M.M.; Wijendran, V.; Shioda, T.; et al. HOXB9, a gene overexpressed in breast cancer, promotes tumorigenicity and lung metastasis. Proc. Natl. Acad. Sci. USA 2010, 107, 1100–1105. [Google Scholar] [CrossRef] [PubMed]
- Eltarhouny, S.A.; Elsawy, W.H.; Radpour, R.; Hahn, S.; Holzgreve, W.; Zhong, X.Y. Genes controlling spread of breast cancer to lung “gang of 4”. Exp. Oncol. 2008, 30, 91–95. [Google Scholar] [PubMed]
- Xie, X.; Tang, S.C.; Cai, Y.; Pi, W.; Deng, L.; Wu, G.; Chavanieu, A.; Teng, Y. Suppression of breast cancer metastasis through the inactivation of ADP-ribosylation factor 1. Oncotarget 2016, 7, 58111–58120. [Google Scholar] [CrossRef] [PubMed]
- Follain, G.; Osmani, N.; Fuchs, C.; Allio, G.; Harlepp, S.; Goetz, J.G. Using the Zebrafish Embryo to Dissect the Early Steps of the Metastasis Cascade. Methods Mol. Biol. 2018, 1749, 195–211. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, R.A.; Keow, J.Y.; Harris, N.D.; Hache, C.A.; Li, D.H.; Crawford, B.D. The zebrafish embryo: A powerful model system for investigating matrix remodeling. Zebrafish 2009, 6, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, K.M.; Streuli, C.H.; Anderson, N.G. Autocrine signalling through ERBB receptors promotes constitutive activation of protein kinase B/Akt in breast cancer cell lines. Breast Cancer Res. Treat. 2003, 81, 117–128. [Google Scholar] [CrossRef] [PubMed]
- D’Amato, N.C.; Rogers, T.J.; Gordon, M.A.; Greene, L.I.; Cochrane, D.R.; Spoelstra, N.S.; Nemkov, T.G.; D’Alessandro, A.; Hansen, K.C.; Richer, J.K. A TDO2-AhR signaling axis facilitates anoikis resistance and metastasis in triple-negative breast cancer. Cancer Res. 2015, 75, 4651–4664. [Google Scholar] [CrossRef] [PubMed]
- De Araujo, E.F.; Feriotti, C.; Galdino, N.A.L.; Preite, N.W.; Calich, V.L.G.; Loures, F.V. The IDO-AhR Axis Controls Th17/Treg Immunity in a Pulmonary Model of Fungal Infection. Front. Immunol. 2017, 8, 880. [Google Scholar] [CrossRef] [PubMed]
- Mezrich, J.D.; Fechner, J.H.; Zhang, X.; Johnson, B.P.; Burlingham, W.J.; Bradfield, C.A. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J. Immunol. 2010, 185, 3190–3198. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.T.; Kimura, A.; Nakahama, T.; Chinen, I.; Masuda, K.; Nohara, K.; Fujii-Kuriyama, Y.; Kishimoto, T. Aryl hydrocarbon receptor negatively regulates dendritic cell immunogenicity via a kynurenine-dependent mechanism. Proc. Natl. Acad. Sci. USA 2010, 107, 19961–19966. [Google Scholar] [CrossRef] [PubMed]
- Hackett, A.J.; Smith, H.S.; Springer, E.L.; Owens, R.B.; Nelson-Rees, W.A.; Riggs, J.L.; Gardner, M.B. Two syngeneic cell lines from human breast tissue: The aneuploid mammary epithelial (Hs578T) and the diploid myoepithelial (Hs578Bst) cell lines. J. Natl. Cancer Inst. 1977, 58, 1795–1806. [Google Scholar] [CrossRef] [PubMed]
- Chu, I.M.; Michalowski, A.M.; Hoenerhoff, M.; Szauter, K.M.; Luger, D.; Sato, M.; Flanders, K.; Oshima, A.; Csiszar, K.; Green, J.E. GATA3 inhibits lysyl oxidase-mediated metastases of human basal triple-negative breast cancer cells. Oncogene 2011. [Google Scholar] [CrossRef] [PubMed]
