The Interplay between Two Transcriptional Repressors and Chaperones Orchestrates Helicobacter pylori Heat-Shock Response



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
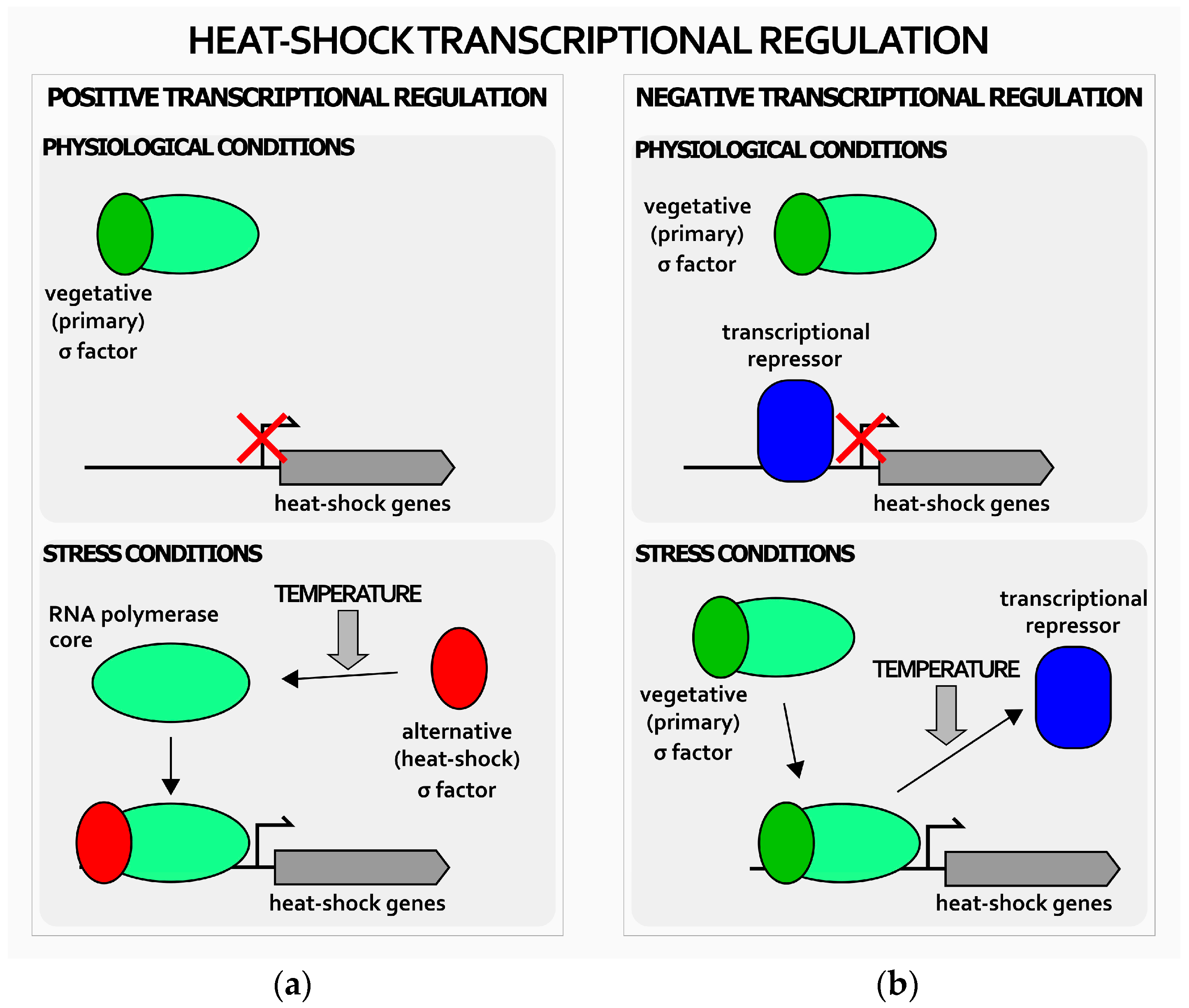
2. Transcriptional Regulation of Heat-Shock Genes in H. pylori
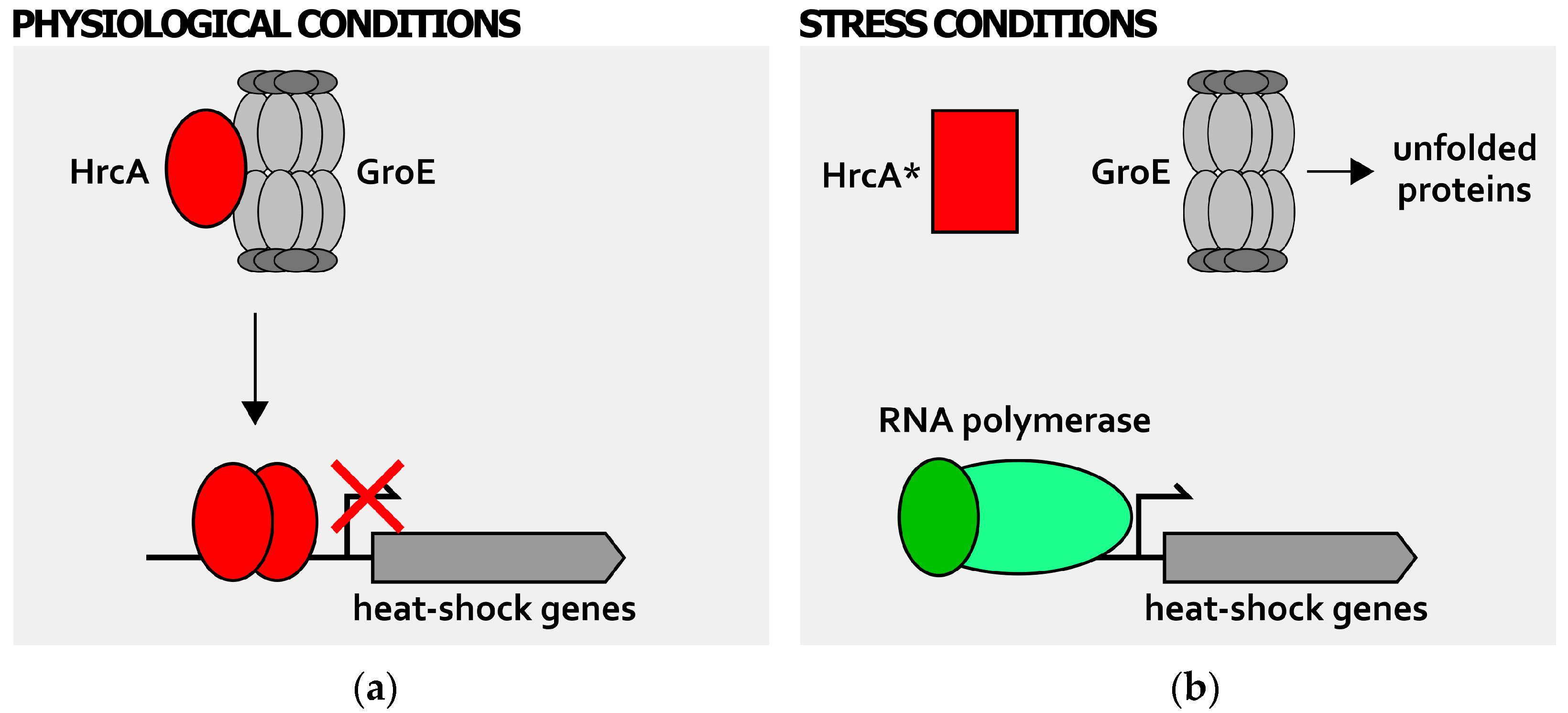
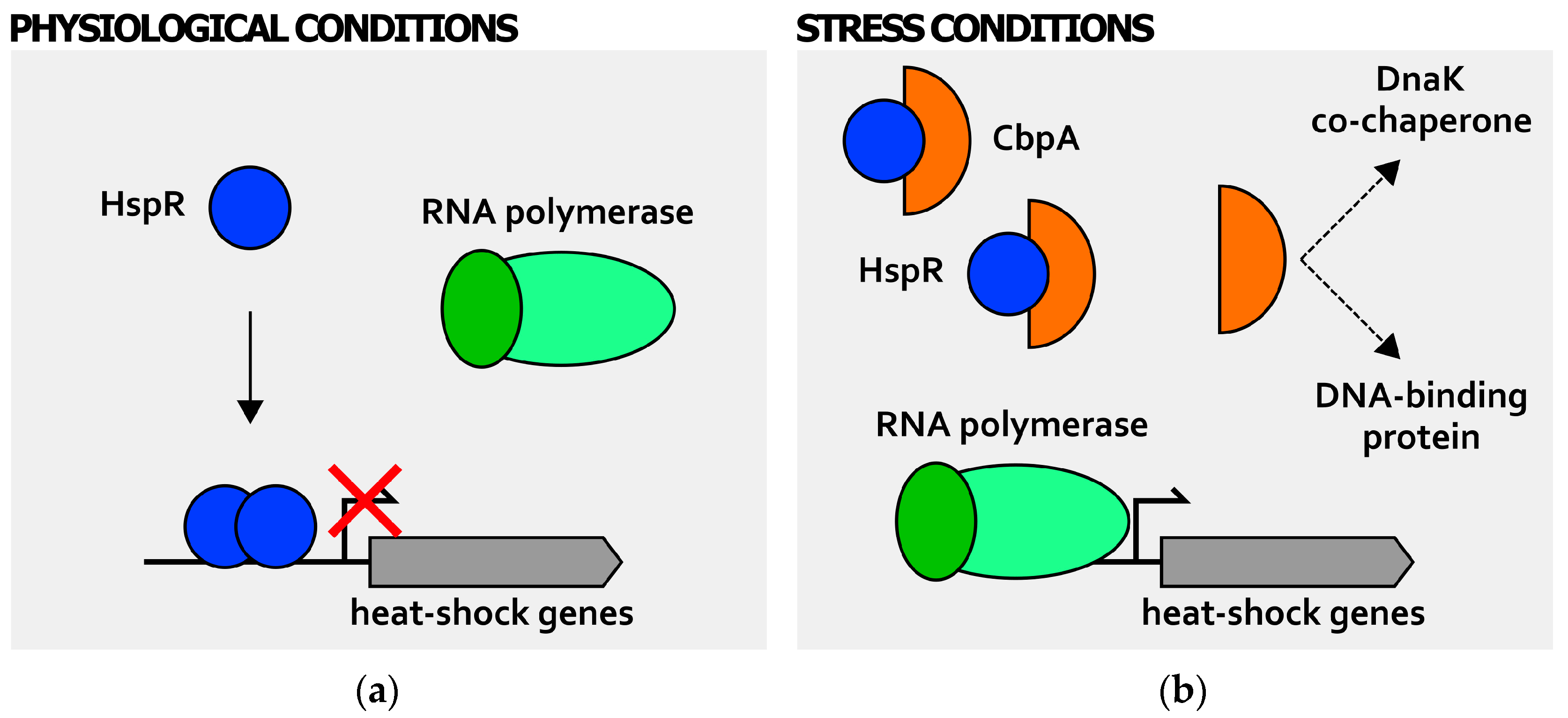
3. Posttranscriptional Regulation
4. Detection of Stress Signals
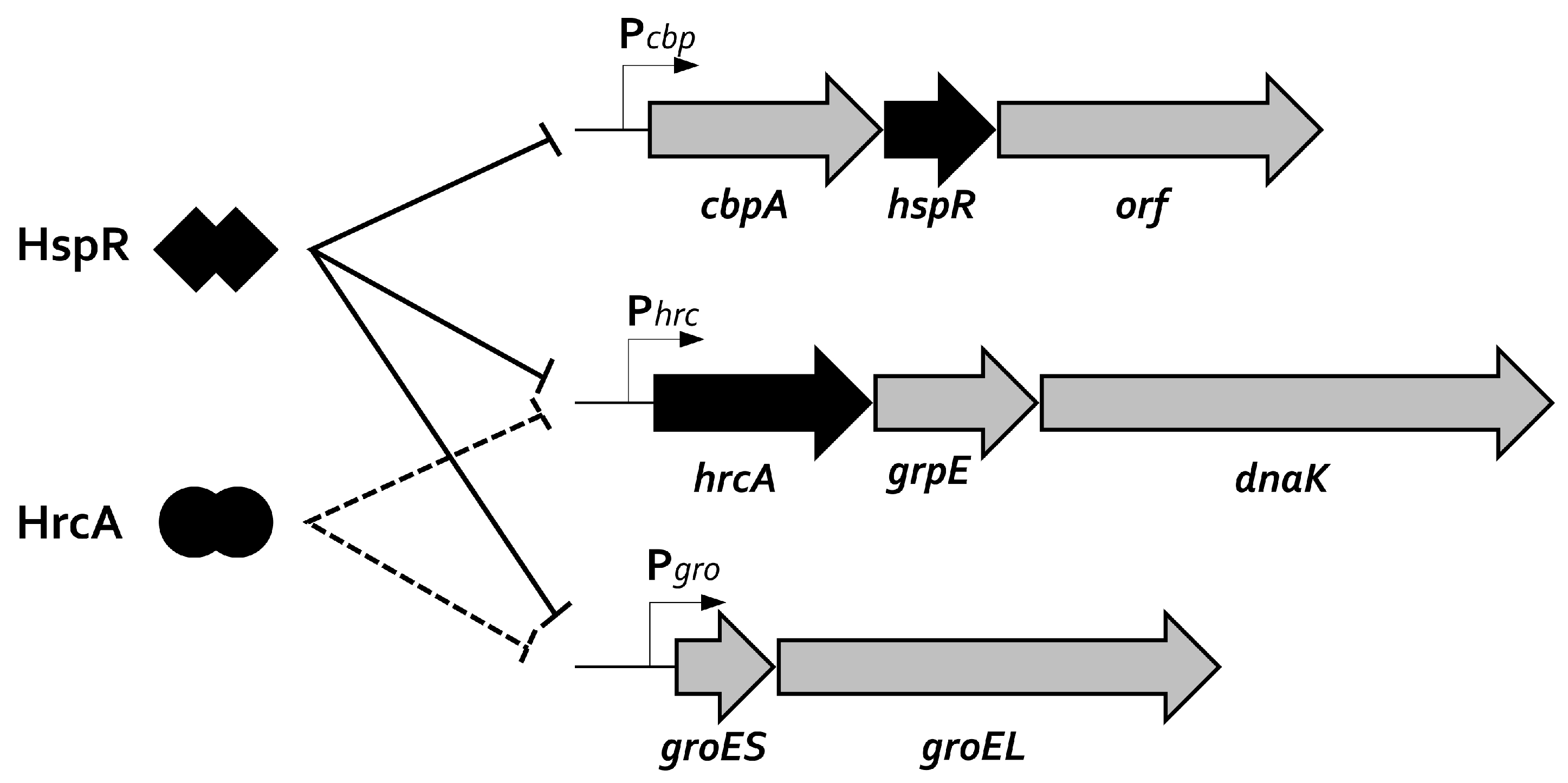
5. Genome-Wide Studies: HspR and HrcA Regulons
6. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| HSP | Heat-Shock Proteins |
| HAIR | HspR Associated Inverted Repeat |
| CIRCE | Controlling Inverted Repeat of Chaperone Expression |
