High Affinity Promotes Internalization of Engineered Antibodies Targeting FGFR1

 , ,
, ,
Abstract
:1. Introduction
2. Results
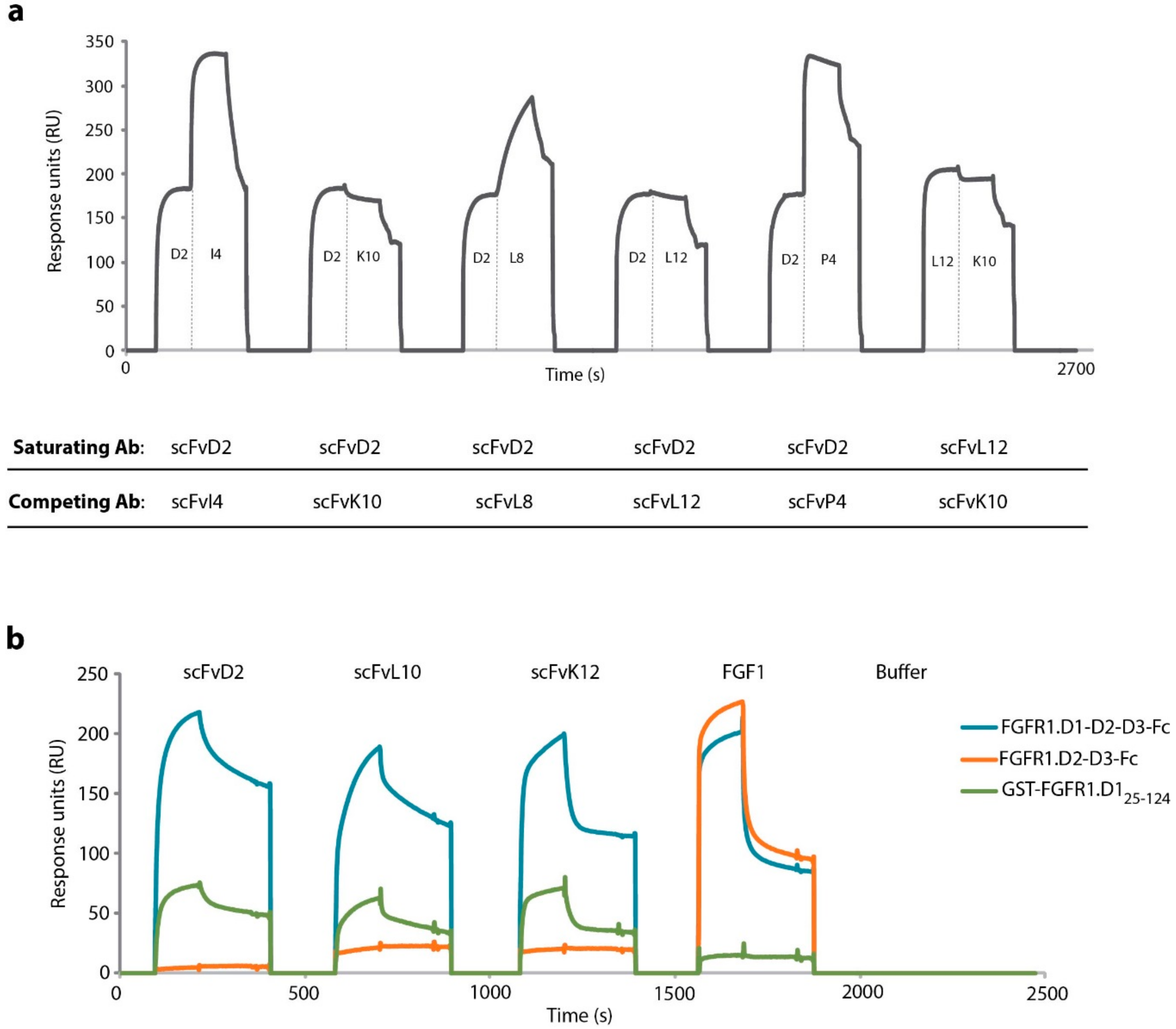
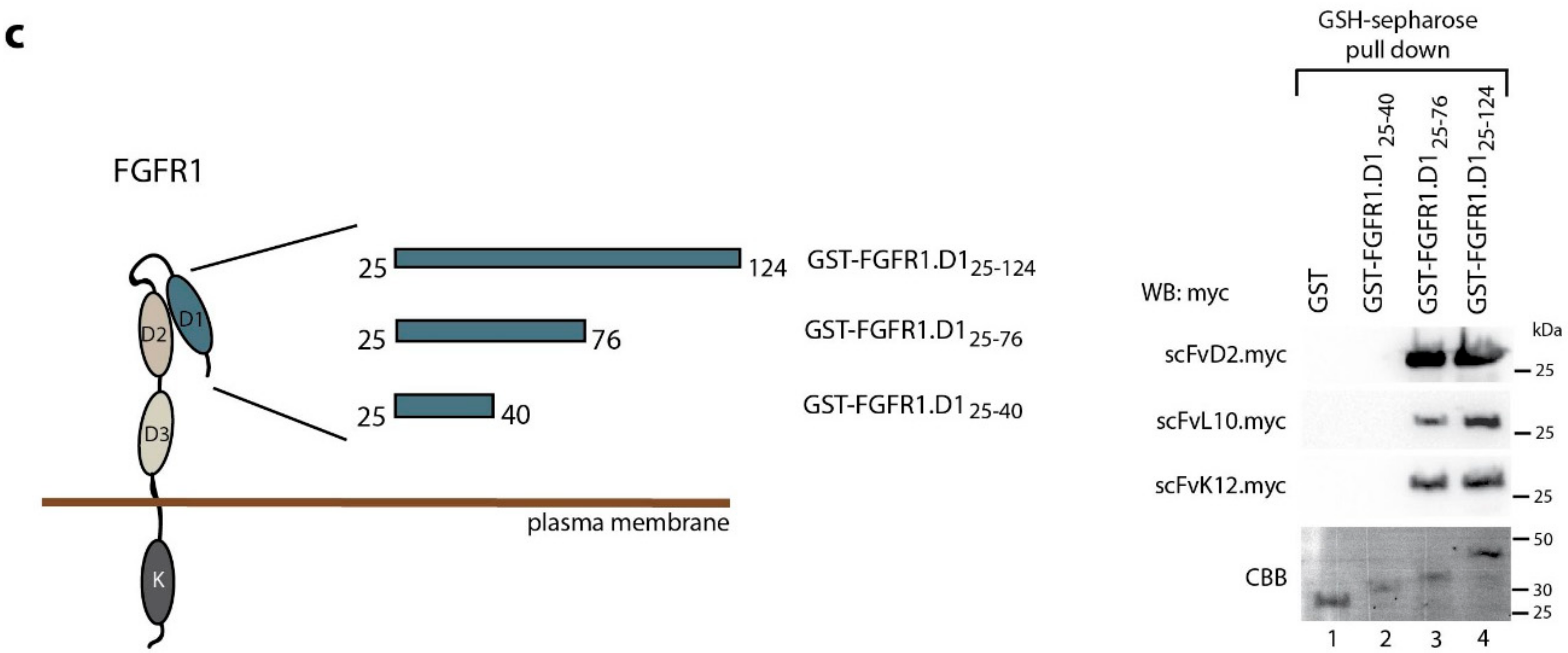
2.1. Engineered Antibody Fragments Recognize the Same Epitope within D1 Domain of the FGFR1
2.2. Engineered Antibodies Differ in the Affinity Towards FGFR1
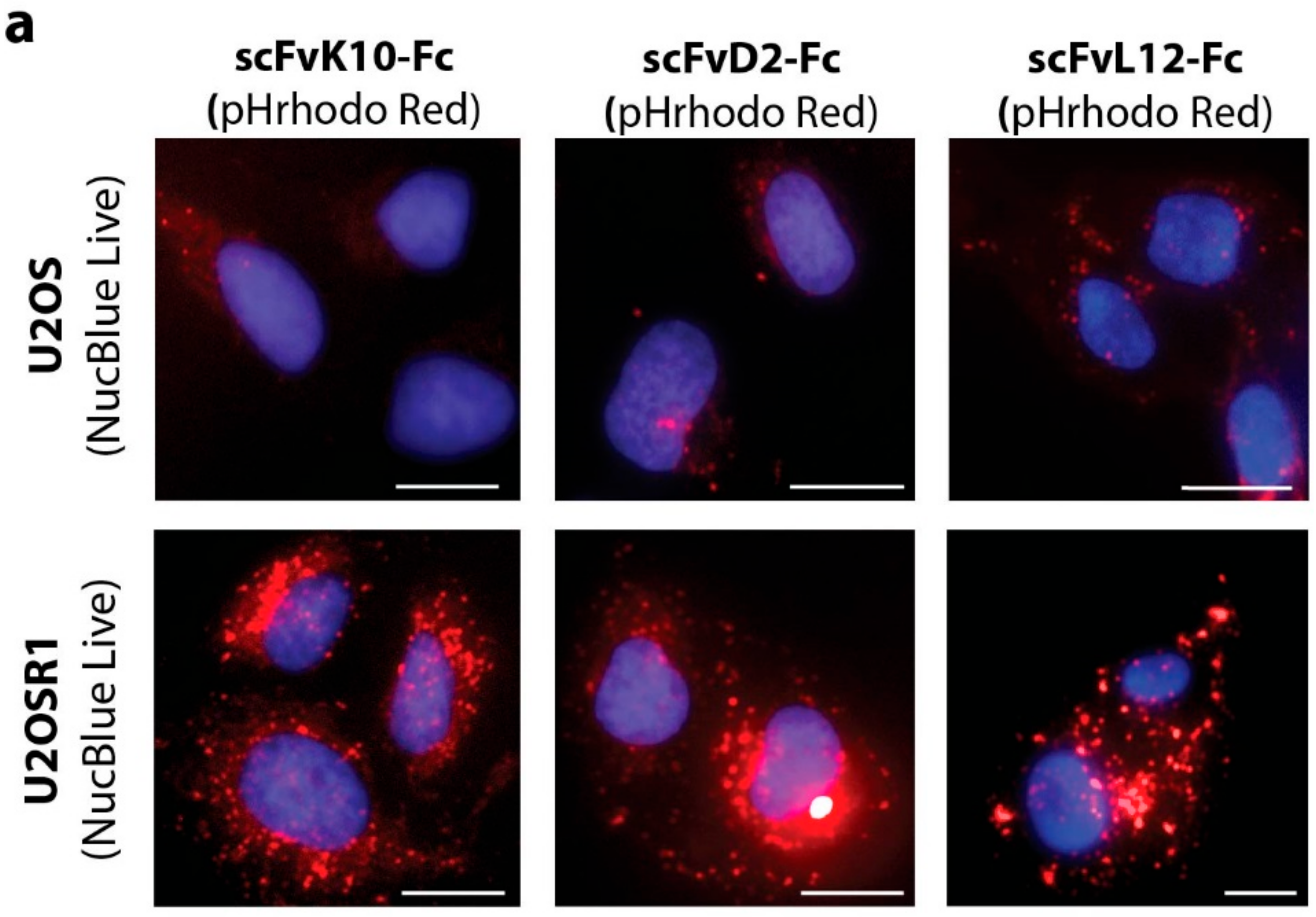
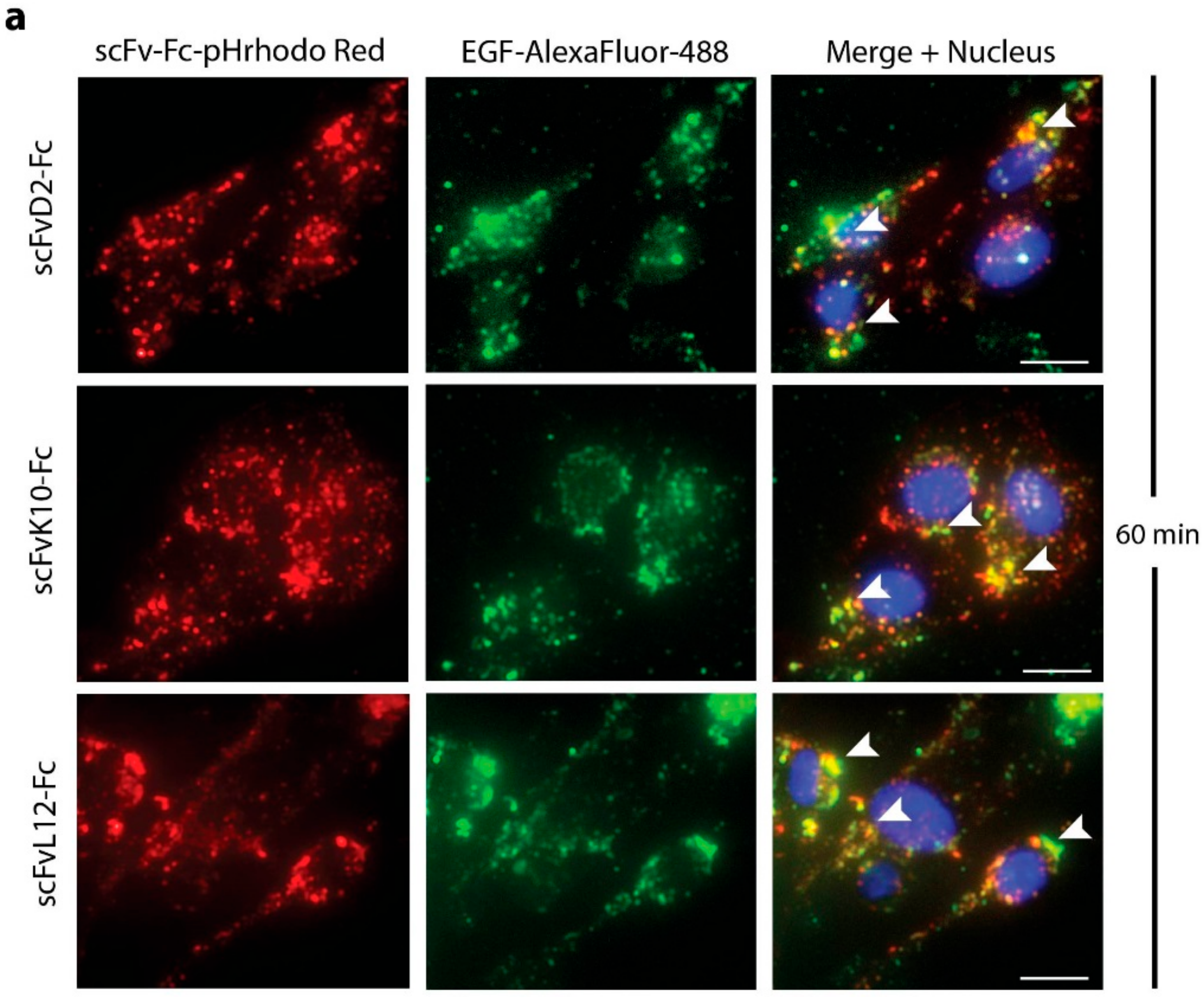
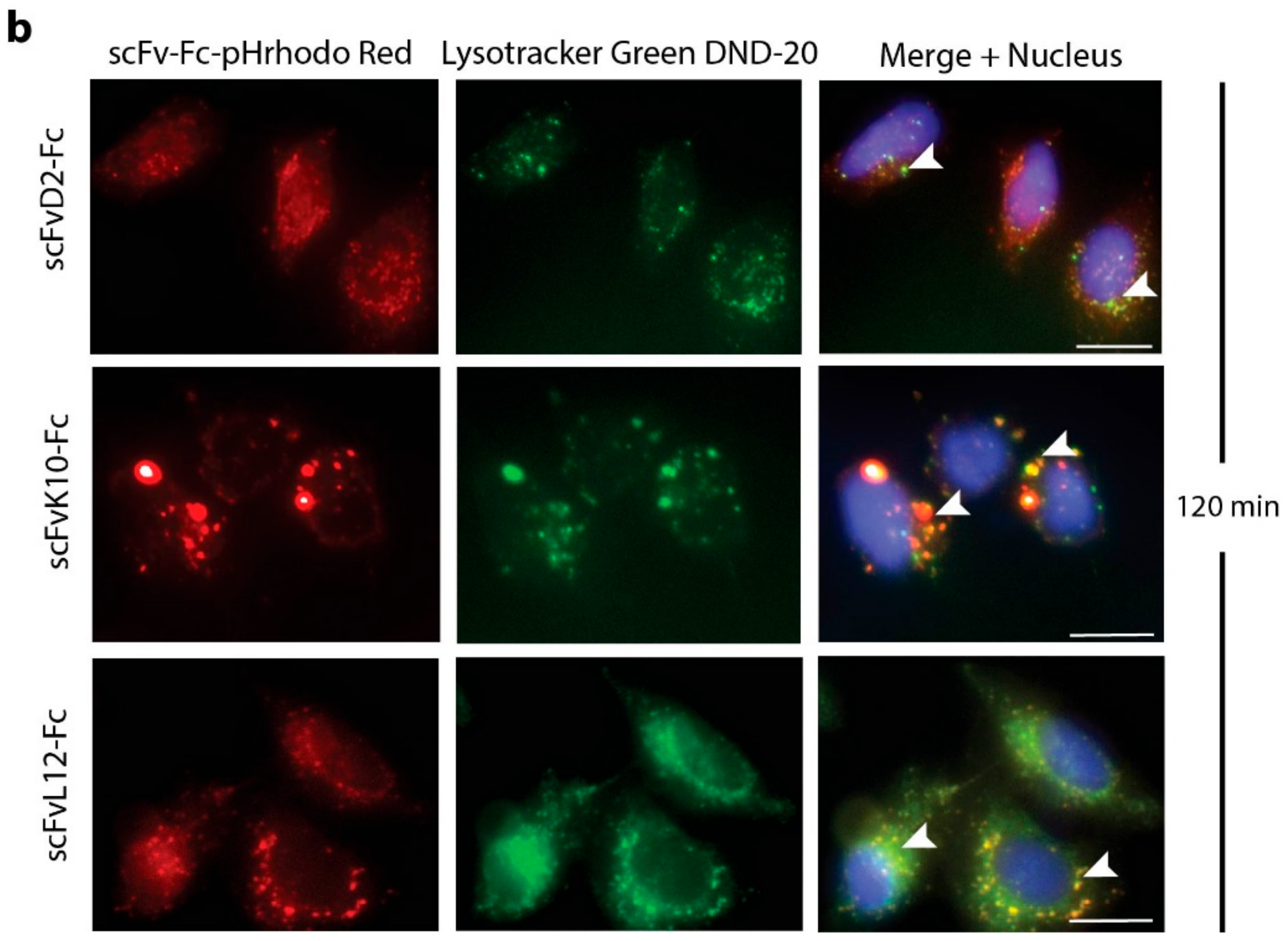
2.3. Antibody Fragments Are Internalized via Receptor Mediated Endocytosis
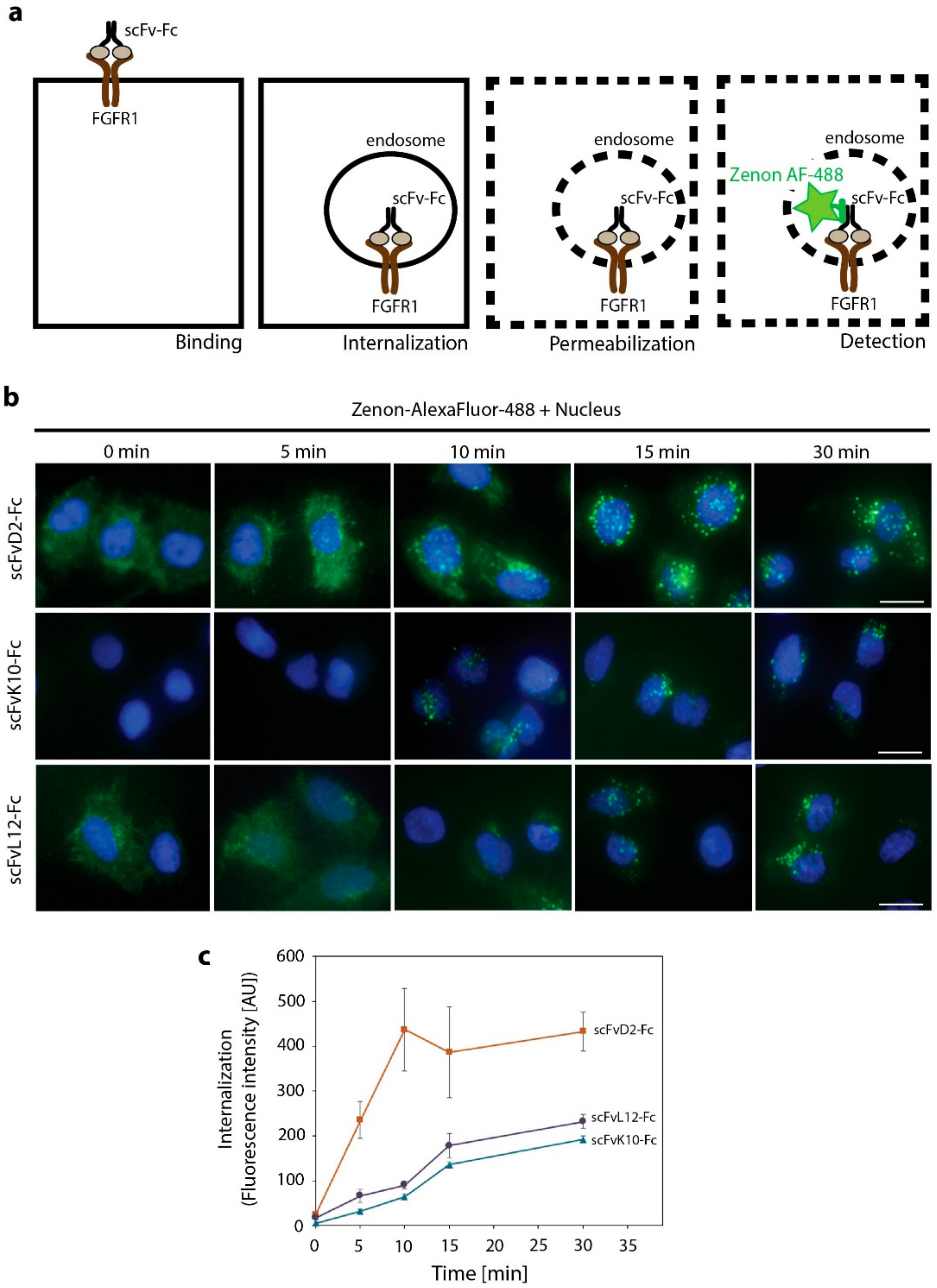
2.4. High Affinity Promotes Internalization of Engineered Antibodies
3. Discussion
4. Materials and Methods
4.1. Antibodies and Recombinant Proteins
4.2. Cells
4.3. Selection of Antibody Fragments against FGFR1 by Phage Display Technology
4.4. SPR-Based Interaction Studies and Affinity Measurements
4.5. Pull-Down
4.6. Fluorescence Microscopy
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ADC | Antibody Drug Conjugates |
| FGFR1 | Fibroblast Growth Factor Receptor 1 |
