Extension of Tissue Plasminogen Activator Treatment Window by Granulocyte-Colony Stimulating Factor in a Thromboembolic Rat Model of Stroke

 ,
, 

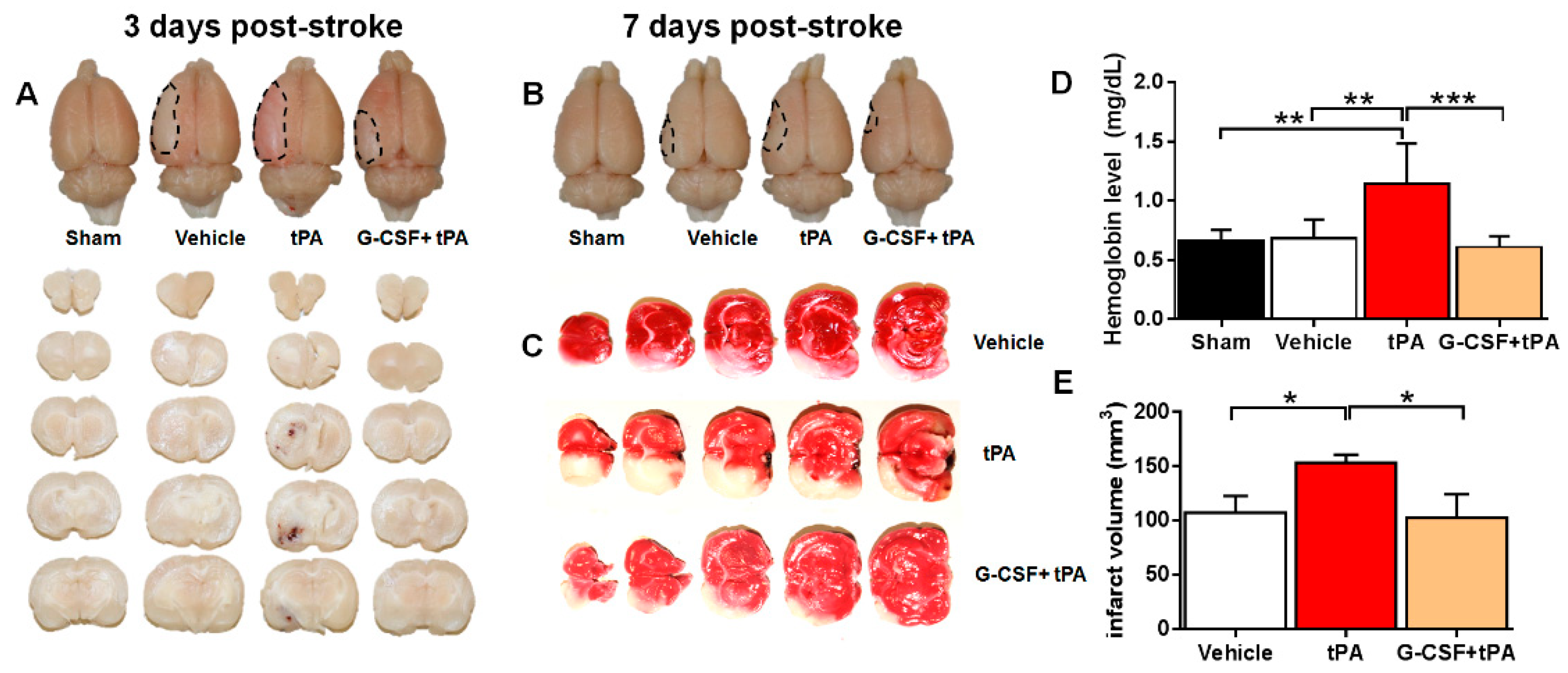{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. G-CSF Attenuated Delayed tPA-Induced Hemorrhage and Cerebral Infarction in a TE Stroke Model
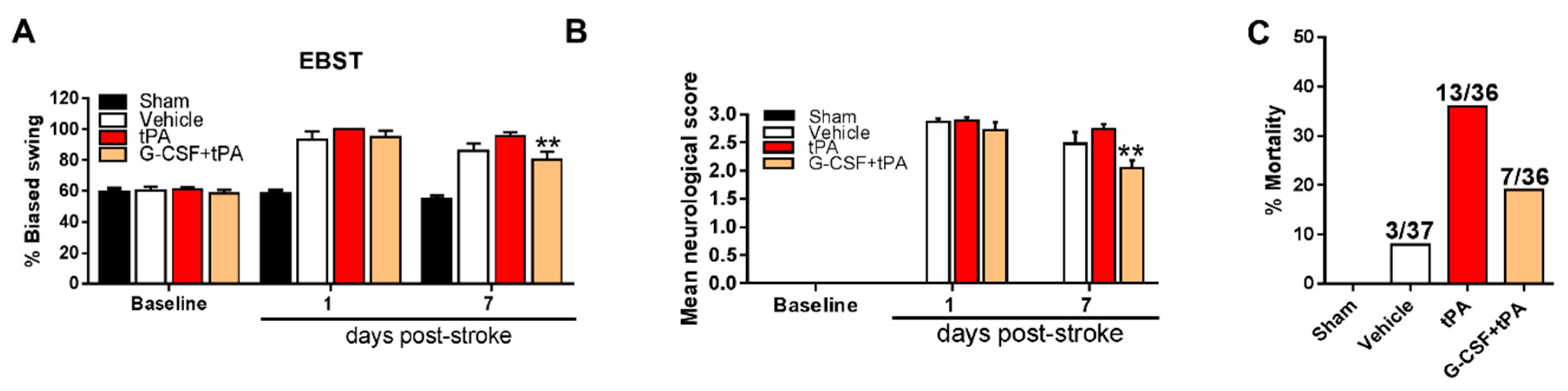
2.2. G-CSF Decreased Delayed tPA-Induced Neurological Deficits and Mortality
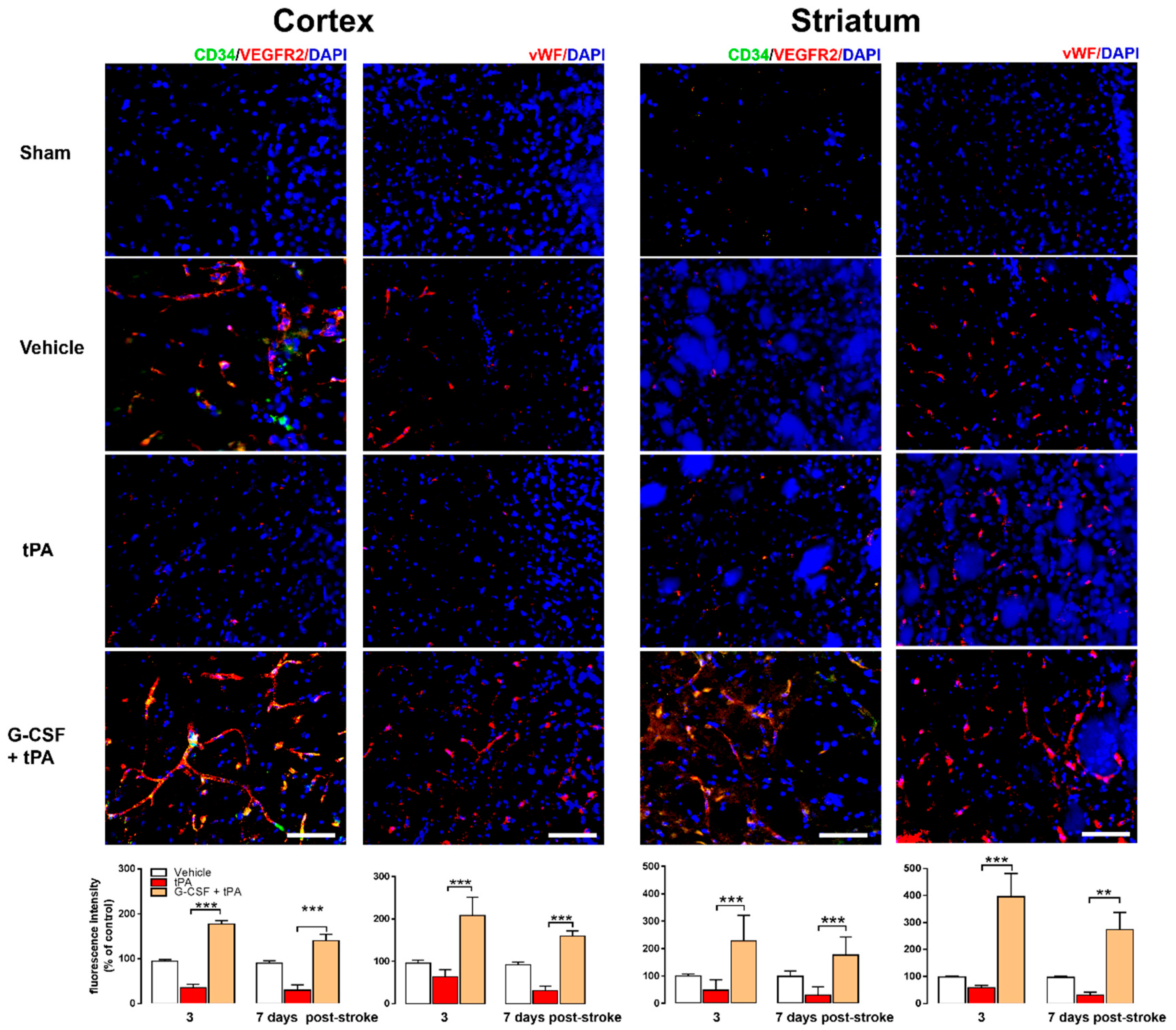
2.3. Increased CD34+ and VEGFR-2 and vWF Expression in G-CSF+tPA-Treated Rats
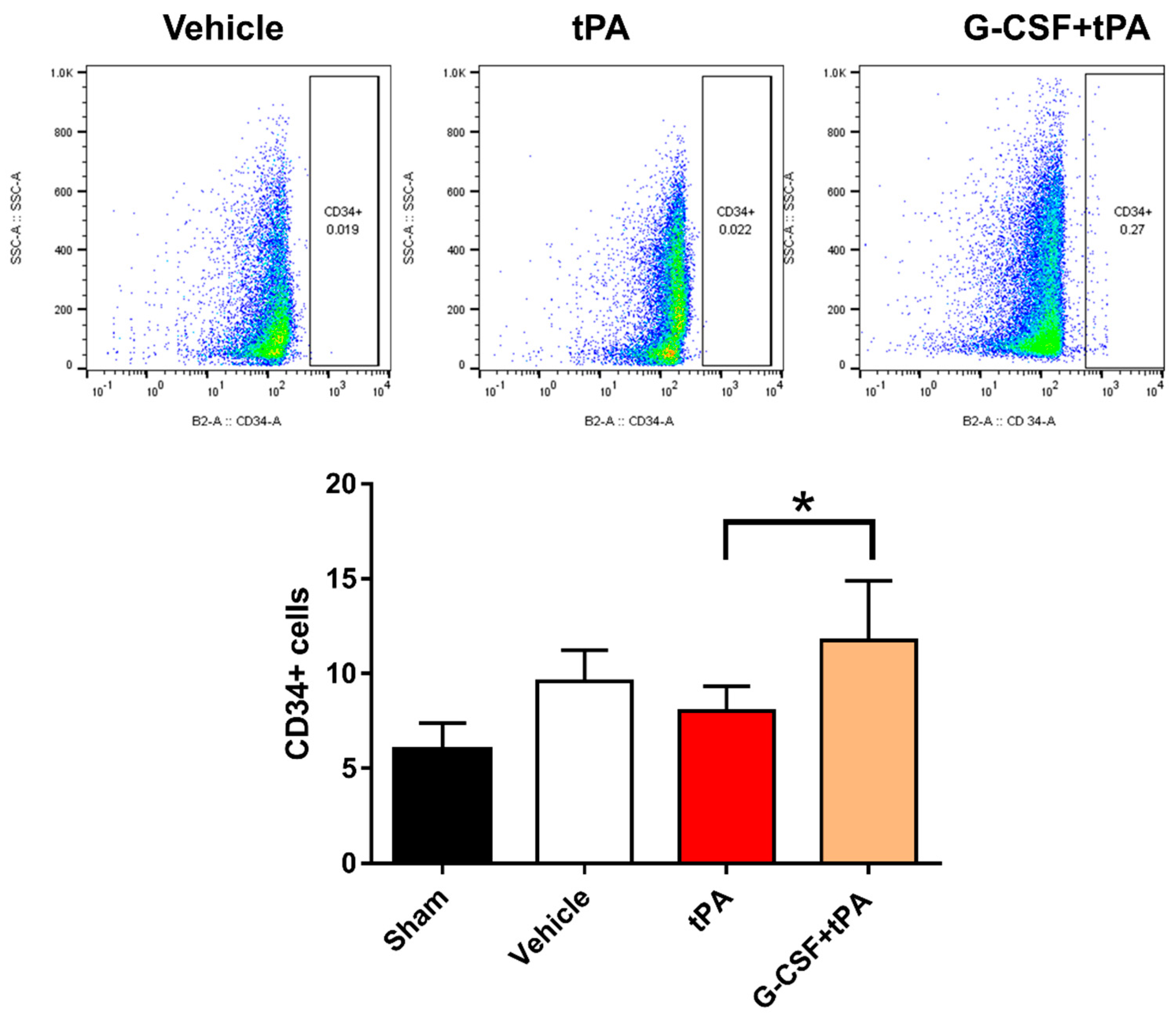
2.4. Increased CD34-Positive Cells in the Peripheral Blood of G-CSF+tPA-Treated Rats
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Study Design and Treatment Groups
4.3. Thromboembolic (TE) Stroke
4.4. Measurement of Brain Hemorrhage and Infarction
4.5. Motor and Neurological Tests
4.6. Histology and Immunohistochemistry
4.7. Flow Cytometry
4.8. Statistical Analysis
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| G-CSF | granulocyte-colony stimulating factor |
| HT | hemorrhagic transformation |
| tPA | tissue plasminogen activator |
| EPC | endothelial progenitor cell |
| CD34 | cluster of differentiation 4 |
| VEGFR | vascular endothelial growth factor receptor |
| vWF | von Willebrand factor |
References
- Benjamin, E.J.; Blaha, M.J.; Chiuve, S.E.; Cushman, M.; Das, S.R.; Deo, R.; Floyd, J.; Fornage, M.; Gillespie, C.; Isasi, C.R.; et al. Heart disease and stroke statistics—2017 update: A report from the American Heart Association. Circulation 2017, 135, e229–e445. [Google Scholar] [CrossRef] [PubMed]
- The NINDS t-PA Stroke Study Group. Intracerebral hemorrhage after intravenous t-PA therapy for ischemic stroke. Stroke 1997, 28, 2109–2118. [Google Scholar]
