The Role of High-Density Lipoproteins in Diabetes and Its Vascular Complications

 and
and
Abstract
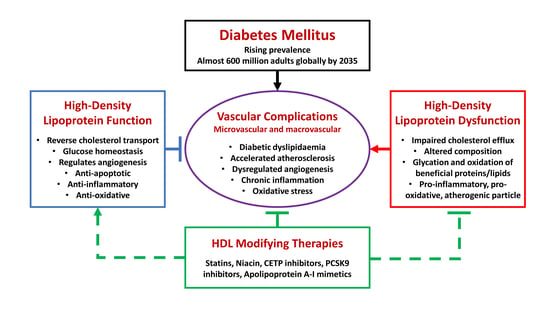
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Diabetic Complications and High-Density Lipoproteins (HDL) Levels
3. Molecular Functions of HDL in Diabetes
3.1. HDL and Glucose Metabolism
3.2. HDL and Atherosclerosis in Diabetes
3.3. HDL and Angiogenesis in Diabetes
4. Dysfunctional HDL in Diabetes
5. HDL-Modifying Treatment Approaches in Diabetes
6. Conclusions
Conflicts of Interest
References
- World Health Organization. Global Report on Diabetes. World Health Organization. Available online: http://www.who.int/iris/handle/10665/204871 (accessed on 22 April 2018).
- Guariguata, L.; Whiting, D.R.; Hambleton, I.; Beagley, J.; Linnenkamp, U.; Shaw, J.E. Global estimates of diabetes prevalence for 2013 and projections for 2035. Diabetes Res. Clin. Pract. 2014, 103, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Gregg, E.W.; Sattar, N.; Ali, M.K. The changing face of diabetes complications. Lancet Diabetes Endocrinol. 2016, 4, 537–547. [Google Scholar] [CrossRef]
- Costa, P.Z.; Soares, R. Neovascularization in diabetes and its complications. Unraveling the angiogenic paradox. Life Sci. 2013, 92, 1037–1045. [Google Scholar] [CrossRef] [PubMed]
- Fowler, M.J. Microvascular and macrovascular complications of diabetes. Clin. Diabetes 2011, 29, 116. [Google Scholar] [CrossRef]
- Aronson, D.; Edelman, E.R. Coronary artery disease and diabetes mellitus. Cardiol. Clin. 2014, 32, 439–455. [Google Scholar] [CrossRef] [PubMed]
- Mohammedi, K.; Woodward, M.; Hirakawa, Y.; Zoungas, S.; Williams, B.; Lisheng, L.; Rodgers, A.; Mancia, G.; Neal, B.; Harrap, S.; et al. Microvascular and macrovascular disease and risk for major peripheral arterial disease in patients with type 2 diabetes. Diabetes Care 2016, 39, 1796–1803. [Google Scholar] [CrossRef] [PubMed]
- Levelt, E.; Piechnik, S.K.; Liu, A.; Wijesurendra, R.S.; Mahmod, M.; Ariga, R.; Francis, J.M.; Greiser, A.; Clarke, K.; Neubauer, S.; et al. Adenosine stress CMR T1-mappi.ng detects early microvascular dysfunction in patients with type 2 diabetes mellitus without obstructive coronary artery disease. J. Cardiovasc. Magn. Reson. 2017, 19, 81. [Google Scholar] [CrossRef] [PubMed]
- Gerstein, H.C.; Miller, M.E.; Byington, R.P.; Goff, D.C., Jr.; Bigger, J.T.; Buse, J.B.; Cushman, W.C.; Genuth, S.; Ismail-Beigi, F.; Grimm, R.H., Jr.; et al. Effects of intensive glucose lowering in type 2 diabetes. N. Engl. J. Med. 2008, 358, 2545–2559. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; MacMahon, S.; Chalmers, J.; Neal, B.; Billot, L.; Woodward, M.; Marre, M.; Cooper, M.; Glasziou, P.; Grobbee, D.; et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N. Engl. J. Med. 2008, 358, 2560–2572. [Google Scholar] [CrossRef] [PubMed]
- Nathan, D.M.; Cleary, P.A.; Backlund, J.Y.; Genuth, S.M.; Lachin, J.M.; Orchard, T.J.; Raskin, P.; Zinman, B. Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N. Engl. J. Med. 2005, 353, 2643–2653. [Google Scholar] [CrossRef] [PubMed]
- Ginsberg, H.N.; MacCallum, P.R. The obesity, metabolic syndrome, and type 2 diabetes mellitus pandemic: Part i. Increased cardiovascular disease risk and the importance of atherogenic dyslipidemia in persons with the metabolic syndrome and type 2 diabetes mellitus. J. Cardiometab. Syndr. 2009, 4, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Schofield, J.D.; Liu, Y.; Rao-Balakrishna, P.; Malik, R.A.; Soran, H. Diabetes dyslipidemia. Diabetes Ther. 2016, 7, 203–219. [Google Scholar] [CrossRef] [PubMed]
- Rana, J.S.; Liu, J.Y.; Moffet, H.H.; Solomon, M.D.; Go, A.S.; Jaffe, M.G.; Karter, A.J. Metabolic dyslipidemia and risk of coronary heart disease in 28,318 adults with diabetes mellitus and low-density lipoprotein cholesterol <100 mg/dL. Am. J. Cardiol. 2015, 116, 1700–1704. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, M.; Schinske, A.; Pop-Busui, R. Lipids and lipid management in diabetes. Best Pract. Res. Clin. Endocrinol. Metab. 2014, 28, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Chait, A.; Goldberg, I. Treatment of dyslipidemia in diabetes: Recent advances and remaining questions. Curr. Diabetes Rep. 2017, 17, 112. [Google Scholar] [CrossRef] [PubMed]
- Ogita, M.; Miyauchi, K.; Miyazaki, T.; Naito, R.; Konishi, H.; Tsuboi, S.; Dohi, T.; Kasai, T.; Yokoyama, T.; Okazaki, S.; et al. Low high-density lipoprotein cholesterol is a residual risk factor associated with long-term clinical outcomes in diabetic patients with stable coronary artery disease who achieve optimal control of low-density lipoprotein cholesterol. Heart Vessels 2014, 29, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Soran, H.; Hama, S.; Yadav, R.; Durrington, P.N. HDL functionality. Curr. Opin. Lipidol. 2012, 23, 353–366. [Google Scholar] [CrossRef] [PubMed]
- Rye, K.A.; Barter, P.J. Cardioprotective functions of HDLs. J. Lipid. Res. 2014, 55, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Abbasi, A.; Corpeleijn, E.; Gansevoort, R.T.; Gans, R.O.; Hillege, H.L.; Stolk, R.P.; Navis, G.; Bakker, S.J.; Dullaart, R.P. Role of HDL cholesterol and estimates of HDL particle composition in future development of type 2 diabetes in the general population: The prevend study. J. Clin. Endocrinol. Metab. 2013, 98, E1352–E1359. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, H.M.; Miller, M.; Nasir, K.; McEvoy, J.W.; Herrington, D.; Blumenthal, R.S.; Blaha, M.J. Primary low level of high-density lipoprotein cholesterol and risks of coronary heart disease, cardiovascular disease, and death: Results from the multi-ethnic study of atherosclerosis. Am. J. Epidemiol. 2016, 183, 875–883. [Google Scholar] [CrossRef] [PubMed]