| FGF | Fibroblast Growth Factor |
| EGF | Epidermal Growth Factor |
References
- Ferlay, J.; Soerjomataram, I.; Ervik, M.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. GLOBOCAN 2012 v1.0, Cancer Incidence and Mortality Worldwide: IARC CancerBase; No. 11; International Agency for Research on Cancer: Lyon, France, 2013. [Google Scholar]
- Almagro, J.C.; Daniels-Wells, T.R.; Perez-Tapia, S.M.; Penichet, M.L. Progress and Challenges in the Design and Clinical Development of Antibodies for Cancer Therapy. Front. Immunol. 2018, 8, 1751–1770. [Google Scholar] [CrossRef] [PubMed]
- Nasiri, H.; Valedkarimi, Z.; Aghebati-Maleki, L.; Majidi, J. Antibody-drug conjugates: Promising and efficient tools for targeted cancer therapy. J. Cell. Physiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Sochaj, A.M.; Świderska, K.W.; Otlewski, J. Current methods for the synthesis of homogeneous antibody-drug conjugates. Biotechnol. Adv. 2015, 33, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Chalouni, C.; Doll, S. Fate of Antibody-Drug Conjugates in Cancer Cells. J. Exp. Clin. Cancer Res. 2018, 37, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Fujimori, K.; Covell, D.G.; Fletcher, J.E.; Weinstein, J.N. A modeling analysis of monoclonal antibody percolation through tumors: A binding-site barrier. J. Nucl. Med. 1990, 31, 1191–1198. [Google Scholar] [PubMed]
- Van Osdol, W.; Fujimori, K.; Weinstein, J.N. An analysis of monoclonal antibody distribution in microscopic tumor nodules: Consequences of a “binding site barrier”. Cancer Res. 1991, 51, 4776–4786. [Google Scholar] [PubMed]
- Juweid, M.; Neumann, R.; Paik, C.; Perez-Bacete, M.J.; Sato, J.; van Osdol, W.; Weinstein, J.N. Micropharmacology of monoclonal antibodies in solid tumors: Direct experimental evidence for a binding site barrier. Cancer Res. 1992, 52, 5144–5143. [Google Scholar] [PubMed]
- Adams, G.P.; Schier, R.; Marshall, K.; Wolf, E.J.; McCall, A.M.; Marks, J.D.; Weiner, L.M. Increased affinity leads to improved selective tumor delivery of single-chain Fv antibodies. Cancer Res. 1998, 58, 485–490. [Google Scholar] [PubMed]