- Dela Peña, I.; Borlongan, C.V.; Shen, G.; Davis, W. Strategies to Extend Thrombolytic Time Window for Ischemic Stroke Treatment: An Unmet Clinical Need. J. Stroke 2017, 19, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Knecht, T.; Story, J.; Liu, J.; Davis, W.; Borlongan, C.V.; dela Peña, I.C. Adjunctive Therapy Approaches for Ischemic Stroke: Innovations to Expand Time Window of Treatment. Int. J. Mol. Sci. 2017, 18, 2756. [Google Scholar] [CrossRef] [PubMed]
- Hartung, T. Anti-inflammatory effects of granulocyte colony-stimulating factor. Curr. Opin. Hematol. 1998, 5, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Schabitz, W.R.; Kollmar, R.; Schwaninger, M.; Juettler, E.; Bardutzky, J.; Scholzke, M.N.; Sommer, C.; Schwab, S. Neuroprotective effect of granulocyte colony-stimulating factor after focal cerebral ischemia. Stroke 2003, 34, 745–751. [Google Scholar] [CrossRef] [PubMed]
- Han, J.L.; Blank, T.; Schwab, S.; Kollmar, R. Inhibited glutamate release by granulocyte-colony stimulating factor after experimental stroke. Neurosci. Lett. 2008, 432, 167–169. [Google Scholar] [CrossRef] [PubMed]
- Shyu, W.C.; Lin, S.Z.; Yang, H.I.; Tzeng, Y.S.; Pang, C.Y.; Yen, P.S.; Li, H. Functional recovery of stroke rats induced by granulocyte colony-stimulating factor-stimulated stem cells. Circulation 2004, 110, 1847–1854. [Google Scholar] [CrossRef] [PubMed]
- Bussolino, F.; Ziche, M.; Wang, J.M.; Alessi, D.; Morbidellim, L.; Cremona, O.; Bosia, A.; Marchisio, P.C.; Mantovani, A. In vitro and in vivo activation of endothelial cells by colony-stimulating factors. J. Clin. Investig. 1991, 87, 986–995. [Google Scholar] [CrossRef] [PubMed]
