Prioritization of Variants Detected by Next Generation Sequencing According to the Mutation Tolerance and Mutational Architecture of the Corresponding Genes



Abstract
:1. Introduction
2. Mutation Tolerance
2.1. Introduction
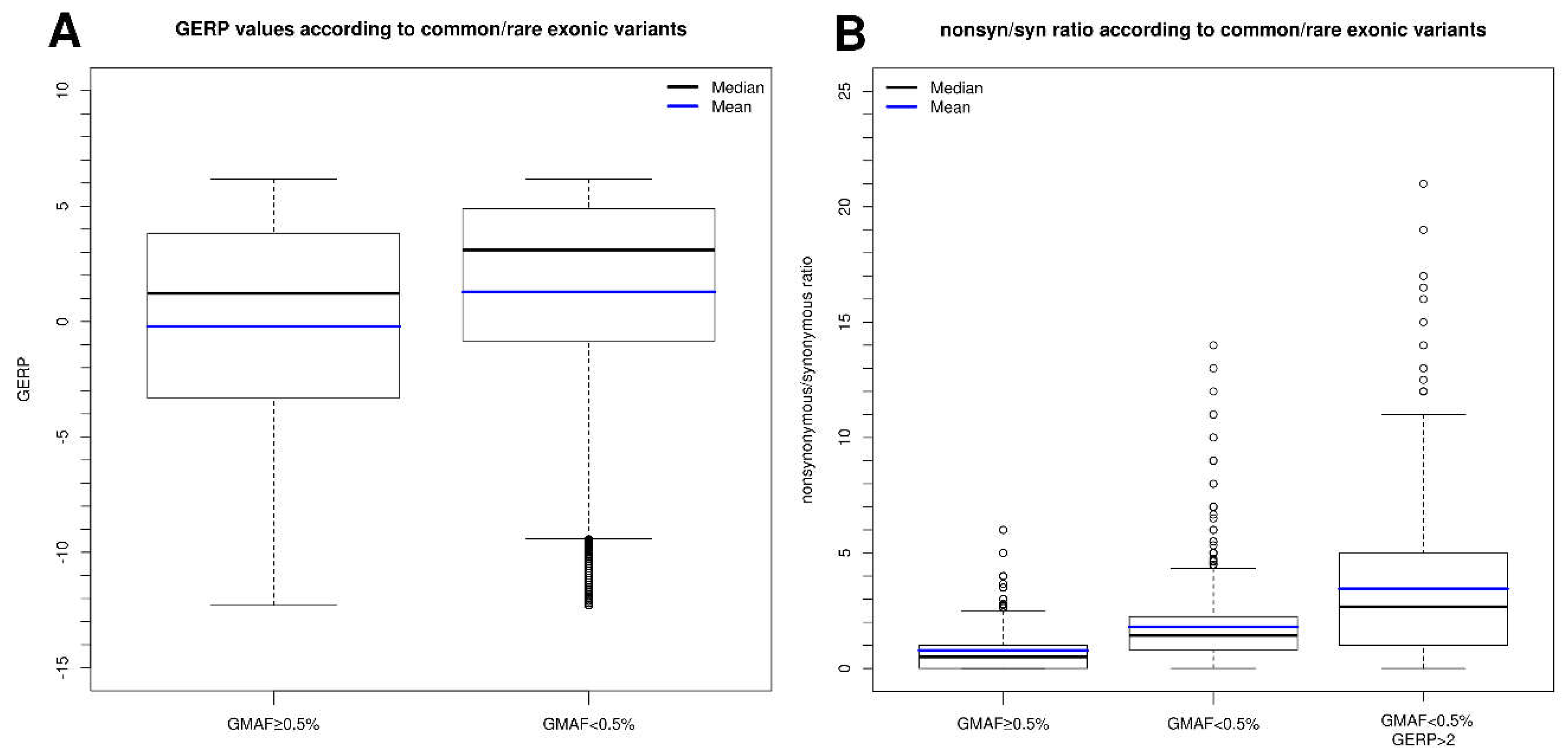
2.2. Information That Can Be Extracted from Common and Rare Variants in Control Sample Databases to Evaluate Mutation Tolerance
2.3. Mutation Tolerance of Genes Involved in Metabolic/Neurologic Diseases: Examples
2.4. Information Extracted from Rare Variant Spectrum for Genes Implicated in Metabolic/Neurologic Diseases: Examples
2.5. Conservation Score for Nucleotide(s) Affected in the Variant and Relationship with Variant Frequency
3. Mutational Architecture
3.1. KCNQ2: Disease Severity Depends on the Mutation Type (Truncating vs. Missense)
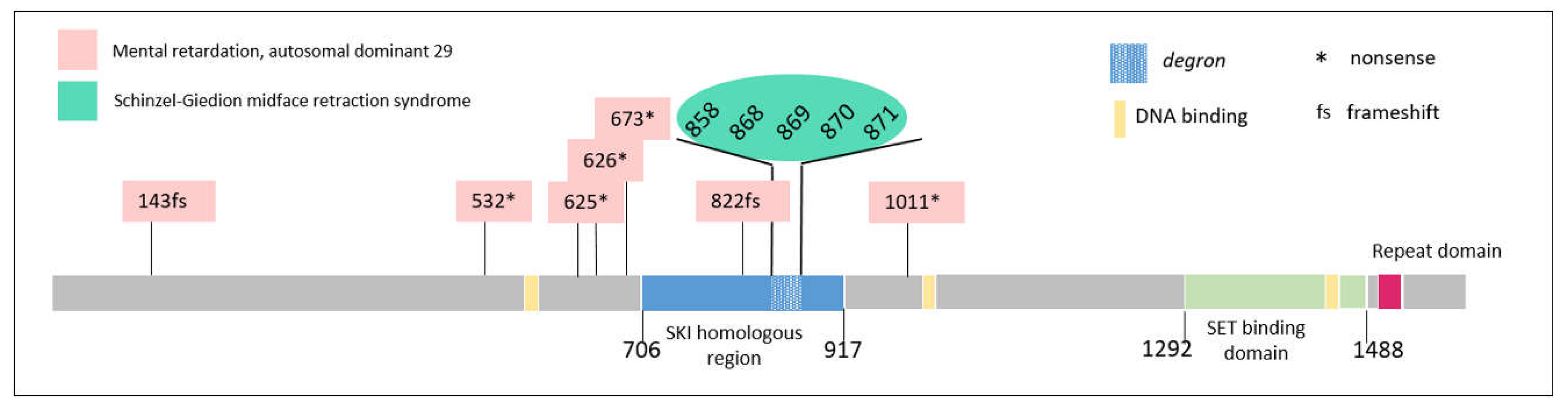
3.2. SETBP1: Phenotypes Differ Markedly Depending on the Type of Mutation, the Location Thereof, and the Percentage of Cells Affected
3.3. TCF4: Phenotype Depends on the Longitudinal Location of the Mutation in the Gene
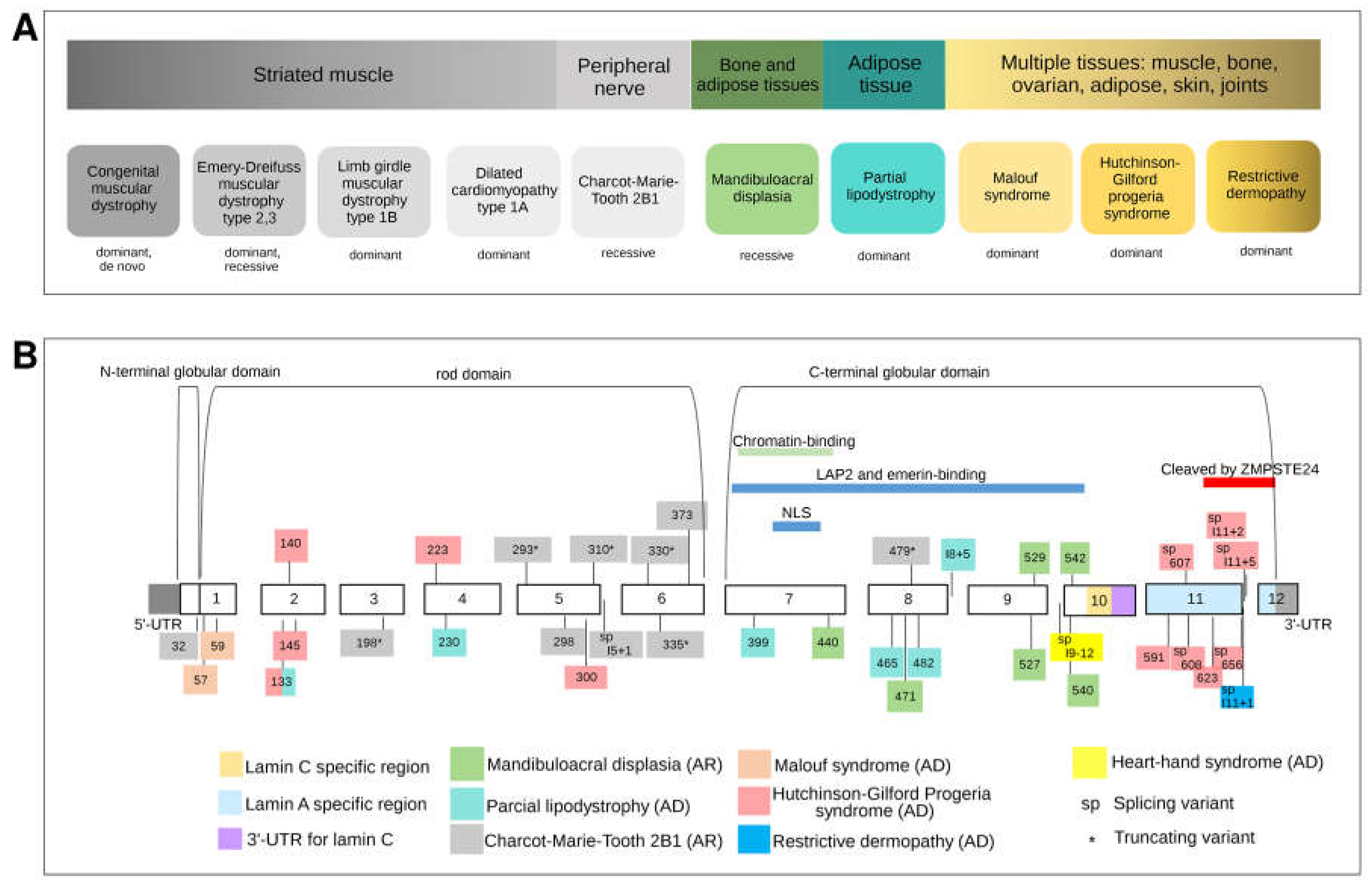
3.4. LMNA: The Malleable Gene Paradigm
3.4.1. Striated Muscle Diseases
3.4.2. Heart-Hand-Foot Involvement
3.4.3. Adipose Tissue Involvement
3.4.4. Peripheral Nerve Involvement
3.4.5. Mandibuloacral Dysplasia: Bone and Adipose Tissue Involvement
3.4.6. Multiple Tissue Involvement (Progeroid Syndromes)
Hutchinson–Gilford Progeria Syndrome
Malouf Syndrome
Restrictive Dermopathy
3.4.7. Combination of Phenotypes in Isolated Patients
3.4.8. Mutational Architecture of LMNA
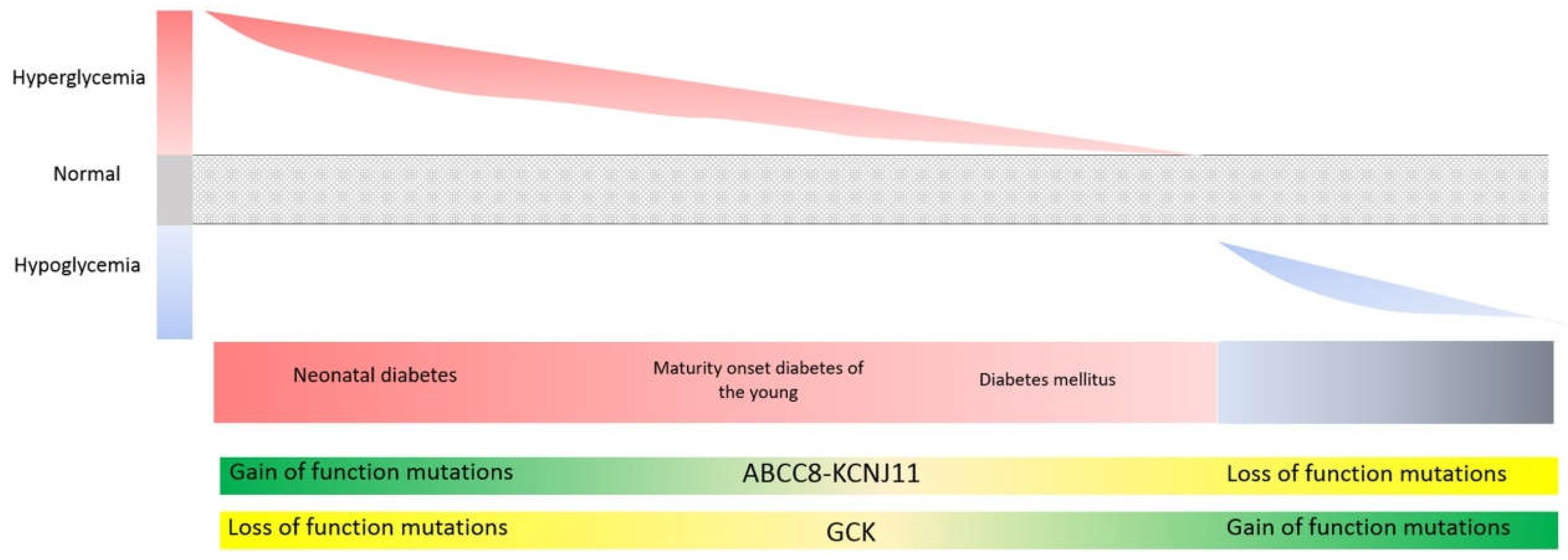
3.5. Genes Involved in Glycemic Control (ABCC8, KCNJ11, GCK, HNF1A, HNF4A): Different Mutation Types (Loss vs. Gain-of-Function) Can Give Rise to Opposing Phenotypes, While Identical Mutations Can Give Rise to Opposing Phenotypes at Different Stages of Life
KATP Potassium Channel
4. Inheritance
5. Conclusions
- to first prioritize genes based on their tolerance to the mutation (z-score) and the a priori probability of finding one (for dominant) or two (for recessive) variants in this gene by chance (proportional to its length);
- to characterize the mutational architecture of the gene in which the variants are located, i.e., how a given gene behaves in response to damage of different types (loss vs. gain-of-function mutations) or in different locations (e.g., protein domains);
- to perform familial analyses after variant prioritization in order to determine the mode of inheritance of the variants and assess their pathogenicity.
Acknowledgments
Conflicts of Interest
References
- Tennessen, J.A.; Bigham, A.W.; O’Connor, T.D.; Fu, W.; Kenny, E.E.; Gravel, S.; McGee, S.; Do, R.; Liu, X.; Jun, G.; et al. Exome Sequencing Project Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science 2012, 337, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Davydov, E.V.; Goode, D.L.; Sirota, M.; Cooper, G.M.; Sidow, A.; Batzoglou, S. Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput. Biol. 2010, 6, e1001025. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013. [Google Scholar] [CrossRef] [PubMed]
- Shihab, H.A.; Gough, J.; Cooper, D.N.; Stenson, P.D.; Barker, G.L.A.; Edwards, K.J.; Day, I.N.M.; Gaunt, T.R. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum. Mutat. 2013, 34, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Chun, S.; Fay, J.C. Identification of deleterious mutations within three human genomes. Genome Res. 2009, 19, 1553–1561. [Google Scholar] [CrossRef] [PubMed]
- Reva, B.; Antipin, Y.; Sander, C. Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic Acids Res. 2011, 39, e118. [Google Scholar] [CrossRef] [PubMed]
- González-Pérez, A.; López-Bigas, N. Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel. Am. J. Hum. Genet. 2011, 88, 440–449. [Google Scholar] [CrossRef] [PubMed]
- Li, M.-X.; Gui, H.-S.; Kwan, J.S.H.; Bao, S.-Y.; Sham, P.C. A comprehensive framework for prioritizing variants in exome sequencing studies of Mendelian diseases. Nucleic Acids Res. 2012, 40, e53. [Google Scholar] [CrossRef] [PubMed]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Knecht, C.; Mort, M.; Junge, O.; Cooper, D.N.; Krawczak, M.; Caliebe, A. IMHOTEP-a composite score integrating popular tools for predicting the functional consequences of non-synonymous sequence variants. Nucleic Acids Res. 2017, 45, e13. [Google Scholar] [CrossRef] [PubMed]
- Yeo, G.; Burge, C.B. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. 2004, 11, 377–394. [Google Scholar] [CrossRef] [PubMed]
- Desmet, F.-O.; Hamroun, D.; Lalande, M.; Collod-Béroud, G.; Claustres, M.; Béroud, C. Human Splicing Finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009, 37, e67. [Google Scholar] [CrossRef] [PubMed]
- 1000 Genomes Project Consortium; Abecasis, G.R.; Auton, A.; Brooks, L.D.; DePristo, M.A.; Durbin, R.M.; Handsaker, R.E.; Kang, H.M.; Marth, G.T.; McVean, G.A. An integrated map of genetic variation from 1092 human genomes. Nature 2012, 491, 56–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, N.G.C.; Webster, M.T.; Ellegren, H. Deterministic mutation rate variation in the human genome. Genome Res. 2002, 12, 1350–1356. [Google Scholar] [CrossRef] [PubMed]
- Nachman, M.W.; Crowell, S.L. Estimate of the mutation rate per nucleotide in humans. Genetics 2000, 156, 297–304. [Google Scholar] [PubMed]