- Reina, S.A.; Llabre, M.M.; Allison, M.A.; Wilkins, J.T.; Mendez, A.J.; Arnan, M.K.; Schneiderman, N.; Sacco, R.L.; Carnethon, M.; Delaney, J.A. HDL cholesterol and stroke risk: The multi-ethnic study of atherosclerosis. Atherosclerosis 2015, 243, 314–319. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Kawashima, S.; Itoh, H.; Yamada, N.; Sone, H.; Watanabe, H.; Hattori, Y.; Ohrui, T.; Yokote, K.; Nomura, H.; et al. Low HDL cholesterol is associated with the risk of stroke in elderly diabetic individuals: Changes in the risk for atherosclerotic diseases at various ages. Diabetes Care 2009, 32, 1221–1223. [Google Scholar] [CrossRef] [PubMed]
- Ikura, K.; Hanai, K.; Shinjyo, T.; Uchigata, Y. HDL cholesterol as a predictor for the incidence of lower extremity amputation and wound-related death in patients with diabetic foot ulcers. Atherosclerosis 2015, 239, 465–469. [Google Scholar] [CrossRef] [PubMed]
- Pei, E.; Li, J.; Lu, C.; Xu, J.; Tang, T.; Ye, M.; Zhang, X.; Li, M. Effects of lipids and lipoproteins on diabetic foot in people with type 2 diabetes mellitus: A meta-analysis. J. Diabetes Complicat. 2014, 28, 559–564. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhi, Y.; Li, C.; Liu, Y.; Zhang, L.; Wang, Y.; Che, K. HDL cholesterol and risk of diabetic nephropathy in patient with type 1 diabetes: A meta-analysis of cohort studies. Diabetes Res. Clin. Pract. 2016, 122, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Sacks, F.M.; Hermans, M.P.; Fioretto, P.; Valensi, P.; Davis, T.; Horton, E.; Wanner, C.; Al-Rubeaan, K.; Aronson, R.; Barzon, I.; et al. Association between plasma triglycerides and high-density lipoprotein cholesterol and microvascular kidney disease and retinopathy in type 2 diabetes mellitus: A global case-control study in 13 countries. Circulation 2014, 129, 999–1008. [Google Scholar] [CrossRef] [PubMed]
- Russo, G.T.; De Cosmo, S.; Viazzi, F.; Pacilli, A.; Ceriello, A.; Genovese, S.; Guida, P.; Giorda, C.; Cucinotta, D.; Pontremoli, R.; et al. Plasma triglycerides and HDL-c levels predict the development of diabetic kidney disease in subjects with type 2 diabetes: The amd annals initiative. Diabetes Care 2016, 39, 2278–2287. [Google Scholar] [CrossRef] [PubMed]
- Morton, J.; Zoungas, S.; Li, Q.; Patel, A.A.; Chalmers, J.; Woodward, M.; Celermajer, D.S.; Beulens, J.W.; Stolk, R.P.; Glasziou, P.; et al. Low HDL cholesterol and the risk of diabetic nephropathy and retinopathy: Results of the advance study. Diabetes Care 2012, 35, 2201–2206. [Google Scholar] [CrossRef] [PubMed]
- Barter, P.J. The causes and consequences of low levels of high density lipoproteins in patients with diabetes. Diabetes Metab. J. 2011, 35, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Drew, B.G.; Rye, K.A.; Duffy, S.J.; Barter, P.; Kingwell, B.A. The emerging role of HDL in glucose metabolism. Nat. Rev. Endocrinol. 2012, 8, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Stancu, C.S.; Toma, L.; Sima, A.V. Dual role of lipoproteins in endothelial cell dysfunction in atherosclerosis. Cell Tissue Res. 2012, 349, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Qi, Q.; Liang, L.; Doria, A.; Hu, F.B.; Qi, L. Genetic predisposition to dyslipidemia and type 2 diabetes risk in two prospective cohorts. Diabetes 2012, 61, 745–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haase, C.L.; Tybjaerg-Hansen, A.; Nordestgaard, B.G.; Frikke-Schmidt, R. HDL cholesterol and risk of type 2 diabetes: A mendelian randomization study. Diabetes 2015, 64, 3328–3333. [Google Scholar] [CrossRef] [PubMed]
- Voight, B.F.; Peloso, G.M.; Orho-Melander, M.; Frikke-Schmidt, R.; Barbalic, M.; Jensen, M.K.; Hindy, G.; Hólm, H.; Ding, E.L.; Johnson, T.; et al. Plasma HDL cholesterol and risk of myocardial infarction: A mendelian randomisation study. Lancet 2012, 380, 572–580. [Google Scholar] [CrossRef]
- Holmes, M.V.; Asselbergs, F.W.; Palmer, T.M.; Drenos, F.; Lanktree, M.B.; Nelson, C.P.; Dale, C.E.; Padmanabhan, S.; Finan, C.; Swerdlow, D.I.; et al. Mendelian randomization of blood lipids for coronary heart disease. Eur. Heart J. 2015, 36, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Bernard, S.; Moulin, P.; Lagrost, L.; Picard, S.; Elchebly, M.; Ponsin, G.; Chapuis, F.; Berthezène, F. Association between plasma HDL-cholesterol concentration and Taq1B CETP gene polymorphism in non-insulin-dependent diabetes mellitus. J. Lipid Res. 1998, 39, 59–65. [Google Scholar] [PubMed]
- Thompson, A.; di Angelantonio, E.; Sarwar, N.; Erqou, S.; Saleheen, D.; Dullaart, R.P.; Keavney, B.; Ye, Z.; Danesh, J. Association of cholesteryl ester transfer protein genotypes with CETP mass and activity, lipid levels, and coronary risk. JAMA 2008, 299, 2777–2788. [Google Scholar] [CrossRef] [PubMed]
- Niu, W.; Qi, Y. Circulating cholesteryl ester transfer protein and coronary heart disease: Mendelian randomization meta-analysis. Circ. Cardiovasc. Genet. 2015, 8, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Lou, Y.; Qiu, X.; Liu, Y.; Lu, L.; Chen, Q.; Jin, W. Association of cholesteryl ester transfer protein (CETP) gene polymorphism, high density lipoprotein cholesterol and risk of coronary artery disease: A meta-analysis using a mendelian randomization approach. BMC Med. Genet. 2014, 15, 118. [Google Scholar] [CrossRef] [PubMed]
- Von Eckardstein, A.; Widmann, C. High-density lipoprotein, beta cells, and diabetes. Cardiovasc. Res. 2014, 103, 384–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fryirs, M.A.; Barter, P.J.; Appavoo, M.; Tuch, B.E.; Tabet, F.; Heather, A.K.; Rye, K.A. Effects of high-density lipoproteins on pancreatic beta-cell insulin secretion. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1642–1648. [Google Scholar] [CrossRef] [PubMed]
- Cochran, B.J.; Bisoendial, R.J.; Hou, L.; Glaros, E.N.; Rossy, J.; Thomas, S.R.; Barter, P.J.; Rye, K.A. Apolipoprotein A-I increases insulin secretion and production from pancreatic β-cells via a G-protein-cAMP-PKA-FoxO1-dependent mechanism. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2261–2267. [Google Scholar] [CrossRef] [PubMed]