- Kawada, H.; Takizawa, S.; Takanashim, T.; Morita, Y.; Fujita, J.; Fukuda, K.; Takagi, S.; Okano, H.; Ando, K.; Hotta, T. Administration of hematopoietic cytokines in the subacute phase after cerebral infarction is effective for functional recovery facilitating proliferation of intrinsic neural stem/progenitor cells and transition of bone marrow-derived neuronal cells. Circulation 2006, 113, 701–710. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.T.; Chu, K.; Jung, K.H.; Ko, S.Y.; Kim, E.H.; Sinn, D.I.; Lee, Y.S.; Lo, E.H.; Kim, M.; Roh, J.K. Granulocyte colony-stimulating factor enhances angiogenesis after focal cerebral ischemia. Brain Res. 2005, 1058, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Dela Peña, I.C.; Yoo, A.; Tajiri, N.; Acosta, S.A.; Ji, X.; Kaneko, Y.; Borlongan, C.V. Granulocyte colony-stimulating factor attenuates delayed tPA-induced hemorrhagic transformation in ischemic stroke rats by enhancing angiogenesis and vasculogenesis. J. Cereb. Blood Flow Metab. 2015, 35, 338–346. [Google Scholar] [CrossRef] [PubMed]
- Fisher, M.; Feuerstein, G.; Howells, D.W.; Hurn, P.D.; Kent, T.A.; Savitz, S.I.; Lo, E.H.; STAIR Group. Update of the stroke therapy academic industry roundtable preclinical recommendations. Stroke 2009, 40, 2244–2250. [Google Scholar] [CrossRef] [PubMed]
- Sloan, M.A. Thrombolysis and stroke: Past and future. Arch. Neurol. 1987, 44, 748–768. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, R.L.; Jiang, Q.; Raman, S.B.; Cantwell, L.; Chopp, M. A new rat model of thrombotic focal cerebral ischemia. J. Cereb. Blood Flow Metab. 