- Petrovski, S.; Wang, Q.; Heinzen, E.L.; Allen, A.S.; Goldstein, D.B. Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet. 2013, 9, e1003709. [Google Scholar] [CrossRef]
- Feng, B.-J. PERCH: A Unified Framework for Disease Gene Prioritization. Hum. Mutat. 2017, 38, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, B.; Kronenberg, Z.; Hu, H.; Moore, B.; Flygare, S.; Reese, M.G.; Jorde, L.B.; Yandell, M.; Huff, C. Using VAAST to Identify Disease-Associated Variants in Next-Generation Sequencing Data. Curr. Protoc. Hum. Genet. 2014, 81, 6.14.1–6.14.25. [Google Scholar] [CrossRef] [PubMed]
- Rauch, A.; Wieczorek, D.; Graf, E.; Wieland, T.; Endele, S.; Schwarzmayr, T.; Albrecht, B.; Bartholdi, D.; Beygo, J.; Di Donato, N.; et al. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: An exome sequencing study. Lancet 2012, 380, 1674–1682. [Google Scholar] [CrossRef]
- Epi4K Consortium; Epilepsy Phenome/Genome Project; Allen, A.S.; Berkovic, S.F.; Cossette, P.; Delanty, N.; Dlugos, D.; Eichler, E.E.; Epstein, M.P.; Glauser, T.; et al. De novo mutations in epileptic encephalopathies. Nature 2013, 501, 217–221. [Google Scholar] [CrossRef] [PubMed]
- LeWinter, M.M.; Granzier, H.L. Titin is a major human disease gene. Circulation 2013, 127, 938–944. [Google Scholar] [CrossRef] [PubMed]
- Chauveau, C.; Rowell, J.; Ferreiro, A. A rising titan: TTN review and mutation update. Hum. Mutat. 2014, 35, 1046–1059. [Google Scholar] [CrossRef] [PubMed]
- Hackman, P.; Vihola, A.; Haravuori, H.; Marchand, S.; Sarparanta, J.; De Seze, J.; Labeit, S.; Witt, C.; Peltonen, L.; Richard, I.; et al. Tibial muscular dystrophy is a titinopathy caused by mutations in TTN, the gene encoding the giant skeletal-muscle protein titin. Am. J. Hum. Genet. 2002, 71, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, G.; Elliott, H.R.; Griffin, H.; Barresi, R.; Miller, J.; Marsh, J.; Evilä, A.; Vihola, A.; Hackman, P.; Straub, V.; et al. Titin mutation segregates with hereditary myopathy with early respiratory failure. Brain 2012, 135, 1695–1713. [Google Scholar] [CrossRef] [PubMed]
- Pénisson-Besnier, I.; Hackman, P.; Suominen, T.; Sarparanta, J.; Huovinen, S.; Richard-Crémieux, I.; Udd, B. Myopathies caused by homozygous titin mutations: Limb-girdle muscular dystrophy 2J and variations of phenotype. J. Neurol. Neurosurg. Psychiatry 2010, 81, 1200–1202. [Google Scholar] [CrossRef] [PubMed]
- Ceyhan-Birsoy, O.; Agrawal, P.B.; Hidalgo, C.; Schmitz-Abe, K.; DeChene, E.T.; Swanson, L.C.; Soemedi, R.; Vasli, N.; Iannaccone, S.T.; Shieh, P.B.; et al. Recessive truncating titin gene, TTN, mutations presenting as centronuclear myopathy. Neurology 2013, 81, 1205–1214. [Google Scholar] [CrossRef] [PubMed]
- Herman, D.S.; Lam, L.; Taylor, M.R.G.; Wang, L.; Teekakirikul, P.; Christodoulou, D.; Conner, L.; DePalma, S.R.; McDonough, B.; Sparks, E.; et al. Truncations of titin causing dilated cardiomyopathy. N. Engl. J. Med. 2012, 366, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Chauveau, C.; Bonnemann, C.G.; Julien, C.; Kho, A.L.; Marks, H.; Talim, B.; Maury, P.; Arne-Bes, M.C.; Uro-Coste, E.; Alexandrovich, A.; et al. Recessive TTN truncating mutations define novel forms of core myopathy with heart disease. Hum. Mol. Genet. 2014, 23, 980–991. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Marmiesse, A.; Carrascosa-Romero, M.C.; Alfaro Ponce, B.; Nascimento, A.; Ortez, C.; Romero, N.; Palacios, L.; Jimenez-Mallebrera, C.; Jou, C.; Gouveia, S.; et al. Homozygous truncating mutation in prenatally expressed skeletal isoform of TTN gene results in arthrogryposis multiplex congenita and myopathy without cardiac involvement. Neuromuscul. Disord. 2017, 27, 188–192. [Google Scholar] [CrossRef] [PubMed]
- Gussow, A.B.; Petrovski, S.; Wang, Q.; Allen, A.S.; Goldstein, D.B. The intolerance to functional genetic variation of protein domains predicts the localization of pathogenic mutations within genes. Genome Biol. 2016, 17, 9. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.S.; Pan, Z.; Shi, W.; Brown, B.S.; Wymore, R.S.; Cohen, I.S.; Dixon, J.E.; McKinnon, D. KCNQ2 and KCNQ3 potassium channel subunits: Molecular correlates of the M-channel. Science 1998, 282, 1890–1893. [Google Scholar] [CrossRef] [PubMed]
- Biervert, C.; Schroeder, B.C.; Kubisch, C.; Berkovic, S.F.; Propping, P.; Jentsch, T.J.; Steinlein, O.K. A potassium channel mutation in neonatal human epilepsy. Science 1998, 279, 403–406. [Google Scholar] [CrossRef] [PubMed]
- Charlier, C.; Singh, N.A.; Ryan, S.G.; Lewis, T.B.; Reus, B.E.; Leach, R.J.; Leppert, M. A pore mutation in a novel KQT-like potassium channel gene in an idiopathic epilepsy family. Nat. Genet. 1998, 18, 53–55. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.A.; Charlier, C.; Stauffer, D.; DuPont, B.R.; Leach, R.J.; Melis, R.; Ronen, G.M.; Bjerre, I.; Quattlebaum, T.; Murphy, J.V.; et al. A novel potassium channel gene, KCNQ2, is mutated in an inherited epilepsy of newborns. Nat. Genet. 1998, 18, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Yamagata, T.; Kubota, M.; Arai, H.; Yamashita, S.; Nakagawa, T.; Fujii, T.; Sugai, K.; Imai, K.; Uster, T.; et al. Clinical spectrum of early onset epileptic encephalopathies caused by KCNQ2 mutation. Epilepsia 2013, 54, 1282–1287. [Google Scholar] [CrossRef] [PubMed]