- Rutti, S.; Ehses, J.A.; Sibler, R.A.; Prazak, R.; Rohrer, L.; Georgopoulos, S.; Meier, D.T.; Niclauss, N.; Berney, T.; Donath, M.Y.; et al. Low- and high-density lipoproteins modulate function, apoptosis, and proliferation of primary human and murine pancreatic beta-cells. Endocrinology 2009, 150, 4521–4530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pétremand, J.; Puyal, J.; Chatton, J.Y.; Duprez, J.; Allagnat, F.; Frias, M.; James, R.W.; Waeber, G.; Jonas, J.C.; Widmann, C. HDLs protect pancreatic β-cells against er stress by restoring protein folding and trafficking. Diabetes 2012, 61, 1100–1111. [Google Scholar] [CrossRef] [PubMed]
- Puyal, J.; Petremand, J.; Dubuis, G.; Rummel, C.; Widmann, C. HDLs protect the min6 insulinoma cell line against tunicamycin-induced apoptosis without inhibiting er stress and without restoring er functionality. Mol. Cell. Endocrinol. 2013, 381, 291–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, R.; Lai, R.; Ding, Q.; Wang, Z.; Luo, X.; Zhang, Y.; Cui, G.; He, J.; Liu, W.; Chen, Y. Apolipoprotein A-I stimulates AMP-activated protein kinase and improves glucose metabolism. Diabetologia 2007, 50, 1960–1968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stenkula, K.G.; Lindahl, M.; Petrlova, J.; Dalla-Riva, J.; Goransson, O.; Cushman, S.W.; Krupinska, E.; Jones, H.A.; Lagerstedt, J.O. Single injections of apoA-I acutely improve in vivo glucose tolerance in insulin-resistant mice. Diabetologia 2014, 57, 797–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drew, B.G.; Duffy, S.J.; Formosa, M.F.; Natoli, A.K.; Henstridge, D.C.; Penfold, S.A.; Thomas, W.G.; Mukhamedova, N.; de Courten, B.; Forbes, J.M.; et al. High-density lipoprotein modulates glucose metabolism in patients with type 2 diabetes mellitus. Circulation 2009, 119, 2103–2111. [Google Scholar] [CrossRef] [PubMed]
- Annema, W.; von Eckardstein, A. High-density lipoproteins. Circ. J. 2013, 77, 2432–2448. [Google Scholar] [CrossRef] [PubMed]
- Tangirala, R.K.; Tsukamoto, K.; Chun, S.H.; Usher, D.; Pure, E.; Rader, D.J. Regression of atherosclerosis induced by liver-directed gene transfer of apolipoprotein A-I in mice. Circulation 1999, 100, 1816–1822. [Google Scholar] [CrossRef] [PubMed]
- Sirtori, C.R.; Calabresi, L.; Franceschini, G.; Baldassarre, D.; Amato, M.; Johansson, J.; Salvetti, M.; Monteduro, C.; Zulli, R.; Muiesan, M.L.; et al. Cardiovascular status of carriers of the apolipoprotein A-I(milano) mutant: The limone sul garda study. Circulation 2001, 103, 1949–1954. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.K.; Nilsson, J.; Kaul, S.; Fishbein, M.C.; Ageland, H.; Hamsten, A.; Johansson, J.; Karpe, F.; Cercek, B. Effects of recombinant apolipoprotein A-I(milano) on aortic atherosclerosis in apolipoprotein e-deficient mice. Circulation 1998, 97, 780–785. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.K.; Yano, J.; Reyes, O.; Chyu, K.Y.; Kaul, S.; Bisgaier, C.L.; Drake, S.; Cercek, B. High-dose recombinant apolipoprotein A-I(milano) mobilizes tissue cholesterol and rapidly reduces plaque lipid and macrophage content in apolipoprotein e-deficient mice. Potential implications for acute plaque stabilization. Circulation 2001, 103, 3047–3050. [Google Scholar] [CrossRef] [PubMed]
- Parolini, C.; Marchesi, M.; Lorenzon, P.; Castano, M.; Balconi, E.; Miragoli, L.; Chaabane, L.; Morisetti, A.; Lorusso, V.; Martin, B.J.; et al. Dose-related effects of repeated etc-216 (recombinant apolipoprotein A-I milano/1-palmitoyl-2-oleoyl phosphatidylcholine complexes) administrations on rabbit lipid-rich soft plaques: In vivo assessment by intravascular ultrasound and magnetic resonance imaging. J. Am. Coll. Cardiol. 2008, 51, 1098–1103. [Google Scholar] [CrossRef] [PubMed]
- Ibanez, B.; Vilahur, G.; Cimmino, G.; Speidl, W.S.; Pinero, A.; Choi, B.G.; Zafar, M.U.; Santos-Gallego, C.G.; Krause, B.; Badimon, L.; et al. Rapid change in plaque size, composition, and molecular footprint after recombinant apolipoprotein A-I milano (ETC-216) administration: Magnetic resonance imaging study in an experimental model of atherosclerosis. J. Am. Coll. Cardiol. 2008, 51, 1104–1109. [Google Scholar] [CrossRef] [PubMed]
- Nissen, S.E.; Tsunoda, T.; Tuzcu, E.M.; Schoenhagen, P.; Cooper, C.J.; Yasin, M.; Eaton, G.M.; Lauer, M.A.; Sheldon, W.S.; Grines, C.L.; et al. Effect of recombinant apoA-I milano on coronary atherosclerosis in patients with acute coronary syndromes: A randomized controlled trial. JAMA 2003, 290, 2292–2300. [Google Scholar] [CrossRef] [PubMed]
- Tran-Dinh, A.; Diallo, D.; Delbosc, S.; Varela-Perez, L.M.; Dang, Q.B.; Lapergue, B.; Burillo, E.; Michel, J.B.; Levoye, A.; Martin-Ventura, J.L.; et al. HDL and endothelial protection. Br. J. Pharmacol. 2013, 169, 493–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieuwdorp, M.; Vergeer, M.; Bisoendial, R.J.; op't Roodt, J.; Levels, H.; Birjmohun, R.S.; Kuivenhoven, J.A.; Basser, R.; Rabelink, T.J.; Kastelein, J.J.; et al. Reconstituted HDL infusion restores endothelial function in patients with type 2 diabetes mellitus. Diabetologia 2008, 51, 1081–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whetzel, A.M.; Bolick, D.T.; Srinivasan, S.; Macdonald, T.L.; Morris, M.A.; Ley, K.; Hedrick, C.C. Sphingosine-1 phosphate prevents monocyte/endothelial interactions in type 1 diabetic nod mice through activation of the s1p1 receptor. Circ. Res. 2006, 99, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Brinck, J.W.; Thomas, A.; Lauer, E.; Jornayvaz, F.R.; Brulhart-Meynet, M.C.; Prost, J.C.; Pataky, Z.; Lofgren, P.; Hoffstedt, J.; Eriksson, M.; et al. Diabetes mellitus is associated with reduced high-density lipoprotein sphingosine-1-phosphate content and impaired high-density lipoprotein cardiac cell protection. Arterioscler Thromb. Vasc. Biol. 2016, 36, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Muller, C.; Salvayre, R.; Negre-Salvayre, A.; Vindis, C. HDLs inhibit endoplasmic reticulum stress and autophagic response induced by oxidized ldls. Cell. Death Differ. 2011, 18, 817–828. [Google Scholar] [CrossRef] [PubMed]