1997, 17, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Yebenes, I.; Sobrado, M.; Zarruk, J.G.; Castellanos, M.; de la Ossa, N.P.; Davalos, A.; Serena, J.; Lizasoain, I.; Moro, M.A. A mouse model of hemorrhagic transformation by delayed tissue plasminogen activator administration after in situ thromboembolic stroke. Stroke 2011, 42, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, R.L.; Jiang, Q.; Ding, G.; Chopp, M.; Zhang, Z.G. Focal embolic cerebral ischemia in the rat. Nat. Protoc. 2015, 10, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Tajiri, N.; Shinozuka, K.; Vasconcellos, J.; Kaneko, Y.; Lee, H.J.; Mimura, O.; Dezawa, M.; Kim, S.U.; Borlongan, C.V. Vasculogenesis in experimental stroke after human cerebral endothelial cell transplantation. Stroke 2013, 4, 3473–3481. [Google Scholar] [CrossRef] [PubMed]
- Asahara, T.; Murohara, T.; Sullivan, A.; Silver, M.; van der Zee, R.; Li, T.; Witzenbichler, B.; Schatteman, G.; Isner, J.M. Isolation of putative progenitor endothelial cells for angiogenesis. Science 1997, 175, 964–967. [Google Scholar] [CrossRef]
- Borlongan, C.V. Bone marrow stem cell mobilization in stroke: A “bonehead” may be good after all! Leukemia 2011, 25, 1674–1686. [Google Scholar] [CrossRef] [PubMed]
- Urbich, C.; Dimmeler, S. Endothelial progenitor cells: Characterization and role in vascular biology. Circ. Res. 2004, 95, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Liman, T.G.; Endres, M. New vessels after stroke: Postischemic neovascularization and regeneration. Cerebrovasc. Dis. 2012, 33, 492–999. [Google Scholar] [CrossRef] [PubMed]
- Toth, Z.E.; Leker, R.R.; Shahar, T.; Pastorino, S.; Szalayova, I.; Asemenew, B.; Key, S.; Parmelee, A.; Mayer, B.; Nemeth, K.; et al. The combination of granulocyte colony-stimulating factor and stem cell factor significantly increases the number of bone marrow-derived endothelial cells in brains of mice following cerebral ischemia. Blood 2008, 111, 5544–5552. [Google Scholar] [CrossRef] [PubMed]