- Serino, D.; Specchio, N.; Pontrelli, G.; Vigevano, F.; Fusco, L. Video/EEG findings in a KCNQ2 epileptic encephalopathy: A case report and revision of literature data. Epileptic Disord. 2013, 15, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Saitsu, H.; Kato, M.; Koide, A.; Goto, T.; Fujita, T.; Nishiyama, K.; Tsurusaki, Y.; Doi, H.; Miyake, N.; Hayasaka, K.; et al. Whole exome sequencing identifies KCNQ2 mutations in Ohtahara syndrome. Ann. Neurol. 2012, 72, 298–300. [Google Scholar] [CrossRef] [PubMed]
- Weckhuysen, S.; Mandelstam, S.; Suls, A.; Audenaert, D.; Deconinck, T.; Claes, L.R.F.; Deprez, L.; Smets, K.; Hristova, D.; Yordanova, I.; et al. KCNQ2 encephalopathy: Emerging phenotype of a neonatal epileptic encephalopathy. Ann. Neurol. 2012, 71, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Weckhuysen, S.; Ivanovic, V.; Hendrickx, R.; Van Coster, R.; Hjalgrim, H.; Møller, R.S.; Grønborg, S.; Schoonjans, A.-S.; Ceulemans, B.; Heavin, S.B.; et al. Extending the KCNQ2 encephalopathy spectrum: Clinical and neuroimaging findings in 17 patients. Neurology 2013, 81, 1697–1703. [Google Scholar] [CrossRef] [PubMed]
- Filges, I.; Shimojima, K.; Okamoto, N.; Röthlisberger, B.; Weber, P.; Huber, A.R.; Nishizawa, T.; Datta, A.N.; Miny, P.; Yamamoto, T. Reduced expression by SETBP1 haploinsufficiency causes developmental and expressive language delay indicating a phenotype distinct from Schinzel-Giedion syndrome. J. Med. Genet. 2011, 48, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Marseglia, G.; Scordo, M.R.; Pescucci, C.; Nannetti, G.; Biagini, E.; Scandurra, V.; Gerundino, F.; Magi, A.; Benelli, M.; Torricelli, F. 372 kb microdeletion in 18q12.3 causing SETBP1 haploinsufficiency associated with mild mental retardation and expressive speech impairment. Eur. J. Med. Genet. 2012, 55, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Coe, B.P.; Witherspoon, K.; Rosenfeld, J.A.; van Bon, B.W.M.; Vulto-van Silfhout, A.T.; Bosco, P.; Friend, K.L.; Baker, C.; Buono, S.; Vissers, L.E.L.M.; et al. Refining analyses of copy number variation identifies specific genes associated with developmental delay. Nat. Genet. 2014, 46, 1063–1071. [Google Scholar] [CrossRef] [PubMed]
- Hamdan, F.F.; Srour, M.; Capo-Chichi, J.-M.; Daoud, H.; Nassif, C.; Patry, L.; Massicotte, C.; Ambalavanan, A.; Spiegelman, D.; Diallo, O.; et al. De novo mutations in moderate or severe intellectual disability. PLoS Genet. 2014, 10, e1004772. [Google Scholar] [CrossRef] [PubMed]
- Hoischen, A.; van Bon, B.W.M.; Gilissen, C.; Arts, P.; van Lier, B.; Steehouwer, M.; de Vries, P.; de Reuver, R.; Wieskamp, N.; Mortier, G.; et al. De novo mutations of SETBP1 cause Schinzel-Giedion syndrome. Nat. Genet. 2010, 42, 483–485. [Google Scholar] [CrossRef] [PubMed]
- Piazza, R.; Valletta, S.; Winkelmann, N.; Redaelli, S.; Spinelli, R.; Pirola, A.; Antolini, L.; Mologni, L.; Donadoni, C.; Papaemmanuil, E.; et al. Recurrent SETBP1 mutations in atypical chronic myeloid leukemia. Nat. Genet. 2013, 45, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, H.; Okuno, Y.; Muramatsu, H.; Yoshida, K.; Shiraishi, Y.; Takahashi, M.; Kon, A.; Sanada, M.; Chiba, K.; Tanaka, H.; et al. Exome sequencing identifies secondary mutations of SETBP1 and JAK3 in juvenile myelomonocytic leukemia. Nat. Genet. 2013, 45, 937–941. [Google Scholar] [CrossRef] [PubMed]
- Makishima, H.; Yoshida, K.; Nguyen, N.; Przychodzen, B.; Sanada, M.; Okuno, Y.; Ng, K.P.; Gudmundsson, K.O.; Vishwakarma, B.A.; Jerez, A.; et al. Somatic SETBP1 mutations in myeloid malignancies. Nat. Genet. 2013, 45, 942–946. [Google Scholar] [CrossRef] [PubMed]
- Hoischen, A.; Krumm, N.; Eichler, E.E. Prioritization of neurodevelopmental disease genes by discovery of new mutations. Nat. Neurosci. 2014, 17, 764–772. [Google Scholar] [CrossRef] [PubMed]
- Agha, M.M.; Williams, J.I.; Marrett, L.; To, T.; Zipursky, A.; Dodds, L. Congenital abnormalities and childhood cancer. Cancer 2005, 103, 1939–1948. [Google Scholar] [CrossRef] [PubMed]
- Bjørge, T.; Cnattingius, S.; Lie, R.T.; Tretli, S.; Engeland, A. Cancer risk in children with birth defects and in their families: A population based cohort study of 5.2 million children from Norway and Sweden. Cancer Epidemiol. Biomark. Prev. 2008, 17, 500–506. [Google Scholar] [CrossRef] [PubMed]
- Merks, J.H.M.; Ozgen, H.M.; Koster, J.; Zwinderman, A.H.; Caron, H.N.; Hennekam, R.C.M. Prevalence and patterns of morphological abnormalities in patients with childhood cancer. JAMA 2008, 299, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Durmaz, A.; Durmaz, B.; Kadioglu, B.; Aksoylar, S.; Karapinar, D.; Koturoglu, G.; Orman, M.N.; Ozkinay, F.; Cogulu, O. The Association of minor congenital anomalies and childhood cancer. Pediatr. Blood Cancer 2011, 56, 1098–1102. [Google Scholar] [CrossRef] [PubMed]
- Kelleher, F.C.; Fennelly, D.; Rafferty, M. Common critical pathways in embryogenesis and cancer. Acta Oncol. 2006, 45, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Acuna-Hidalgo, R.; Sengul, H.; Steehouwer, M.; van de Vorst, M.; Vermeulen, S.H.; Kiemeney, L.A.L.M.; Veltman, J.A.; Gilissen, C.; Hoischen, A. Ultra-sensitive Sequencing Identifies High Prevalence of Clonal Hematopoiesis-Associated Mutations throughout Adult Life. Am. J. Hum. Genet. 2017, 101, 50–64. [Google Scholar] [CrossRef] [PubMed]
- Marangi, G.; Ricciardi, S.; Orteschi, D.; Lattante, S.; Murdolo, M.; Dallapiccola, B.; Biscione, C.; Lecce, R.; Chiurazzi, P.; Romano, C.; et al. The Pitt-Hopkins syndrome: Report of 16 new patients and clinical diagnostic criteria. Am. J. Med. Genet. A 2011, 155, 1536–1545. [Google Scholar] [CrossRef] [PubMed]