- White, C.R.; Datta, G.; Giordano, S. High-density lipoprotein regulation of mitochondrial function. Adv. Exp. Med. Biol. 2017, 982, 407–429. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Ding, F.H.; Sun, J.T.; Pu, L.J.; Zhang, R.Y.; Zhang, Q.; Chen, Q.J.; Shen, W.F.; Lu, L. Association of elevated apoA-I glycation and reduced HDL-associated paraoxonase1, 3 activity, and their interaction with angiographic severity of coronary artery disease in patients with type 2 diabetes mellitus. Cardiovasc. Diabetol. 2015, 14, 52. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Drew, B.G.; Nakhla, S.; Duffy, S.J.; Murphy, A.J.; Barter, P.J.; Rye, K.A.; Chin-Dusting, J.; Hoang, A.; Sviridov, D.; et al. Reconstituted high-density lipoprotein increases plasma high-density lipoprotein anti-inflammatory properties and cholesterol efflux capacity in patients with type 2 diabetes. J. Am. Coll. Cardiol. 2009, 53, 962–971. [Google Scholar] [CrossRef] [PubMed]
- Kakouros, N.; Rade, J.J.; Kourliouros, A.; Resar, J.R. Platelet function in patients with diabetes mellitus: From a theoretical to a practical perspective. Int. J. Endocrinol. 2011, 2011, 742719. [Google Scholar] [CrossRef] [PubMed]
- Calkin, A.C.; Drew, B.G.; Ono, A.; Duffy, S.J.; Gordon, M.V.; Schoenwaelder, S.M.; Sviridov, D.; Cooper, M.E.; Kingwell, B.A.; Jackson, S.P. Reconstituted high-density lipoprotein attenuates platelet function in individuals with type 2 diabetes mellitus by promoting cholesterol efflux. Circulation 2009, 120, 2095–2104. [Google Scholar] [CrossRef] [PubMed]
- Brodde, M.F.; Korporaal, S.J.; Herminghaus, G.; Fobker, M.; Van Berkel, T.J.; Tietge, U.J.; Robenek, H.; van Eck, M.; Kehrel, B.E.; Nofer, J.R. Native high-density lipoproteins inhibit platelet activation via scavenger receptor BI: Role of negatively charged phospholipids. Atherosclerosis 2011, 215, 374–382. [Google Scholar] [CrossRef] [PubMed]
- Sumi, M.; Sata, M.; Miura, S.; Rye, K.A.; Toya, N.; Kanaoka, Y.; Yanaga, K.; Ohki, T.; Saku, K.; Nagai, R. Reconstituted high-density lipoprotein stimulates differentiation of endothelial progenitor cells and enhances ischemia-induced angiogenesis. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 813–818. [Google Scholar] [CrossRef] [PubMed]
- Van Oostrom, O.; Nieuwdorp, M.; Westerweel, P.E.; Hoefer, I.E.; Basser, R.; Stroes, E.S.; Verhaar, M.C. Reconstituted HDL increases circulating endothelial progenitor cells in patients with type 2 diabetes. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1864–1865. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.T.; Prosser, H.C.; Vanags, L.Z.; Monger, S.A.; Ng, M.K.; Bursill, C.A. High-density lipoproteins augment hypoxia-induced angiogenesis via regulation of post-translational modulation of hypoxia-inducible factor 1alpha. FASEB J. 2014, 28, 206–217. [Google Scholar] [CrossRef] [PubMed]
- Prosser, H.C.; Tan, J.T.; Dunn, L.L.; Patel, S.; Vanags, L.Z.; Bao, S.; Ng, M.K.; Bursill, C.A. Multifunctional regulation of angiogenesis by high-density lipoproteins. Cardiovasc. Res. 2014, 101, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.T.; Ng, M.K.; Bursill, C.A. The role of high-density lipoproteins in the regulation of angiogenesis. Cardiovasc. Res. 2015, 106, 184–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tahergorabi, Z.; Khazaei, M. Imbalance of angiogenesis in diabetic complications: The mechanisms. Int. J. Prev. Med. 2012, 3, 827–838. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.T.; Prosser, H.C.; Dunn, L.L.; Vanags, L.Z.; Ridiandries, A.; Tsatralis, T.; Lecce, L.; Clayton, Z.E.; Yuen, S.C.; Robertson, S.; et al. High-density lipoproteins rescue diabetes-impaired angiogenesis via scavenger receptor class b type i. Diabetes 2016, 65, 3091–3103. [Google Scholar] [CrossRef] [PubMed]
- Femlak, M.; Gluba-Brzozka, A.; Cialkowska-Rysz, A.; Rysz, J. The role and function of HDL in patients with diabetes mellitus and the related cardiovascular risk. Lipids Health Dis. 2017, 16, 207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenson, R.S.; Brewer, H.B., Jr.; Ansell, B.J.; Barter, P.; Chapman, M.J.; Heinecke, J.W.; Kontush, A.; Tall, A.R.; Webb, N.R. Dysfunctional HDL and atherosclerotic cardiovascular disease. Nat. Rev. Cardiol. 2016, 13, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Asztalos, B.F.; Tani, M.; Schaefer, E.J. Metabolic and functional relevance of HDL subspecies. Cur. Opin. Lipidol. 2011, 22, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Hwang, Y.C.; Hayashi, T.; Fujimoto, W.Y.; Kahn, S.E.; Leonetti, D.L.; McNeely, M.J.; Boyko, E.J. Differential association between HDL subclasses and the development of type 2 diabetes in a prospective study of japanese americans. Diabetes Care 2015, 38, 2100–2105. [Google Scholar] [CrossRef] [PubMed]
- Tabara, Y.; Arai, H.; Hirao, Y.; Takahashi, Y.; Setoh, K.; Kawaguchi, T.; Kosugi, S.; Ito, Y.; Nakayama, T.; Matsuda, F.; et al. Different inverse association of large high-density lipoprotein subclasses with exacerbation of insulin resistance and incidence of type 2 diabetes: The nagahama study. Diabetes Res. Clin. Pract. 2017, 127, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Long, S.; Li, C.; Liu, Y.; Chen, Y.; Zeng, Z.; Fu, M. High-density lipoprotein subclass and particle size in coronary heart disease patients with or without diabetes. Lipids Health Dis. 2012, 11, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermans, M.P.; Amoussou-Guenou, K.D.; Bouenizabila, E.; Sadikot, S.S.; Ahn, S.A.; Rousseau, M.F. Size, density and cholesterol load of HDL predict microangiopathy, coronary artery disease and β-cell function in men with T2DM. Diabetes Metab. Syndr. 2017, 11, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Farbstein, D.; Levy, A.P. HDL dysfunction in diabetes: Causes and possible treatments. Expert Rev. Cardiovasc. Ther. 2012, 10, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.C. Reverse cholesterol transport in type 2 diabetes mellitus. Diabetes Obes. Metab. 2009, 11, 534–543. [Google Scholar] [CrossRef] [PubMed]