- Gautier, S.; Ouk, T.; Tagzirt, M.; Lefebvre, C.; Laprais, M.; Pétrault, O.; Dupont, A.; Leys, D.; Bordet, R. Impact of the neutrophil response to granulocyte colony-stimulating factor on the risk of hemorrhage when used in combination with tissue plasminogen activator during the acute phase of experimental stroke. J. Neuroinflamm. 2014, 11, 96. [Google Scholar] [CrossRef] [PubMed]
- Lees, K.R.; Bluhmki, E.; von Kummer, R.; Brott, T.G.; Toni, D.; Grotta, J.C.; Albers, G.W.; Kaste, M.; Marler, J.R.; Hamilton, S.A.; et al. Time to treatment with intravenous alteplase and outcome in stroke: An updated pooled analysis of ECASS, ATLANTIS, NINDS, and EPITHET trials. Lancet 2010, 375, 1695–1703. [Google Scholar] [CrossRef]
- Balseanu, A.T.; Buga, A.M.; Catalin, B.; Wagner, D.C.; Boltze, J.; Zagrean, A.M.; Reymann, K.; Schaebitz, W.; Popa-Wagner, A. Multimodal approaches for regenerative stroke therapies: Combination of granulocyte colony-stimulating factor with bone marrow mesenchymal stem cells is not superior to G-CSF alone. Front. Aging Neurosci. 2014, 23, 130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popa-Wagner, A.; Buga, A.M.; Kokaia, Z. Perturbed cellular response to brain injury during aging. Ageing Res. Rev. 2011, 10, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Tsuji, K.; Lee, S.R.; Ning, M.; Furie, K.L.; Buchan, A.M.; Lo, E.H. Mechanisms of hemorrhagic transformation after tissue plasminogen activator reperfusion therapy for ischemic stroke. Stroke 2004, 35, 2726–2730. [Google Scholar] [CrossRef] [PubMed]