- Whalen, S.; Héron, D.; Gaillon, T.; Moldovan, O.; Rossi, M.; Devillard, F.; Giuliano, F.; Soares, G.; Mathieu-Dramard, M.; Afenjar, A.; et al. Novel comprehensive diagnostic strategy in Pitt-Hopkins syndrome: Clinical score and further delineation of the TCF4 mutational spectrum. Hum. Mutat. 2012, 33, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Van Balkom, I.D.C.; Vuijk, P.J.; Franssens, M.; Hoek, H.W.; Hennekam, R.C.M. Development, cognition, and behaviour in Pitt-Hopkins syndrome. Dev. Med. Child Neurol. 2012, 54, 925–931. [Google Scholar] [CrossRef] [PubMed]
- Sweatt, J.D. Pitt-Hopkins Syndrome: Intellectual disability due to loss of TCF4-regulated gene transcription. Exp. Mol. Med. 2013, 45, e21. [Google Scholar] [CrossRef] [PubMed]
- De Winter, C.F.; Baas, M.; Bijlsma, E.K.; van Heukelingen, J.; Routledge, S.; Hennekam, R.C.M. Phenotype and natural history in 101 individuals with Pitt-Hopkins syndrome through an internet questionnaire system. Orphanet J. Rare Dis. 2016, 11, 37. [Google Scholar] [CrossRef] [PubMed]
- Bedeschi, M.F.; Marangi, G.; Calvello, M.R.; Ricciardi, S.; Leone, F.P.C.; Baccarin, M.; Guerneri, S.; Orteschi, D.; Murdolo, M.; Lattante, S.; et al. Impairment of different protein domains causes variable clinical presentation within Pitt-Hopkins syndrome and suggests intragenic molecular syndromology of TCF4. Eur. J. Med. Genet. 2017, 60, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Sepp, M.; Kannike, K.; Eesmaa, A.; Urb, M.; Timmusk, T. Functional diversity of human basic helix-loop-helix transcription factor TCF4 isoforms generated by alternative 5′ exon usage and splicing. PLoS ONE 2011, 6, e22138. [Google Scholar] [CrossRef] [PubMed]
- Forrest, M.P.; Hill, M.J.; Quantock, A.J.; Martin-Rendon, E.; Blake, D.J. The emerging roles of TCF4 in disease and development. Trends Mol. Med. 2014, 20, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Kalscheuer, V.M.; Feenstra, I.; Van Ravenswaaij-Arts, C.M.A.; Smeets, D.F.C.M.; Menzel, C.; Ullmann, R.; Musante, L.; Ropers, H.-H. Disruption of the TCF4 gene in a girl with mental retardation but without the classical Pitt-Hopkins syndrome. Am. J. Med. Genet. A 2008, 146, 2053–2059. [Google Scholar] [CrossRef] [PubMed]
- Maduro, V.; Pusey, B.N.; Cherukuri, P.F.; Atkins, P.; du Souich, C.; Rupps, R.; Limbos, M.; Adams, D.R.; Bhatt, S.S.; Eydoux, P.; et al. Complex translocation disrupting TCF4 and altering TCF4 isoform expression segregates as mild autosomal dominant intellectual disability. Orphanet J. Rare Dis. 2016, 11, 62. [Google Scholar] [CrossRef] [PubMed]
- Kharbanda, M.; Kannike, K.; Lampe, A.; Berg, J.; Timmusk, T.; Sepp, M. Partial deletion of TCF4 in three generation family with non-syndromic intellectual disability, without features of Pitt-Hopkins syndrome. Eur. J. Med. Genet. 2016, 59, 310–314. [Google Scholar] [CrossRef] [PubMed]
- Raffaele Di Barletta, M.; Ricci, E.; Galluzzi, G.; Tonali, P.; Mora, M.; Morandi, L.; Romorini, A.; Voit, T.; Orstavik, K.H.; Merlini, L.; et al. Different mutations in the LMNA gene cause autosomal dominant and autosomal recessive Emery-Dreifuss muscular dystrophy. Am. J. Hum. Genet. 2000, 66, 1407–1412. [Google Scholar] [CrossRef] [PubMed]
- Manilal, S.; Recan, D.; Sewry, C.A.; Hoeltzenbein, M.; Llense, S.; Leturcq, F.; Deburgrave, N.; Barbot, J.; Man, N.; Muntoni, F.; et al. Mutations in Emery-Dreifuss muscular dystrophy and their effects on emerin protein expression. Hum. Mol. Genet. 1998, 7, 855–864. [Google Scholar] [CrossRef] [PubMed]
- Yates, J.R.; Wehnert, M. The Emery-Dreifuss Muscular Dystrophy Mutation Database. Neuromuscul. Disord. 1999, 9, 199. [Google Scholar] [PubMed]
- Brown, C.A.; Lanning, R.W.; McKinney, K.Q.; Salvino, A.R.; Cherniske, E.; Crowe, C.A.; Darras, B.T.; Gominak, S.; Greenberg, C.R.; Grosmann, C.; et al. Novel and recurrent mutations in lamin A/C in patients with Emery-Dreifuss muscular dystrophy. Am. J. Med. Genet. 2001, 102, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Muchir, A.; Bonne, G.; van der Kooi, A.J.; van Meegen, M.; Baas, F.; Bolhuis, P.A.; de Visser, M.; Schwartz, K. Identification of mutations in the gene encoding lamins A/C in autosomal dominant limb girdle muscular dystrophy with atrioventricular conduction disturbances (LGMD1B). Hum. Mol. Genet. 2000, 9, 1453–1459. [Google Scholar] [CrossRef] [PubMed]
- Jacob, K.N.; Garg, A. Laminopathies: Multisystem dystrophy syndromes. Mol. Genet. Metab. 2006, 87, 289–302. [Google Scholar] [CrossRef] [PubMed]
- Todorova, A.; Halliger-Keller, B.; Walter, M.C.; Dabauvalle, M.-C.; Lochmüller, H.; Müller, C.R. A synonymous codon change in the LMNA gene alters mRNA splicing and causes limb girdle muscular dystrophy type 1B. J. Med. Genet. 2003, 40, e115. [Google Scholar] [CrossRef] [PubMed]
- Fatkin, D.; MacRae, C.; Sasaki, T.; Wolff, M.R.; Porcu, M.; Frenneaux, M.; Atherton, J.; Vidaillet, H.J.; Spudich, S.; De Girolami, U.; et al. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N. Engl. J. Med. 1999, 341, 1715–1724. [Google Scholar] [CrossRef] [PubMed]
- Brodsky, G.L.; Muntoni, F.; Miocic, S.; Sinagra, G.; Sewry, C.; Mestroni, L. Lamin A/C gene mutation associated with dilated cardiomyopathy with variable skeletal muscle involvement. Circulation 2000, 101, 473–476. [Google Scholar] [CrossRef] [PubMed]