- Coleman, S.J.; Bruce, C.; Chioni, A.M.; Kocher, H.M.; Grose, R.P. The ins and outs of fibroblast growth factor receptor signalling. Clin. Sci. 2014, 127, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Carter, E.P.; Fearon, A.E.; Grose, R.P. Careless talk costs lives: Fibroblast growth factor receptor signalling and the consequences of pathway malfunction. Trends Cell Biol. 2015, 25, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Wesche, J.; Haglund, K.; Haugsten, E.M. Fibroblast growth factors and their receptors in cancer. Biochem. J. 2011, 437, 199–213. [Google Scholar] [CrossRef] [PubMed]
- Helsten, T.; Elkin, S.; Arthur, E.; Thomson, B.J.; Carter, J.; Kurzrock, R. The FGFR Landscape in Cancer: Analysis of 4853 Tumors by Next-Generation Sequencing. Clin. Cancer Res. 2016, 22, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Behrens, C.; Lin, H.Y.; Lee, J.J.; Raso, M.G.; Hong, W.K.; Wistuba, I.I.; Lotan, R. Immunohistochemical expression of basic fibroblast growth factor and fibroblast growth factor receptors 1 and 2 in the pathogenesis of lung cancer. Clin. Cancer Res. 2008, 14, 6014–6022. [Google Scholar] [CrossRef] [PubMed]