- Dela Peña, I.; Sanberg, P.R.; Acosta, S.; Lin, S.Z.; Borlongan, C.V. G-CSF as an adjunctive therapy with umbilical cord blood cell transplantation for traumatic brain injury. Cell Transplant. 2015, 24, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Liao, S.; Luo, C.; Cao, B.; Hu, H.; Wang, S.; Yue, H.; Chen, L.; Zhou, Z. Endothelial Progenitor Cells for Ischemic Stroke: Update on Basic Research and Application. Stem Cells Int. 2017, 2017, 2193432. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Sobrino, M.; Rosell, A.; Hernandez-Guillamon, M.; Penalba, A.; Ribó, M.; Alvarez-Sabín, J.; Montaner, J. Mobilization, endothelial differentiation and functional capacity of endothelial progenitor cells after ischemic stroke. Microvasc. Res. 2010, 80, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Tuttolomondo, A.; Di Sciacca, R.; Di Raimondo, D.; Pedone, C.; La Placa, S.; Pinto, A.; Licata, G. Effects of clinical and laboratory variables and of pretreatment with cardiovascular drugs in acute ischaemic stroke: A retrospective chart review from the GIFA study. Int. J. Cardiol. 2011, 51, 318–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuttolomondo, A.; Pecoraro, R.; Casuccio, A.; Di Raimondo, D.; Buttà, C.; Clemente, G.; Della Corte, V.; Guggino, G.; Arnao, V.; Maida, C.; et al. Peripheral frequency of CD4+ CD28- cells in acute ischemic stroke: Relationship with stroke subtype and severity markers. Medicine (Baltimore) 2015, 94, e81. [Google Scholar] [CrossRef] [PubMed]
- García-Culebras, A.; Palma-Tortosa, S.; Moraga, A.; García-Yébenes, I.; Durán-Laforet, V.; Cuartero, M.I.; de la Parra, J.; Barrios-Muñoz, A.L.; Díaz-Guzmán, J.; Pradillo, J.M.; et al. Toll-like receptor 4 mediates hemorrhagic transformation after delayed tissue plasminogen activator administration in in situ thromboembolic stroke. Stroke 2017, 48, 1695–1699. [Google Scholar] [CrossRef] [PubMed]