- Quijano-Roy, S.; Mbieleu, B.; Bönnemann, C.G.; Jeannet, P.-Y.; Colomer, J.; Clarke, N.F.; Cuisset, J.-M.; Roper, H.; De Meirleir, L.; D’Amico, A.; et al. De novo LMNA mutations cause a new form of congenital muscular dystrophy. Ann. Neurol. 2008, 64, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Makri, S.; Clarke, N.F.; Richard, P.; Maugenre, S.; Demay, L.; Bonne, G.; Guicheney, P. Germinal mosaicism for LMNA mimics autosomal recessive congenital muscular dystrophy. Neuromuscul. Disord. 2009, 19, 26–28. [Google Scholar] [CrossRef] [PubMed]
- Renou, L.; Stora, S.; Yaou, R.B.; Volk, M.; Sinkovec, M.; Demay, L.; Richard, P.; Peterlin, B.; Bonne, G. Heart-hand syndrome of Slovenian type: A new kind of laminopathy. J. Med. Genet. 2008, 45, 666–671. [Google Scholar] [CrossRef] [PubMed]
- Dunnigan, M.G.; Cochrane, M.A.; Kelly, A.; Scott, J.W. Familial lipoatrophic diabetes with dominant transmission. A new syndrome. Q. J. Med. 1974, 43, 33–48. [Google Scholar] [PubMed]
- Cao, H.; Hegele, R.A. Nuclear lamin A/C R482Q mutation in canadian kindreds with Dunnigan-type familial partial lipodystrophy. Hum. Mol. Genet. 2000, 9, 109–112. [Google Scholar] [CrossRef] [PubMed]
- Shackleton, S.; Lloyd, D.J.; Jackson, S.N.; Evans, R.; Niermeijer, M.F.; Singh, B.M.; Schmidt, H.; Brabant, G.; Kumar, S.; Durrington, P.N.; et al. LMNA, encoding lamin A/C, is mutated in partial lipodystrophy. Nat. Genet. 2000, 24, 153–156. [Google Scholar] [CrossRef] [PubMed]
- Speckman, R.A.; Garg, A.; Du, F.; Bennett, L.; Veile, R.; Arioglu, E.; Taylor, S.I.; Lovett, M.; Bowcock, A.M. Mutational and haplotype analyses of families with familial partial lipodystrophy (Dunnigan variety) reveal recurrent missense mutations in the globular C-terminal domain of lamin A/C. Am. J. Hum. Genet. 2000, 66, 1192–1198. [Google Scholar] [CrossRef] [PubMed]
- Dhe-Paganon, S.; Werner, E.D.; Chi, Y.-I.; Shoelson, S.E. Structure of the globular tail of nuclear lamin. J. Biol. Chem. 2002, 277, 17381–17384. [Google Scholar] [CrossRef] [PubMed]
- Krimm, I.; Ostlund, C.; Gilquin, B.; Couprie, J.; Hossenlopp, P.; Mornon, J.-P.; Bonne, G.; Courvalin, J.-C.; Worman, H.J.; Zinn-Justin, S. The Ig-like structure of the C-terminal domain of lamin A/C, mutated in muscular dystrophies, cardiomyopathy, and partial lipodystrophy. Structure 2002, 10, 811–823. [Google Scholar] [CrossRef]
- De Sandre-Giovannoli, A.; Chaouch, M.; Kozlov, S.; Vallat, J.-M.; Tazir, M.; Kassouri, N.; Szepetowski, P.; Hammadouche, T.; Vandenberghe, A.; Stewart, C.L.; et al. Homozygous defects in LMNA, encoding lamin A/C nuclear-envelope proteins, cause autosomal recessive axonal neuropathy in human (Charcot-Marie-Tooth disorder type 2) and mouse. Am. J. Hum. Genet. 2002, 70, 726–736. [Google Scholar] [CrossRef] [PubMed]
- Tazir, M.; Azzedine, H.; Assami, S.; Sindou, P.; Nouioua, S.; Zemmouri, R.; Hamadouche, T.; Chaouch, M.; Feingold, J.; Vallat, J.M.; et al. Phenotypic variability in autosomal recessive axonal Charcot-Marie-Tooth disease due to the R298C mutation in lamin A/C. Brain 2004, 127, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Chaouch, M.; Allal, Y.; De Sandre-Giovannoli, A.; Vallat, J.M.; Amer-el-Khedoud, A.; Kassouri, N.; Chaouch, A.; Sindou, P.; Hammadouche, T.; Tazir, M.; et al. The phenotypic manifestations of autosomal recessive axonal Charcot-Marie-Tooth due to a mutation in Lamin A/C gene. Neuromuscul. Disord. 2003, 13, 60–67. [Google Scholar] [CrossRef]
- Novelli, G.; Muchir, A.; Sangiuolo, F.; Helbling-Leclerc, A.; D’Apice, M.R.; Massart, C.; Capon, F.; Sbraccia, P.; Federici, M.; Lauro, R.; et al. Mandibuloacral dysplasia is caused by a mutation in LMNA-encoding lamin A/C. Am. J. Hum. Genet. 2002, 71, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.; Cogulu, O.; Ozkinay, F.; Onay, H.; Agarwal, A.K. A novel homozygous Ala529Val LMNA mutation in Turkish patients with mandibuloacral dysplasia. J. Clin. Endocrinol. Metab. 2005, 90, 5259–5264. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.K.; Kazachkova, I.; Ten, S.; Garg, A. Severe mandibuloacral dysplasia-associated lipodystrophy and progeria in a young girl with a novel homozygous Arg527Cys LMNA mutation. J. Clin. Endocrinol. Metab. 2008, 93, 4617–4623. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.K.; Fryns, J.-P.; Auchus, R.J.; Garg, A. Zinc metalloproteinase, ZMPSTE24, is mutated in mandibuloacral dysplasia. Hum. Mol. Genet. 2003, 12, 1995–2001. [Google Scholar] [CrossRef] [PubMed]
- Filesi, I.; Gullotta, F.; Lattanzi, G.; D’Apice, M.R.; Capanni, C.; Nardone, A.M.; Columbaro, M.; Scarano, G.; Mattioli, E.; Sabatelli, P.; et al. Alterations of nuclear envelope and chromatin organization in mandibuloacral dysplasia, a rare form of laminopathy. Physiol. Genom. 2005, 23, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, J. Illustrations of Exceptional Symptoms and Examples of Rare Forms of Disease. Br. Med. J. 1886, 2, 148–149. [Google Scholar] [CrossRef] [PubMed]
- McKusick, V.A. The clinical observations of Jonathan Hutchinson. Am. J. Syph. Gonorrhea Vener. Dis. 1952, 36, 101–126. [Google Scholar] [PubMed]
- DeBusk, F.L. The Hutchinson-Gilford progeria syndrome. Report of 4 cases and review of the literature. J. Pediatr. 1972, 80, 697–724. [Google Scholar] [CrossRef]