- Haugsten, E.M.; Sorensen, V.; Brech, A.; Olsnes, S.; Wesche, J. Different intracellular trafficking of FGF1 endocytosed by the four homologous FGF receptors. J. Cell Sci. 2005, 118, 3869–3881. [Google Scholar] [CrossRef] [PubMed]
- Sokolowska-Wedzina, A.; Chodaczek, G.; Chudzian, J.; Borek, A.; Zakrzewska, M.; Otlewski, J. High-Affinity Internalizing Human scFv-Fc Antibody for Targeting FGFR1-Overexpressing Lung Cancer. Mol. Cancer Res. 2017, 15, 1040–1050. [Google Scholar] [CrossRef] [PubMed]
- Opaliński, Ł.; Sokołowska-Wędzina, A.; Szczepara, M.; Zakrzewska, M.; Otlewski, J. Antibody-induced dimerization of FGFR1 promotes receptor endocytosis independently of its kinase activity. Sci. Rep. 2017, 7, 7121. [Google Scholar] [CrossRef] [PubMed]
- Sokolowska-Wedzina, A.; Borek, A.; Chudzian, J.; Jakimowicz, P.; Zakrzewska, M.; Otlewski, J. Efficient production and purification of extracellular domain of human FGFR-Fc fusion proteins from Chinese hamster ovary cells. Protein Expr. Purif. 