- Mauldin, J.P.; Nagelin, M.H.; Wojcik, A.J.; Srinivasan, S.; Skaflen, M.D.; Ayers, C.R.; McNamara, C.A.; Hedrick, C.C. Reduced expression of atp-binding cassette transporter G1 increases cholesterol accumulation in macrophages of patients with type 2 diabetes mellitus. Circulation 2008, 117, 2785–2792. [Google Scholar] [CrossRef] [PubMed]
- Gantman, A.; Fuhrman, B.; Aviram, M.; Hayek, T. High glucose stimulates macrophage SR-BI expression and induces a switch in its activity from cholesterol efflux to cholesterol influx. Biochem. Biophys. Res. Commun. 2010, 391, 523–528. [Google Scholar] [CrossRef] [PubMed]
- Brown, B.E.; Nobecourt, E.; Zeng, J.; Jenkins, A.J.; Rye, K.A.; Davies, M.J. Apolipoprotein A-I glycation by glucose and reactive aldehydes alters phospholipid affinity but not cholesterol export from lipid-laden macrophages. PLoS ONE 2013, 8, e65430. [Google Scholar] [CrossRef] [PubMed]
- Vaisar, T.; Couzens, E.; Hwang, A.; Russell, M.; Barlow, C.E.; DeFina, L.F.; Hoofnagle, A.N.; Kim, F. Type 2 diabetes is associated with loss of HDL endothelium protective functions. PLoS ONE 2018, 13, e0192616. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, S.A.; Besler, C.; Rohrer, L.; Meyer, M.; Heinrich, K.; Bahlmann, F.H.; Mueller, M.; Horvath, T.; Doerries, C.; Heinemann, M.; et al. Endothelial-vasoprotective effects of high-density lipoprotein are impaired in patients with type 2 diabetes mellitus but are improved after extended-release niacin therapy. Circulation 2010, 121, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Tong, X.; Peng, H.; Liu, D.; Ji, L.; Niu, C.; Ren, J.; Pan, B.; Hu, J.; Zheng, L.; Huang, Y. High-density lipoprotein of patients with type 2 diabetes mellitus upregulates cyclooxgenase-2 expression and prostacyclin i-2 release in endothelial cells: Relationship with HDL-associated sphingosine-1-phosphate. Cardiovasc. Diabetol. 2013, 12, 27. [Google Scholar] [CrossRef] [PubMed]
- Tong, X.; Lv, P.; Mathew, A.V.; Liu, D.; Niu, C.; Wang, Y.; Ji, L.; Li, J.; Fu, Z.; Pan, B.; et al. The compensatory enrichment of sphingosine-1-phosphate harbored on glycated high-density lipoprotein restores endothelial protective function in type 2 diabetes mellitus. Cardiovasc. Diabetol. 2014, 13, 82. [Google Scholar] [CrossRef] [PubMed]
- Ng, M.L.; Wadham, C.; Sukocheva, O.A. The role of sphingolipid signalling in diabetesassociated pathologies (review). Int. J. Mol. Med. 2017, 39, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Pan, B.; Ma, Y.; Ren, H.; He, Y.; Wang, Y.; Lv, X.; Liu, D.; Ji, L.; Yu, B.; Wang, Y.; et al. Diabetic HDL is dysfunctional in stimulating endothelial cell migration and proliferation due to down regulation of sr-bi expression. PLoS ONE 2012, 7, e48530. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; He, Z.; Gao, X.; Wu, F.; Ding, R.; Ren, Y.; Jiang, Q.; Fan, M.; Liang, C.; Wu, Z. Oxidized high-density lipoprotein impairs endothelial progenitor cells' function by activation of CD36-MAPK-TSP-1 pathways. Antioxid. Redox Signal. 2015, 22, 308–324. [Google Scholar] [CrossRef] [PubMed]
- Lu, N.; Xie, S.; Li, J.; Tian, R.; Peng, Y.Y. Myeloperoxidase-mediated oxidation targets serum apolipoprotein A-I in diabetic patients and represents a potential mechanism leading to impaired anti-apoptotic activity of high density lipoprotein. Clin. Chim. Acta 2015, 441, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Nukuna, B.; Brennan, M.L.; Sun, M.; Goormastic, M.; Settle, M.; Schmitt, D.; Fu, X.; Thomson, L.; Fox, P.L.; et al. Apolipoprotein A-I is a selective target for myeloperoxidase-catalyzed oxidation and functional impairment in subjects with cardiovascular disease. J. Clin. Investig. 2004, 114, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; DiDonato, J.A.; Levison, B.S.; Schmitt, D.; Li, L.; Wu, Y.; Buffa, J.; Kim, T.; Gerstenecker, G.S.; Gu, X.; et al. An abundant dysfunctional apolipoprotein a1 in human atheroma. Nat. Med. 2014, 20, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Hewing, B.; Parathath, S.; Barrett, T.; Chung, W.K.; Astudillo, Y.M.; Hamada, T.; Ramkhelawon, B.; Tallant, T.C.; Yusufishaq, M.S.; Didonato, J.A.; et al. Effects of native and myeloperoxidase-modified apolipoprotein A-I on reverse cholesterol transport and atherosclerosis in mice. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 779–789. [Google Scholar] [CrossRef] [PubMed]
- Undurti, A.; Huang, Y.; Lupica, J.A.; Smith, J.D.; DiDonato, J.A.; Hazen, S.L. Modification of high density lipoprotein by myeloperoxidase generates a pro-inflammatory particle. J. Biol. Chem. 2009, 284, 30825–30835. [Google Scholar] [CrossRef] [PubMed]
- Nobecourt, E.; Tabet, F.; Lambert, G.; Puranik, R.; Bao, S.; Yan, L.; Davies, M.J.; Brown, B.E.; Jenkins, A.J.; Dusting, G.J.; et al. Nonenzymatic glycation impairs the antiinflammatory properties of apolipoprotein A-I. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 766–772. [Google Scholar] [CrossRef] [PubMed]
- Morgantini, C.; Natali, A.; Boldrini, B.; Imaizumi, S.; Navab, M.; Fogelman, A.M.; Ferrannini, E.; Reddy, S.T. Anti-inflammatory and antioxidant properties of HDLs are impaired in type 2 diabetes. Diabetes 2011, 60, 2617–2623. [Google Scholar] [CrossRef] [PubMed]
- Birner-Gruenberger, R.; Schittmayer, M.; Holzer, M.; Marsche, G. Understanding high-density lipoprotein function in disease: Recent advances in proteomics unravel the complexity of its composition and biology. Prog. Lipid Res. 2014, 56, 36–46. [Google Scholar] [CrossRef] [PubMed]