- Calaf, G.; Russo, J. Transformation of human breast epithelial cells by chemical carcinogens. Carcinogenesis 1993, 14, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Karchner, S.I.; Franks, D.G.; Powell, W.H.; Hahn, M.E. Regulatory interactions among three members of the vertebrate aryl hydrocarbon receptor family: AHR repressor, AHR1, and AHR2. J. Biol. Chem. 2002, 277, 6949–6959. [Google Scholar] [CrossRef] [PubMed]
- Sanda, T.; Tyner, J.W.; Gutierrez, A.; Ngo, V.N.; Glover, J.; Chang, B.H.; Yost, A.; Ma, W.; Fleischman, A.G.; Zhou, W.; et al. TYK2-STAT1-BCL2 pathway dependence in T-cell acute lymphoblastic leukemia. Cancer Discov. 2013, 3, 564–577. [Google Scholar] [CrossRef] [PubMed]
- Shalem, O.; Sanjana, N.E.; Zhang, F. High-throughput functional genomics using CRISPR-Cas9. Nat. Rev. Genet. 2015, 16, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Westerfield, M. The Zebrafish Book: A Guide for the Laboratory Use of Zebrafish (Brachydanio rerio); University of Oregon Press: Eugene, OR, USA, 1994. [Google Scholar]
- Nicoli, S.; Presta, M. The zebrafish/tumor xenograft angiogenesis assay. Nat. Protoc. 2007, 2, 2918–2923. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Encoded Protein | Fold Change | p Value |
|---|---|---|---|
| CDH1 | E-cadherin | 5.07 | 0.052 |
| FN1 | Fibronectin 1 | −3.65 | 0.027 |
| VCAM1 | Vascular Cell Adhesion Protein 1 | −2.58 | 0.039 |
| THBS2 | Thrombospondin 2 | −5.24 | 0.033 |
| COL14A1 | Collagen Type XIV, α1 | −2.05 | 0.005 |
| COL15A1 | Collagen Type XV, α1 | −6.65 | 0.022 |
| MMP1 | Matrix Metalloproteinase 1 | −7.71 | 0.029 |
| MMP13 | Matrix Metalloproteinase 13 | −3.28 | 0.052 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Narasimhan, S.; Stanford Zulick, E.; Novikov, O.; Parks, A.J.; Schlezinger, J.J.; Wang, Z.; Laroche, F.; Feng, H.; Mulas, F.; Monti, S.; et al. Towards Resolving the Pro- and Anti-Tumor Effects of the Aryl Hydrocarbon Receptor. Int. J. Mol. Sci. 2018, 19, 1388. https://doi.org/10.3390/ijms19051388
Narasimhan S, Stanford Zulick E, Novikov O, Parks AJ, Schlezinger JJ, Wang Z, Laroche F, Feng H, Mulas F, Monti S, et al. Towards Resolving the Pro- and Anti-Tumor Effects of the Aryl Hydrocarbon Receptor. International Journal of Molecular Sciences. 2018; 19(5):1388. https://doi.org/10.3390/ijms19051388
Chicago/Turabian StyleNarasimhan, Supraja, Elizabeth Stanford Zulick, Olga Novikov, Ashley J. Parks, Jennifer J. Schlezinger, Zhongyan Wang, Fabrice Laroche, Hui Feng, Francesca Mulas, Stefano Monti, and et al. 2018. "Towards Resolving the Pro- and Anti-Tumor Effects of the Aryl Hydrocarbon Receptor" International Journal of Molecular Sciences 19, no. 5: 1388. https://doi.org/10.3390/ijms19051388
APA StyleNarasimhan, S., Stanford Zulick, E., Novikov, O., Parks, A. J., Schlezinger, J. J., Wang, Z., Laroche, F., Feng, H., Mulas, F., Monti, S., & Sherr, D. H. (2018). Towards Resolving the Pro- and Anti-Tumor Effects of the Aryl Hydrocarbon Receptor. International Journal of Molecular Sciences, 19(5), 1388. https://doi.org/10.3390/ijms19051388