- Merideth, M.A.; Gordon, L.B.; Clauss, S.; Sachdev, V.; Smith, A.C.M.; Perry, M.B.; Brewer, C.C.; Zalewski, C.; Kim, H.J.; Solomon, B.; et al. Phenotype and course of Hutchinson-Gilford progeria syndrome. N. Engl. J. Med. 2008, 358, 592–604. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, M.; Brown, W.T.; Gordon, L.B.; Glynn, M.W.; Singer, J.; Scott, L.; Erdos, M.R.; Robbins, C.M.; Moses, T.Y.; Berglund, P.; et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature 2003, 423, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Lee, L.; Kudlow, B.A.; Dos Santos, H.G.; Sletvold, O.; Shafeghati, Y.; Botha, E.G.; Garg, A.; Hanson, N.B.; Martin, G.M.; et al. LMNA mutations in atypical Werner’s syndrome. Lancet 2003, 362, 440–445. [Google Scholar] [CrossRef]
- Csoka, A.B.; English, S.B.; Simkevich, C.P.; Ginzinger, D.G.; Butte, A.J.; Schatten, G.P.; Rothman, F.G.; Sedivy, J.M. Genome-scale expression profiling of Hutchinson-Gilford progeria syndrome reveals widespread transcriptional misregulation leading to mesodermal/mesenchymal defects and accelerated atherosclerosis. Aging Cell 2004, 3, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Verstraeten, V.L.R.M.; Broers, J.L.V.; van Steensel, M.A.M.; Zinn-Justin, S.; Ramaekers, F.C.S.; Steijlen, P.M.; Kamps, M.; Kuijpers, H.J.H.; Merckx, D.; Smeets, H.J.M.; et al. Compound heterozygosity for mutations in LMNA causes a progeria syndrome without prelamin A accumulation. Hum. Mol. Genet. 2006, 15, 2509–2522. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.; Leistritz, D.F.; Turner, L.; MacGregor, D.; Ohson, K.; Dancey, P.; Martin, G.M.; Oshima, J. Collagen expression in fibroblasts with a novel LMNA mutation. Biochem. Biophys. Res. Commun. 2007, 352, 603–608. [Google Scholar] [CrossRef] [PubMed]
- McPherson, E.; Turner, L.; Zador, I.; Reynolds, K.; Macgregor, D.; Giampietro, P.F. Ovarian failure and dilated cardiomyopathy due to a novel lamin mutation. Am. J. Med. Genet. A 2009, 149, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Malouf, J.; Alam, S.; Kanj, H.; Mufarrij, A.; Der Kaloustian, V.M. Hypergonadotropic hypogonadism with congestive cardiomyopathy: An autosomal-recessive disorder? Am. J. Med. Genet. 1985, 20, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Navarro, C.L.; De Sandre-Giovannoli, A.; Bernard, R.; Boccaccio, I.; Boyer, A.; Geneviève, D.; Hadj-Rabia, S.; Gaudy-Marqueste, C.; Smitt, H.S.; Vabres, P.; et al. Lamin A and ZMPSTE24 (FACE-1) defects cause nuclear disorganization and identify restrictive dermopathy as a lethal neonatal laminopathy. Hum. Mol. Genet. 2004, 13, 2493–2503. [Google Scholar] [CrossRef] [PubMed]
- Navarro, C.L.; Cadiñanos, J.; De Sandre-Giovannoli, A.; Bernard, R.; Courrier, S.; Boccaccio, I.; Boyer, A.; Kleijer, W.J.; Wagner, A.; Giuliano, F.; et al. Loss of ZMPSTE24 (FACE-1) causes autosomal recessive restrictive dermopathy and accumulation of Lamin A precursors. Hum. Mol. Genet. 2005, 14, 1503–1513. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, F.; Gullotta, F.; Columbaro, M.; Filareto, A.; D’Adamo, M.; Vielle, A.; Guglielmi, V.; Nardone, A.M.; Azzolini, V.; Grosso, E.; et al. Compound heterozygosity for mutations in LMNA in a patient with a myopathic and lipodystrophic mandibuloacral dysplasia type A phenotype. J. Clin. Endocrinol. Metab. 2007, 92, 4467–4471. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.J.; Grogan, M.; Ackerman, M.J. LMNA-Mediated Arrhythmogenic Right Ventricular Cardiomyopathy and Charcot-Marie-Tooth Type 2B1: A Patient-Discovered Unifying Diagnosis. J. Cardiovasc. Electrophysiol. 2016, 27, 868–871. [Google Scholar] [CrossRef] [PubMed]
- Iwahara, N.; Hisahara, S.; Hayashi, T.; Kawamata, J.; Shimohama, S. A novel lamin A/C gene mutation causing spinal muscular atrophy phenotype with cardiac involvement: Report of one case. BMC Neurol. 2015, 15, 13. [Google Scholar] [CrossRef] [PubMed]
- Worman, H.J.; Bonne, G. “Laminopathies”: A wide spectrum of human diseases. Exp. Cell Res. 2007, 313, 2121–2133. [Google Scholar] [CrossRef] [PubMed]
- Wiesel, N.; Mattout, A.; Melcer, S.; Melamed-Book, N.; Herrmann, H.; Medalia, O.; Aebi, U.; Gruenbaum, Y. Laminopathic mutations interfere with the assembly, localization, and dynamics of nuclear lamins. Proc. Natl. Acad. Sci. USA 2008, 105, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Shimi, T.; Butin-Israeli, V.; Adam, S.A.; Goldman, R.D. Nuclear lamins in cell regulation and disease. Cold Spring Harb. Symp. Quant. Biol. 2010, 75, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Dubinska-Magiera, M.; Zaremba-Czogalla, M.; Rzepecki, R. Muscle development, regeneration and laminopathies: How lamins or lamina-associated proteins can contribute to muscle development, regeneration and disease. Cell. Mol. Life Sci. 2013, 70, 2713–2741. [Google Scholar] [CrossRef] [PubMed]
- Stoffel, M.; Froguel, P.; Takeda, J.; Zouali, H.; Vionnet, N.; Nishi, S.; Weber, I.T.; Harrison, R.W.; Pilkis, S.J.; Lesage, S. Human glucokinase gene: Isolation, characterization, and identification of two missense mutations linked to early-onset non-insulin-dependent (type 2) diabetes mellitus. Proc. Natl. Acad. Sci. USA 1992, 89, 7698–7702. [Google Scholar] [CrossRef] [PubMed]
- Thornton, P.S.; Satin-Smith, M.S.; Herold, K.; Glaser, B.; Chiu, K.C.; Nestorowicz, A.; Permutt, M.A.; Baker, L.; Stanley, C.A. Familial hyperinsulinism with apparent autosomal dominant inheritance: Clinical and genetic differences from the autosomal recessive variant. J. Pediatr. 1998, 132, 9–14. [Google Scholar] [CrossRef]