- McEneny, J.; Daniels, J.A.; McGowan, A.; Gunness, A.; Moore, K.; Stevenson, M.; Young, I.S.; Gibney, J. A cross-sectional study demonstrating increased serum amyloid a related inflammation in high-density lipoproteins from subjects with type 1 diabetes mellitus and how this association was augmented by poor glycaemic control. J. Diabetes Res. 2015, 2015, 351601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgantini, C.; Meriwether, D.; Baldi, S.; Venturi, E.; Pinnola, S.; Wagner, A.C.; Fogelman, A.M.; Ferrannini, E.; Natali, A.; Reddy, S.T. HDL lipid composition is profoundly altered in patients with type 2 diabetes and atherosclerotic vascular disease. Nutr. Metab. Cardiovasc. Dis. 2014, 24, 594–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganjali, S.; Dallinga-Thie, G.M.; Simental-Mendia, L.E.; Banach, M.; Pirro, M.; Sahebkar, A. HDL functionality in type 1 diabetes. Atherosclerosis 2017, 267, 99–109. [Google Scholar] [CrossRef] [PubMed]
- De Ferranti, S.D.; de Boer, I.H.; Fonseca, V.; Fox, C.S.; Golden, S.H.; Lavie, C.J.; Magge, S.N.; Marx, N.; McGuire, D.K.; Orchard, T.J.; et al. Type 1 diabetes mellitus and cardiovascular disease: A scientific statement from the american heart association and american diabetes association. Circulation 2014, 130, 1110–1130. [Google Scholar] [CrossRef] [PubMed]
- Heier, M.; Borja, M.S.; Brunborg, C.; Seljeflot, I.; Margeirsdottir, H.D.; Hanssen, K.F.; Dahl-Jorgensen, K.; Oda, M.N. Reduced HDL function in children and young adults with type 1 diabetes. Cardiovasc. Diabetol. 2017, 16, 85. [Google Scholar] [CrossRef] [PubMed]
- Manjunatha, S.; Distelmaier, K.; Dasari, S.; Carter, R.E.; Kudva, Y.C.; Nair, K.S. Functional and proteomic alterations of plasma high density lipoproteins in type 1 diabetes mellitus. Metabolism 2016, 65, 1421–1431. [Google Scholar] [CrossRef] [PubMed]
- Sampaio, E.; Barbosa, D.S.; Mazzuco, T.L.; Nunes, V.S.; Passarelli, M.; Nakandakare, E.R.; Carrilho, A.J. Impaired antioxidant action of high density lipoprotein in patients with type 1 diabetes with normoalbuminuria and microalbuminuria. Diabetes. Res. Clin. Pract. 2013, 99, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Denimal, D.; Pais de Barros, J.P.; Petit, J.M.; Bouillet, B.; Verges, B.; Duvillard, L. Significant abnormalities of the HDL phosphosphingolipidome in type 1 diabetes despite normal HDL cholesterol concentration. Atherosclerosis 2015, 241, 752–760. [Google Scholar] [CrossRef] [PubMed]
- Boekholdt, S.M.; Arsenault, B.J.; Hovingh, G.K.; Mora, S.; Pedersen, T.R.; Larosa, J.C.; Welch, K.M.; Amarenco, P.; Demicco, D.A.; Tonkin, A.M.; et al. Levels and changes of HDL cholesterol and apolipoprotein A-I in relation to risk of cardiovascular events among statin-treated patients: A meta-analysis. Circulation 2013, 128, 1504–1512. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto-Sasaki, M.; Yasuda, T.; Monguchi, T.; Nakajima, H.; Mori, K.; Toh, R.; Ishida, T.; Hirata, K. Pitavastatin increases HDL particles functionally preserved with cholesterol efflux capacity and antioxidative actions in dyslipidemic patients. J. Atheroscler. Thromb. 2013, 20, 708–716. [Google Scholar] [CrossRef] [PubMed]
- Niesor, E.J.; Schwartz, G.G.; Perez, A.; Stauffer, A.; Durrwell, A.; Bucklar-Suchankova, G.; Benghozi, R.; Abt, M.; Kallend, D. Statin-induced decrease in ATP-binding cassette transporter A1 expression via microrna33 induction may counteract cholesterol efflux to high-density lipoprotein. Cardiovasc. Drugs Ther. 2015, 29, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Kearney, P.M.; Blackwell, L.; Collins, R.; Keech, A.; Simes, J.; Peto, R.; Armitage, J.; Baigent, C. Efficacy of cholesterol-lowering therapy in 18,686 people with diabetes in 14 randomised trials of statins: A meta-analysis. Lancet 2008, 371, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Stegman, B.; Puri, R.; Cho, L.; Shao, M.; Ballantyne, C.M.; Barter, P.J.; Chapman, M.J.; Erbel, R.; Libby, P.; Raichlen, J.S.; et al. High-intensity statin therapy alters the natural history of diabetic coronary atherosclerosis: Insights from saturn. Diabetes Care 2014, 37, 3114–3120. [Google Scholar] [CrossRef] [PubMed]
- Ganda, O.P. Statin-induced diabetes: Incidence, mechanisms, and implications. F1000Resarch 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Sattar, N.; Preiss, D.; Murray, H.M.; Welsh, P.; Buckley, B.M.; de Craen, A.J.; Seshasai, S.R.; McMurray, J.J.; Freeman, D.J.; Jukema, J.W.; et al. Statins and risk of incident diabetes: A collaborative meta-analysis of randomised statin trials. Lancet 2010, 375, 735–742. [Google Scholar] [CrossRef]
- Thakker, D.; Nair, S.; Pagada, A.; Jamdade, V.; Malik, A. Statin use and the risk of developing diabetes: A network meta-analysis. Pharmacoepidemiol.Drug Saf. 2016, 25, 1131–1149. [Google Scholar] [CrossRef] [PubMed]
- Casula, M.; Mozzanica, F.; Scotti, L.; Tragni, E.; Pirillo, A.; Corrao, G.; Catapano, A.L. Statin use and risk of new-onset diabetes: A meta-analysis of observational studies. Nutr. Metab. Cardiovasc. Dis. 2017, 27, 396–406. [Google Scholar] [CrossRef] [PubMed]
- Collins, R.; Reith, C.; Emberson, J.; Armitage, J.; Baigent, C.; Blackwell, L.; Blumenthal, R.; Danesh, J.; Smith, G.D.; DeMets, D.; et al. Interpretation of the evidence for the efficacy and safety of statin therapy. Lancet 2016, 388, 2532–2561. [Google Scholar] [CrossRef] [Green Version]