- Popa-Wagner, A.; Dinca, I.; Yalikun, S.; Walker, L.; Kroemer, H.; Kessler, C. Accelerated delimitation of the infarct zone by capillary-derived nestin-positive cells in aged rats. Curr. Neurovasc. Res. 2006, 3, 3–13. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dela Peña, I.C.; Yang, S.; Shen, G.; Fang Liang, H.; Solak, S.; Borlongan, C.V. Extension of Tissue Plasminogen Activator Treatment Window by Granulocyte-Colony Stimulating Factor in a Thromboembolic Rat Model of Stroke. Int. J. Mol. Sci. 2018, 19, 1635. https://doi.org/10.3390/ijms19061635
Dela Peña IC, Yang S, Shen G, Fang Liang H, Solak S, Borlongan CV. Extension of Tissue Plasminogen Activator Treatment Window by Granulocyte-Colony Stimulating Factor in a Thromboembolic Rat Model of Stroke. International Journal of Molecular Sciences. 2018; 19(6):1635. https://doi.org/10.3390/ijms19061635
Chicago/Turabian StyleDela Peña, Ike C., Samuel Yang, Guofang Shen, Hsiao Fang Liang, Sara Solak, and Cesar V. Borlongan. 2018. "Extension of Tissue Plasminogen Activator Treatment Window by Granulocyte-Colony Stimulating Factor in a Thromboembolic Rat Model of Stroke" International Journal of Molecular Sciences 19, no. 6: 1635. https://doi.org/10.3390/ijms19061635
APA StyleDela Peña, I. C., Yang, S., Shen, G., Fang Liang, H., Solak, S., & Borlongan, C. V. (2018). Extension of Tissue Plasminogen Activator Treatment Window by Granulocyte-Colony Stimulating Factor in a Thromboembolic Rat Model of Stroke. International Journal of Molecular Sciences, 19(6), 1635. https://doi.org/10.3390/ijms19061635