- Glaser, B.; Kesavan, P.; Heyman, M.; Davis, E.; Cuesta, A.; Buchs, A.; Stanley, C.A.; Thornton, P.S.; Permutt, M.A.; Matschinsky, F.M.; et al. Familial hyperinsulinism caused by an activating glucokinase mutation. N. Engl. J. Med. 1998, 338, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Njølstad, P.R.; Søvik, O.; Cuesta-Muñoz, A.; Bjørkhaug, L.; Massa, O.; Barbetti, F.; Undlien, D.E.; Shiota, C.; Magnuson, M.A.; Molven, A.; et al. Neonatal diabetes mellitus due to complete glucokinase deficiency. N. Engl. J. Med. 2001, 344, 1588–1592. [Google Scholar] [CrossRef] [PubMed]
- Pearson, E.R.; Boj, S.F.; Steele, A.M.; Barrett, T.; Stals, K.; Shield, J.P.; Ellard, S.; Ferrer, J.; Hattersley, A.T. Macrosomia and hyperinsulinaemic hypoglycaemia in patients with heterozygous mutations in the HNF4A gene. PLoS Med. 2007, 4, e118. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, R.R.; Locke, J.; Colclough, K.; Wales, J.; Conn, J.J.; Hattersley, A.T.; Ellard, S.; Hussain, K. Persistent hyperinsulinemic hypoglycemia and maturity-onset diabetes of the young due to heterozygous HNF4A mutations. Diabetes 2008, 57, 1659–1663. [Google Scholar] [CrossRef] [PubMed]
- Stanescu, D.E.; Hughes, N.; Kaplan, B.; Stanley, C.A.; De León, D.D. Novel presentations of congenital hyperinsulinism due to mutations in the MODY genes: HNF1A and HNF4A. J. Clin. Endocrinol. Metab. 2012, 97, E2026–E2030. [Google Scholar] [CrossRef] [PubMed]
- Tung, J.Y.-L.; Boodhansingh, K.; Stanley, C.A.; De León, D.D. Clinical heterogeneity of hyperinsulinism due to HNF1A and HNF4A mutations. Pediatr. Diabetes 2018. [Google Scholar] [CrossRef] [PubMed]
- Simons, Y.B.; Turchin, M.C.; Pritchard, J.K.; Sella, G. The deleterious mutation load is insensitive to recent population history. Nat. Genet. 2014, 46, 220–224. [Google Scholar] [CrossRef] [PubMed]
- Simons, Y.B.; Sella, G. The impact of recent population history on the deleterious mutation load in humans and close evolutionary relatives. Curr. Opin. Genet. Dev. 2016, 41, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Nei, M. The frequency distribution of lethal chromosomes in finite populations. Proc. Natl. Acad. Sci. USA 1968, 60, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Brandvain, Y.; Wright, S.I. The Limits of Natural Selection in a Nonequilibrium World. Trends Genet. 2016, 32, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Balick, D.J.; Do, R.; Cassa, C.A.; Reich, D.; Sunyaev, S.R. Dominance of Deleterious Alleles Controls the Response to a Population Bottleneck. PLoS Genet. 2015, 11, e1005436. [Google Scholar] [CrossRef] [PubMed]
- Beauchamp, K.A.; Muzzey, D.; Wong, K.K.; Hogan, G.J.; Karimi, K.; Candille, S.I.; Mehta, N.; Mar-Heyming, R.; Kaseniit, K.E.; Kang, H.P.; et al. Systematic design and comparison of expanded carrier screening panels. Genet. Med. 2018, 20, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Conrad, D.F.; Keebler, J.E.M.; DePristo, M.A.; Lindsay, S.J.; Zhang, Y.; Casals, F.; Idaghdour, Y.; Hartl, C.L.; Torroja, C.; Garimella, K.V.; et al. Variation in genome-wide mutation rates within and between human families. Nat. Genet. 2011, 43, 712–714. [Google Scholar] [CrossRef] [PubMed]
- Crow, J.F. The origins, patterns and implications of human spontaneous mutation. Nat. Rev. Genet. 2000, 1, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Eyre-Walker, A.; Keightley, P.D. The distribution of fitness effects of new mutations. Nat. Rev. Genet. 2007, 8, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Veltman, J.A.; Brunner, H.G. De novo mutations in human genetic disease. Nat. Rev. Genet. 2012, 13, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Vissers, L.E.L.M.; de Ligt, J.; Gilissen, C.; Janssen, I.; Steehouwer, M.; de Vries, P.; van Lier, B.; Arts, P.; Wieskamp, N.; del Rosario, M.; et al. A de novo paradigm for mental retardation. Nat. Genet. 2010, 42, 1109–1112. [Google Scholar] [CrossRef] [PubMed]
- De Ligt, J.; Willemsen, M.H.; van Bon, B.W.M.; Kleefstra, T.; Yntema, H.G.; Kroes, T.; Vulto-van Silfhout, A.T.; Koolen, D.A.; de Vries, P.; Gilissen, C.; et al. Diagnostic exome sequencing in persons with severe intellectual disability. N. Engl. J. Med. 2012, 367, 1921–1929. [Google Scholar] [CrossRef] [PubMed]
- O’Roak, B.J.; Deriziotis, P.; Lee, C.; Vives, L.; Schwartz, J.J.; Girirajan, S.; Karakoc, E.; Mackenzie, A.P.; Ng, S.B.; Baker, C.; et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat. Genet. 2011, 43, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Yuen, R.K.C.; Jin, X.; Wang, M.; Chen, N.; Wu, X.; Ju, J.; Mei, J.; Shi, Y.; He, M.; et al. Detection of clinically relevant genetic variants in autism spectrum disorder by whole-genome sequencing. Am. J. Hum. Genet. 2013, 93, 249–263. [Google Scholar] [CrossRef] [PubMed]
- O’Roak, B.J.; Stessman, H.A.; Boyle, E.A.; Witherspoon, K.T.; Martin, B.; Lee, C.; Vives, L.; Baker, C.; Hiatt, J.B.; Nickerson, D.A.; et al. Recurrent de novo mutations implicate novel genes underlying simplex autism risk. Nat. Commun. 2014, 5, 5595. [Google Scholar] [CrossRef] [PubMed]
- Sanders, S.J.; Murtha, M.T.; Gupta, A.R.; Murdoch, J.D.; Raubeson, M.J.; Willsey, A.J.; Ercan-Sencicek, A.G.; DiLullo, N.M.; Parikshak, N.N.; Stein, J.L.; et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 2012, 485, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Neale, B.M.; Kou, Y.; Liu, L.; Ma’ayan, A.; Samocha, K.E.; Sabo, A.; Lin, C.-F.; Stevens, C.; Wang, L.-S.; Makarov, V.; et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 2012, 485, 242–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, B.; Roos, J.L.; Dexheimer, P.; Boone, B.; Plummer, B.; Levy, S.; Gogos, J.A.; Karayiorgou, M. Exome sequencing supports a de novo mutational paradigm for schizophrenia. Nat. Genet. 2011, 43, 864–868. [Google Scholar] [CrossRef] [PubMed]