- Rotella, C.M.; Zaninelli, A.; Le Grazie, C.; Hanson, M.E.; Gensini, G.F. Ezetimibe/simvastatin vs simvastatin in coronary heart disease patients with or without diabetes. Lipids Health Dis. 2010, 9, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leiter, L.A.; Betteridge, D.J.; Farnier, M.; Guyton, J.R.; Lin, J.; Shah, A.; Johnson-Levonas, A.O.; Brudi, P. Lipid-altering efficacy and safety profile of combination therapy with ezetimibe/statin vs. Statin monotherapy in patients with and without diabetes: An analysis of pooled data from 27 clinical trials. Diabetes Obes. Metab. 2011, 13, 615–628. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.; Kawamura, M.; Watanabe, T.; Ashidate, K.; Kohro, T.; Tanaka, A.; Mori, Y.; Tagami, M.; Hirano, T.; Yamazaki, T.; et al. Effect of ezetimibe add-on therapy over 52 weeks extension analysis of prospective randomized trial (research study) in type 2 diabetes subjects. Lipids Health Dis. 2017, 16, 122. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.H.; Wu, L.S.; Lee, C.H.; Kuo, C.T.; Liu, J.R.; Wen, M.S.; Chen, W.J.; See, L.C.; Yeh, Y.H. Simvastatin-ezetimibe combination therapy is associated with a lower rate of major adverse cardiac events in type 2 diabetics than high potency statins alone: A population-based dynamic cohort study. Int. J. Cardiol. 2015, 190, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Mikhailidis, D.P.; Sibbring, G.C.; Ballantyne, C.M.; Davies, G.M.; Catapano, A.L. Meta-analysis of the cholesterol-lowering effect of ezetimibe added to ongoing statin therapy. Cur. Med. Res. Opin. 2007, 23, 2009–2026. [Google Scholar] [CrossRef] [PubMed]
- Ruggenenti, P.; Cattaneo, D.; Rota, S.; Iliev, I.; Parvanova, A.; Diadei, O.; Ene-Iordache, B.; Ferrari, S.; Bossi, A.C.; Trevisan, R.; et al. Effects of combined ezetimibe and simvastatin therapy as compared with simvastatin alone in patients with type 2 diabetes: A prospective randomized double-blind clinical trial. Diabetes Care 2010, 33, 1954–1956. [Google Scholar] [CrossRef] [PubMed]
- Cannon, C.P.; Blazing, M.A.; Giugliano, R.P.; McCagg, A.; White, J.A.; Theroux, P.; Darius, H.; Lewis, B.S.; Ophuis, T.O.; Jukema, J.W.; et al. Ezetimibe added to statin therapy after acute coronary syndromes. N. Engl. J. Med. 2015, 372, 2387–2397. [Google Scholar] [CrossRef] [PubMed]
- Giugliano, R.P.; Cannon, C.P.; Blazing, M.A.; Nicolau, J.C.; Corbalan, R.; Spinar, J.; Park, J.G.; White, J.A.; Bohula, E.A.; Braunwald, E.; et al. Benefit of adding ezetimibe to statin therapy on cardiovascular outcomes and safety in patients with versus without diabetes mellitus: Results from improve-it (improved reduction of outcomes: Vytorin efficacy international trial). Circulation 2018, 137, 1571–1582. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.J.; Choi, J.M.; Kim, L.; Kim, B.J.; Sohn, J.H.; Kim, W.J.; Park, S.E.; Rhee, E.J.; Lee, W.Y.; Oh, K.W.; et al. Chronic administration of ezetimibe increases active glucagon-like peptide-1 and improves glycemic control and pancreatic beta cell mass in a rat model of type 2 diabetes. Biochem. Biophys. Res. Commun. 2011, 407, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Tecson, K.M.; Rocha, N.A.; McCullough, P.A. Usefulness of alirocumab and evolocumab for the treatment of patients with diabetic dyslipidemia. Baylor Univ. Med. Center Proc. 2018, 31, 180–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sattar, N.; Preiss, D.; Robinson, J.G.; Djedjos, C.S.; Elliott, M.; Somaratne, R.; Wasserman, S.M.; Raal, F.J. Lipid-lowering efficacy of the PCSK9 inhibitor evolocumab (AMG 145) in patients with type 2 diabetes: A meta-analysis of individual patient data. Lancet Diabetes Endocrinol. 2016, 4, 403–410. [Google Scholar] [CrossRef]
- Sabatine, M.S.; Giugliano, R.P.; Keech, A.C.; Honarpour, N.; Wiviott, S.D.; Murphy, S.A.; Kuder, J.F.; Wang, H.; Liu, T.; Wasserman, S.M.; et al. Evolocumab and clinical outcomes in patients with cardiovascular disease. N. Engl. J. Med. 2017, 376, 1713–1722. [Google Scholar] [CrossRef] [PubMed]
- Sabatine, M.S.; Leiter, L.A.; Wiviott, S.D.; Giugliano, R.P.; Deedwania, P.; de Ferrari, G.M.; Murphy, S.A.; Kuder, J.F.; Gouni-Berthold, I.; Lewis, B.S.; et al. Cardiovascular safety and efficacy of the PCSK9 inhibitor evolocumab in patients with and without diabetes and the effect of evolocumab on glycaemia and risk of new-onset diabetes: A prespecified analysis of the fourier randomised controlled trial. Lancet Diabetes Endocrinol. 2017, 5, 941–950. [Google Scholar] [CrossRef]
- Leiter, L.A.; Cariou, B.; Muller-Wieland, D.; Colhoun, H.M.; del Prato, S.; Tinahones, F.J.; Ray, K.K.; Bujas-Bobanovic, M.; Domenger, C.; Mandel, J.; et al. Efficacy and safety of alirocumab in insulin-treated individuals with type 1 or type 2 diabetes and high cardiovascular risk: The odyssey dm-insulin randomized trial. Diabetes Obes. Metab. 2017, 19, 1781–1792. [Google Scholar] [CrossRef] [PubMed]
- Ray, K.K.; Leiter, L.A.; Muller-Wieland, D.; Cariou, B.; Colhoun, H.M.; Henry, R.R.; Tinahones, F.J.; Bujas-Bobanovic, M.; Domenger, C.; Letierce, A.; et al. Alirocumab vs usual lipid-lowering care as add-on to statin therapy in individuals with type 2 diabetes and mixed dyslipidaemia: The odyssey dm-dyslipidemia randomized trial. Diabetes Obes. Metab. 2018, 20, 1479–1489. [Google Scholar] [CrossRef] [PubMed]
- Lotta, L.A.; Sharp, S.J.; Burgess, S.; Perry, J.R.B.; Stewart, I.D.; Willems, S.M.; Luan, J.; Ardanaz, E.; Arriola, L.; Balkau, B.; et al. Association between low-density lipoprotein cholesterol-lowering genetic variants and risk of type 2 diabetes: A meta-analysis. JAMA 2016, 316, 1383–1391. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.F.; Swerdlow, D.I.; Holmes, M.V.; Patel, R.S.; Fairhurst-Hunter, Z.; Lyall, D.M.; Hartwig, F.P.; Horta, B.L.; Hyppönen, E.; Power, C.; et al. Pcsk9 genetic variants and risk of type 2 diabetes: A mendelian randomisation study. Lancet Diabetes Endocrinol. 2017, 5, 97–105. [Google Scholar] [CrossRef]
- Colhoun, H.M.; Ginsberg, H.N.; Robinson, J.G.; Leiter, L.A.; Muller-Wieland, D.; Henry, R.R.; Cariou, B.; Baccara-Dinet, M.T.; Pordy, R.; Merlet, L.; et al. No effect of pcsk9 inhibitor alirocumab on the incidence of diabetes in a pooled analysis from 10 odyssey phase 3 studies. Eur. Heart J. 2016, 37, 2981–2989. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.X.; Liu, H.H.; Dong, Q.T.; Li, S.; Li, J.J. Effect of proprotein convertase subtilisin/kexin type 9 (PCSK9) monoclonal antibodies on new-onset diabetes mellitus and glucose metabolism: A systematic review and meta-analysis. Diabetes Obes. Metab. 2018, 20, 1391–1398. [Google Scholar] [CrossRef] [PubMed]