2014, 99, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Borek, A.; Sokolowska-Wedzina, A.; Chodaczek, G.; Otlewski, J. Generation of high-affinity, internalizing anti-FGFR2 single-chain variable antibody fragment fused with Fc for targeting gastrointestinal cancers. PLoS ONE 2018, 13, e0192194. [Google Scholar] [CrossRef] [PubMed]
- Margineanu, M.B.; Julfakyan, K.; Sommer, C.; Perez, J.E.; Contreras, M.F.; Khashab, N.; Kosel, J.; Ravasi, T. Semi-automated quantification of living cells with internalized nanostructures. J. Nanobiotechnol. 2016, 14, 4. [Google Scholar] [CrossRef] [PubMed]
- Haugsten, E.M.; Malecki, J.; Bjorklund, S.M.; Olsnes, S.; Wesche, J. Ubiquitination of fibroblast growth factor receptor 1 is required for its intracellular sorting but not for its endocytosis. Mol. Biol. Cell 2008, 19, 3390–3403. [Google Scholar] [CrossRef] [PubMed]
- Glatt, D.M.; Beckford Vera, D.R.; Parrott, M.C.; Luft, J.C.; Benhabbour, S.R.; Mumper, R.J. The Interplay of Antigen Affinity, Internalization, and Pharmacokinetics on CD44-Positive Tumor Targeting of Monoclonal Antibodies. Mol. Pharm. 2016, 13, 1894–1903. [Google Scholar] [CrossRef] [PubMed]