- Girard, S.L.; Gauthier, J.; Noreau, A.; Xiong, L.; Zhou, S.; Jouan, L.; Dionne-Laporte, A.; Spiegelman, D.; Henrion, E.; Diallo, O.; et al. Increased exonic de novo mutation rate in individuals with schizophrenia. Nat. Genet. 2011, 43, 860–863. [Google Scholar] [CrossRef] [PubMed]
- EuroEPINOMICS-RES Consortium; Epilepsy Phenome/Genome Project; Epi4K Consortium. De novo mutations in synaptic transmission genes including DNM1 cause epileptic encephalopathies. Am. J. Hum. Genet. 2014, 95, 360–370. [Google Scholar] [CrossRef]
- Hamdan, F.F.; Myers, C.T.; Cossette, P.; Lemay, P.; Spiegelman, D.; Laporte, A.D.; Nassif, C.; Diallo, O.; Monlong, J.; Cadieux-Dion, M.; et al. High Rate of Recurrent De Novo Mutations in Developmental and Epileptic Encephalopathies. Am. J. Hum. Genet. 2017, 101, 664–685. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
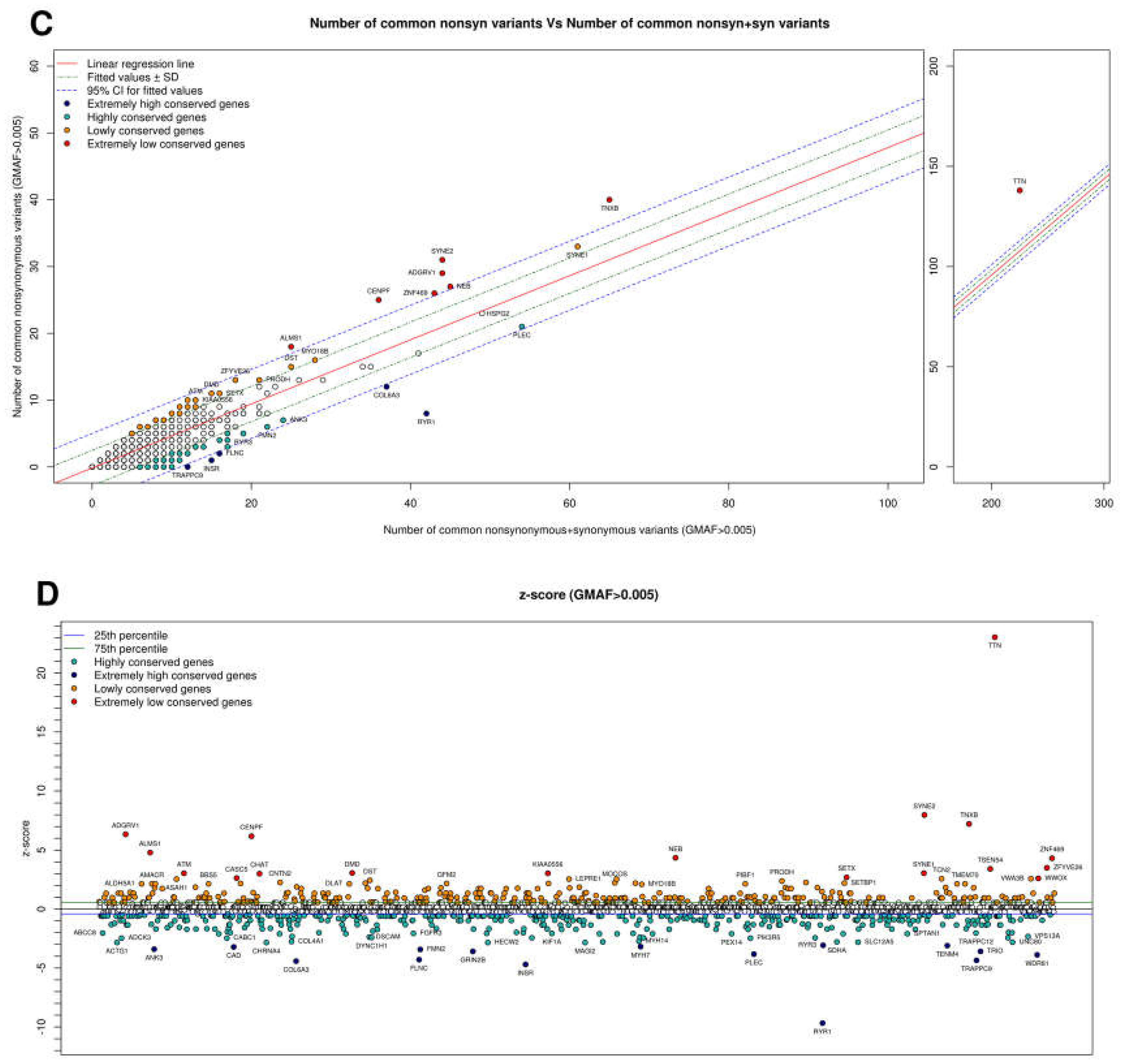
| Most Intolerant Genes | Most Tolerant Genes | ||
|---|---|---|---|
| Gene | z-Score | Gene | z-Score |
| <1000 bp | <1000 bp | ||
| COLEC11 | −1.377098 | TM4SF20 | 2.215303 |
| CAV3 | −1.024795 | TMEM70 | 2.215303 |
| CHCHD10 | −1.024795 | COQ4 | 1.797098 |
| FAS | −1.024795 | HACD1 | 1.797098 |
| MRTO4 | −1.024795 | NDUFAF1 | 1.41199 |
| 1000–2500 bp | 1000–2500 bp | ||
| ACTG1 | −2.953587 | TSEN54 | 3.541998 |
| CHRNA4 | −2.953587 | CHAT | 3.120652 |
| SDHA | −2.889489 | WWOX | 2.700392 |
| ADCK3 | −2.566604 | TCN2 | 2.667196 |
| BRF1 | −2.180352 | ASAH1 | 2.634213 |
| 2500–5000 bp | 2500–5000 bp | ||
| INSR | −4.872409 | KIAA0556 | 3.154369 |
| TRAPPC9 | −4.51124 | VWA3B | 2.667196 |
| GRIN2B | −3.730224 | MOGS | 2.634213 |
| HECW2 | −2.953587 | CNTN2 | 2.347576 |
| MAGI2 | −2.953587 | RECQL4 | 2.347576 |
| >5000 bp | >5000 bp | ||
| RYR1 | −10.002224 | TTN | 24.20914 |
| COL6A3 | −4.54985 | SYNE2 | 8.331247 |
| FLNC | −4.448544 | TNXB | 7.571299 |
| WDR81 | −4.025886 | ADGRV1 | 6.627985 |
| PLEC | −3.909057 | CENPF | 6.455138 |
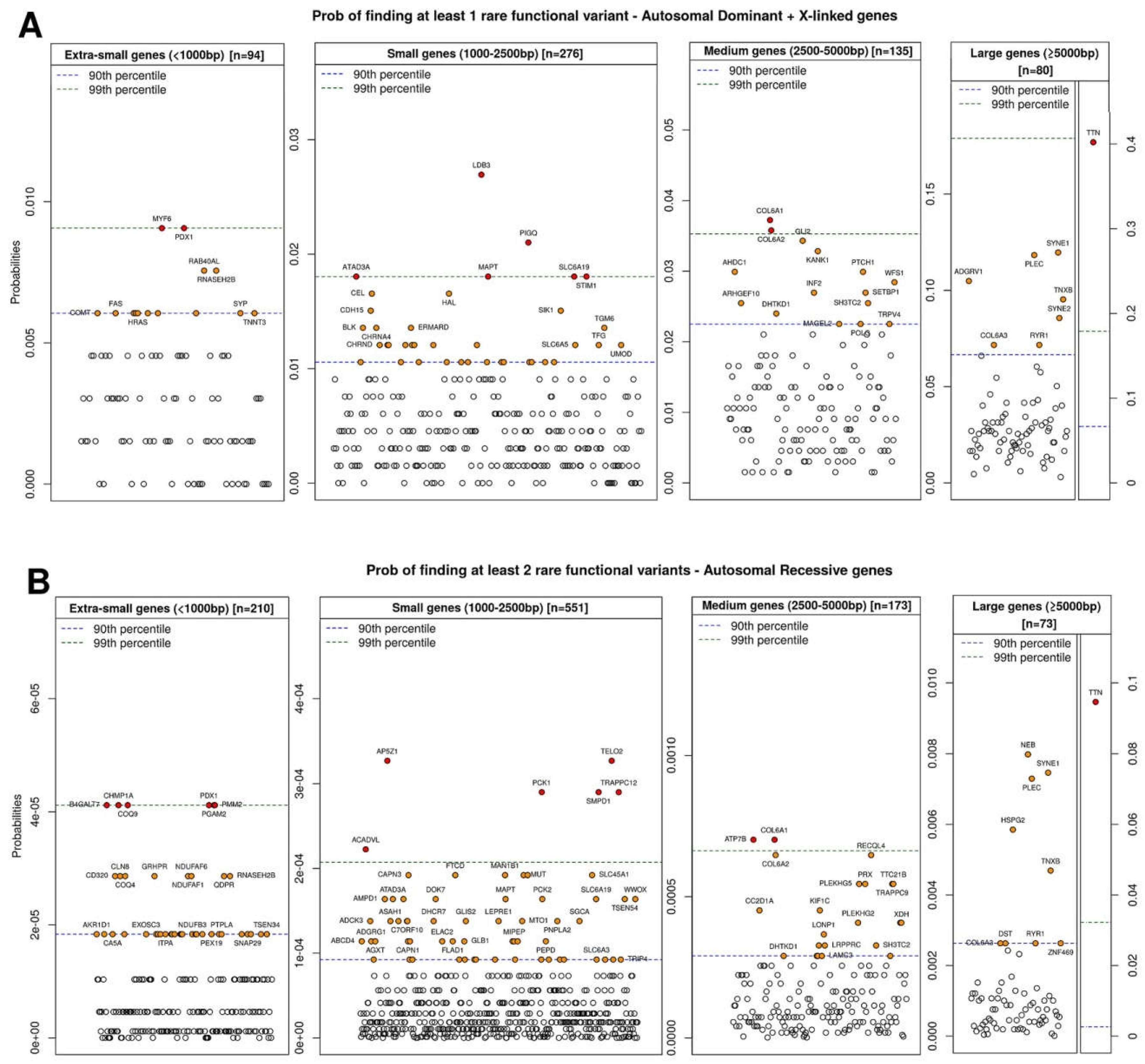
| Dominant Genes | Recessive Genes | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| <1000 bp | p | 1000–2500 bp | p | 2500–5000 bp | p | <1000 bp | p | 1000–2500 bp | p | 2500–5000 bp | p |
| HBA1 | 0 | ACTB | 0 | ATP1A3 | 0.0015 | ARL6IP1 | 0 | ADK | 0 | HK1 | 1.15 × 10−6 |
| HSPB3 | 0 | BRAF | 0 | CNNM2 | 0.0015 | ATP5E | 0 | ALG6 | 0 | ZC3H14 | 4.59 × 10−6 |
| NRAS | 0 | EEF1A2 | 0 | EEF2 | 0.0015 | BBIP1 | 0 | CHST14 | 0 | GRM1 | 1.03 × 10−5 |
| PURA | 0 | GABRG2 | 0 | KIF5C | 0.0015 | BOLA3 | 0 | CLP1 | 0 | ZAK | 1.03 × 10−5 |
| SDHD | 0 | GJC2 | 0 | GRIN1 | 0.0030 | C11ORF73 | 0 | DNAJC3 | 0 | DDHD1 | 1.83 × 10−5 |
| SNAP25 | 0 | GNB1 | 0 | GRIN2D | 0.0030 | CLPP | 0 | ERLIN1 | 0 | HACE1 | 1.83 × 10−5 |
| STX1B | 0 | HSPD1 | 0 | LZTR1 | 0.0030 | COA5 | 0 | GMPPB | 0 | MAGI2 | 1.83 × 10−5 |
| THAP1 | 0 | KIF2A | 0 | MYT1L | 0.0030 | COX14 | 0 | MARS2 | 0 | NDST1 | 1.83 × 10−5 |
| VAMP1 | 0 | LGI1 | 0 | DNM1 | 0.0045 | COX6B1 | 0 | NEU1 | 0 | OGDH | 1.83 × 10−5 |
| YWHAG | 0 | MAT1A | 0 | EFTUD2 | 0.0045 | DNAJB2 | 0 | NHLRC1 | 0 | RAB3GAP1 | 1.83 × 10−5 |
| PPP3CA | 0 | >5000 bp | p | GM2A | 0 | POGLUT1 | 0 | >5000 bp | p | ||
| RAF1 | 0 | CHD2 | 0.0075 | MRPL44 | 0 | SAMHD1 | 0 | CAD | 4.11 × 10−5 | ||
| TPM2 | 0 | SCN2A | 0.0075 | NDUFA12 | 0 | SLC39A14 | 0 | CCDC88A | 5.60 × 10−5 | ||
| TPM3 | 0 | SCN1A | 0.0105 | PARK7 | 0 | ST3GAL5 | 0 | TRIO | 7.30 × 10−5 | ||
| TREX1 | 0 | SMARCA4 | 0.0120 | PCBD1 | 0 | TRMT10C | 0 | ARFGEF2 | 1.37 × 10−4 | ||
| TUBA1A | 0 | CACNA1B | 0.0135 | PCNA | 0 | WDR73 | 0 | CC2D2A | 1.92 × 10−4 | ||
| TUBB | 0 | SCN8A | 0.0135 | PDE6D | 0 | YME1L1 | 0 | DOCK7 | 1.92 × 10−4 | ||
| TUBB2B | 0 | NALCN | 0.0150 | RPIA | 0 | LAMB2 | 1.92 × 10−4 | ||||
| ZIC2 | 0 | ARID1A | 0.0165 | TMEM138 | 0 | ATR | 2.22 × 10−4 | ||||
| ZMYND11 | 0 | DYNC1H1 | 0.0165 | TXN2 | 0 | CIT | 2.22 × 10−4 | ||||
| ITPR1 | 0.0165 | UQCC3 | 0 | ALS2 | 2.90 × 10−4 | ||||||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roca, I.; Fernández-Marmiesse, A.; Gouveia, S.; Segovia, M.; Couce, M.L. Prioritization of Variants Detected by Next Generation Sequencing According to the Mutation Tolerance and Mutational Architecture of the Corresponding Genes. Int. J. Mol. Sci. 2018, 19, 1584. https://doi.org/10.3390/ijms19061584
Roca I, Fernández-Marmiesse A, Gouveia S, Segovia M, Couce ML. Prioritization of Variants Detected by Next Generation Sequencing According to the Mutation Tolerance and Mutational Architecture of the Corresponding Genes. International Journal of Molecular Sciences. 2018; 19(6):1584. https://doi.org/10.3390/ijms19061584
Chicago/Turabian StyleRoca, Iria, Ana Fernández-Marmiesse, Sofía Gouveia, Marta Segovia, and María L. Couce. 2018. "Prioritization of Variants Detected by Next Generation Sequencing According to the Mutation Tolerance and Mutational Architecture of the Corresponding Genes" International Journal of Molecular Sciences 19, no. 6: 1584. https://doi.org/10.3390/ijms19061584
APA StyleRoca, I., Fernández-Marmiesse, A., Gouveia, S., Segovia, M., & Couce, M. L. (2018). Prioritization of Variants Detected by Next Generation Sequencing According to the Mutation Tolerance and Mutational Architecture of the Corresponding Genes. International Journal of Molecular Sciences, 19(6), 1584. https://doi.org/10.3390/ijms19061584