- Hafiane, A.; Kellett, S.; Genest, J. Treatment options for low high-density lipoproteins. Curr. Opin. Endocrinol. Diabetes Obes. 2014, 21, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Santos-Gallego, C.G.; Rosenson, R.S. Role of HDL in those with diabetes. Curr. Cardiol. Rep. 2014, 16, 512. [Google Scholar] [CrossRef] [PubMed]
- Boden, W.E.; Probstfield, J.L.; Anderson, T.; Chaitman, B.R.; Desvignes-Nickens, P.; Koprowicz, K.; McBride, R.; Teo, K.; Weintraub, W. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N. Engl. J. Med. 2011, 365, 2255–2267. [Google Scholar] [CrossRef] [PubMed]
- Group, H.T.C.; Landray, M.J.; Haynes, R.; Hopewell, J.C.; Parish, S.; Aung, T.; Tomson, J.; Wallendszus, K.; Craig, M.; Jiang, L.; et al. Effects of extended-release niacin with laropiprant in high-risk patients. N. Engl. J. Med. 2014, 371, 203–212. [Google Scholar] [CrossRef]
- Khera, A.V.; Patel, P.J.; Reilly, M.P.; Rader, D.J. The addition of niacin to statin therapy improves high-density lipoprotein cholesterol levels but not metrics of functionality. J. Am. Coll. Cardiol. 2013, 62, 1909–1910. [Google Scholar] [CrossRef] [PubMed]
- Goldie, C.; Taylor, A.J.; Nguyen, P.; McCoy, C.; Zhao, X.Q.; Preiss, D. Niacin therapy and the risk of new-onset diabetes: A meta-analysis of randomised controlled trials. Heart 2016, 102, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Barter, P.J.; Caulfield, M.; Eriksson, M.; Grundy, S.M.; Kastelein, J.J.; Komajda, M.; Lopez-Sendon, J.; Mosca, L.; Tardif, J.C.; Waters, D.D.; et al. Effects of torcetrapib in patients at high risk for coronary events. N. Engl. J. Med. 2007, 357, 2109–2122. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, G.G.; Olsson, A.G.; Abt, M.; Ballantyne, C.M.; Barter, P.J.; Brumm, J.; Chaitman, B.R.; Holme, I.M.; Kallend, D.; Leiter, L.A.; et al. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N. Engl. J. Med. 2012, 367, 2089–2099. [Google Scholar] [CrossRef] [PubMed]
- Lincoff, A.M.; Nicholls, S.J.; Riesmeyer, J.S.; Barter, P.J.; Brewer, H.B.; Fox, K.A.A.; Gibson, C.M.; Granger, C.; Menon, V.; Montalescot, G.; et al. Evacetrapib and cardiovascular outcomes in high-risk vascular disease. N. Engl. J. Med. 2017, 376, 1933–1942. [Google Scholar] [CrossRef] [PubMed]
- Group, H.T.R.C.; Bowman, L.; Hopewell, J.C.; Chen, F.; Wallendszus, K.; Stevens, W.; Collins, R.; Wiviott, S.D.; Cannon, C.P.; Braunwald, E.; et al. Effects of anacetrapib in patients with atherosclerotic vascular disease. N. Engl. J. Med. 2017, 377, 1217–1227. [Google Scholar] [CrossRef]
- Yvan-Charvet, L.; Kling, J.; Pagler, T.; Li, H.; Hubbard, B.; Fisher, T.; Sparrow, C.P.; Taggart, A.K.; Tall, A.R. Cholesterol efflux potential and antiinflammatory properties of high-density lipoprotein after treatment with niacin or anacetrapib. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1430–1438. [Google Scholar] [CrossRef] [PubMed]
- Tall, A.R.; Rader, D.J. Trials and tribulations of CETP inhibitors. Circ. Res. 2018, 122, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Sviridov, D.; Remaley, A.T. High-density lipoprotein mimetics: Promises and challenges. Biochem. J. 2015, 472, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Morgantini, C.; Imaizumi, S.; Grijalva, V.; Navab, M.; Fogelman, A.M.; Reddy, S.T. Apolipoprotein A-I mimetic peptides prevent atherosclerosis development and reduce plaque inflammation in a murine model of diabetes. Diabetes 2010, 59, 3223–3228. [Google Scholar] [CrossRef] [PubMed]
- McClelland, A.D.; Kantharidis, P. Microrna in the development of diabetic complications. Clin. Sci 2014, 126, 95–110. [Google Scholar] [CrossRef] [PubMed]
- Shantikumar, S.; Caporali, A.; Emanueli, C. Role of micrornas in diabetes and its cardiovascular complications. Cardiovasc. Res. 2012, 93, 583–593. [Google Scholar] [CrossRef] [PubMed]
- Desgagne, V.; Bouchard, L.; Guerin, R. Micrornas in lipoprotein and lipid metabolism: From biological function to clinical application. Clin. Chem. Lab. Med. 2017, 55, 667–686. [Google Scholar] [CrossRef] [PubMed]
- Regazzi, R. Micrornas as therapeutic targets for the treatment of diabetes mellitus and its complications. Expert Opin. Ther. Targets 2018, 22, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Guay, C.; Regazzi, R. Circulating micrornas as novel biomarkers for diabetes mellitus. Nat. Rev. Endocrinol. 2013, 9, 513–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vickers, K.C.; Palmisano, B.T.; Shoucri, B.M.; Shamburek, R.D.; Remaley, A.T. Micrornas are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nat. Cell. Biol. 2011, 13, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Michell, D.L.; Vickers, K.C. Lipoprotein carriers of micrornas. Biochim. Biophys. Acta 2016, 1861, 2069–2074. [Google Scholar] [CrossRef] [PubMed]
- Canfran-Duque, A.; Lin, C.S.; Goedeke, L.; Suarez, Y.; Fernandez-Hernando, C. Micro-rnas and high-density lipoprotein metabolism. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1076–1084. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wong, N.K.P.; Nicholls, S.J.; Tan, J.T.M.; Bursill, C.A. The Role of High-Density Lipoproteins in Diabetes and Its Vascular Complications. Int. J. Mol. Sci. 2018, 19, 1680. https://doi.org/10.3390/ijms19061680
Wong NKP, Nicholls SJ, Tan JTM, Bursill CA. The Role of High-Density Lipoproteins in Diabetes and Its Vascular Complications. International Journal of Molecular Sciences. 2018; 19(6):1680. https://doi.org/10.3390/ijms19061680
Chicago/Turabian StyleWong, Nathan K. P., Stephen J. Nicholls, Joanne T. M. Tan, and Christina A. Bursill. 2018. "The Role of High-Density Lipoproteins in Diabetes and Its Vascular Complications" International Journal of Molecular Sciences 19, no. 6: 1680. https://doi.org/10.3390/ijms19061680
APA StyleWong, N. K. P., Nicholls, S. J., Tan, J. T. M., & Bursill, C. A. (2018). The Role of High-Density Lipoproteins in Diabetes and Its Vascular Complications. International Journal of Molecular Sciences, 19(6), 1680. https://doi.org/10.3390/ijms19061680