- Rudnick, S.I.; Lou, J.; Shaller, C.C.; Tang, Y.; Klein-Szanto, A.J.; Weiner, L.M.; Marks, J.D.; Adams, G.P. Influence of affinity and antigen internalization on the uptake and penetration of Anti-HER2 antibodies in solid tumors. Cancer Res. 2011, 71, 2250–2289. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.M.; Thurber, G.M.; Wittrup, K.D. Kinetics of anti-carcinoembryonic antigen antibody internalization: Effects of affinity, bivalency, and stability. Cancer Immunol. Immunother. 2008, 57, 1879–1890. [Google Scholar] [CrossRef] [PubMed]
- Zakrzewska, M.; Krowarsch, D.; Wiedlocha, A.; Otlewski, J. Design of fully active FGF-1 variants with increased stability. Protein Eng. Des. Sel. 2004, 17, 603–611. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| scFvD2-Fc * | scFvK10-Fc | scFvL12-Fc | |
|---|---|---|---|
| KD (nmol/L) | 0.59 | 9.41 ± 1.47 | 11.6 ± 1.25 |
| kon (M−1s−1) | 6.47 × 105 | 6.82 × 105 | 4.08 × 105 |
| koff (s−1) | 3.84 × 10−4 | 6.42 × 10−3 | 5.45 × 10−3 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Opaliński, Ł.; Szymczyk, J.; Szczepara, M.; Kucińska, M.; Krowarsch, D.; Zakrzewska, M.; Otlewski, J. High Affinity Promotes Internalization of Engineered Antibodies Targeting FGFR1. Int. J. Mol. Sci. 2018, 19, 1435. https://doi.org/10.3390/ijms19051435
Opaliński Ł, Szymczyk J, Szczepara M, Kucińska M, Krowarsch D, Zakrzewska M, Otlewski J. High Affinity Promotes Internalization of Engineered Antibodies Targeting FGFR1. International Journal of Molecular Sciences. 2018; 19(5):1435. https://doi.org/10.3390/ijms19051435
Chicago/Turabian StyleOpaliński, Łukasz, Jakub Szymczyk, Martyna Szczepara, Marika Kucińska, Daniel Krowarsch, Małgorzata Zakrzewska, and Jacek Otlewski. 2018. "High Affinity Promotes Internalization of Engineered Antibodies Targeting FGFR1" International Journal of Molecular Sciences 19, no. 5: 1435. https://doi.org/10.3390/ijms19051435
APA StyleOpaliński, Ł., Szymczyk, J., Szczepara, M., Kucińska, M., Krowarsch, D., Zakrzewska, M., & Otlewski, J. (2018). High Affinity Promotes Internalization of Engineered Antibodies Targeting FGFR1. International Journal of Molecular Sciences, 19(5), 1435. https://doi.org/10.3390/ijms19051435