| bp | Base-pairs |
| Pgro | Promoter region controlling groES-groEL operon transcription |
| Phrc | Promoter region controlling hrcA-grpE-dnaK operon transcription |
| Pcbp | Promoter region controlling cbpA-hspR-helicase operon transcription |
| DNase I | Deoxyribonuclease I |
| STAT-3 | Signal transducer and activator of transcription 3 |
| COX-2 | Cyclooxygenase 2 |
References
- Narberhaus, F. Negative regulation of bacterial heat shock genes. Mol. Microbiol. 1999, 31, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roncarati, D.; Scarlato, V. Regulation of heat-shock genes in bacteria: From signal sensing to gene expression output. FEMS Microbiol. Rev. 2017, 41, 549–574. [Google Scholar] [CrossRef] [PubMed]
- Mogk, A.; Tomoyasu, T.; Goloubinoff, P.; Rüdiger, S.; Röder, D.; Langen, H.; Bukau, B. Identification of thermolabile Escherichia coli proteins: Prevention and reversion of aggregation by DnaK and ClpB. EMBO J. 1999, 18, 6934–6949. [Google Scholar] [CrossRef] [PubMed]
- Peterson, J.W. Bacterial Pathogenesis. In Medical Microbiology, 4th ed.; Baron, S., Ed.; University of Texas Medical Branch at Galveston: Galveston, TX, USA, 1996; Chapter 7; pp. 100–110, ISBN-10 0-9631172-1-1. [Google Scholar]
- Gu, H. Role of Flagella in the Pathogenesis of Helicobacter pylori. Curr. Microbiol. 2017, 74, 863–869. [Google Scholar] [CrossRef] [PubMed]
- Mobley, H.L. The role of Helicobacter pylori urease in the pathogenesis of gastritis and peptic ulceration. Aliment. Pharmacol. Ther. 1996, 1, 57–64. [Google Scholar] [CrossRef]
- Posselt, G.; Backert, S.; Wessler, S. The functional interplay of Helicobacter pylori factors with gastric epithelial cells induces a multi-step process in pathogenesis. Cell Commun. Signal. 2013, 11, 77. [Google Scholar] [CrossRef] [PubMed]
- Covacci, A.; Falkow, S.; Berg, D.E.; Rappuoli, R. Did the inheritance of a pathogenicity island modify the virulence of Helicobacter pylori? Trends Microbiol. 1997, 5, 205–208. [Google Scholar] [CrossRef]
- De Bernard, M.; Papini, E.; de Filippis, V.; Gottardi, E.; Telford, J.; Manetti, R.; Fontana, A.; Rappuoli, R.; Montecucco, C. Low pH activates the vacuolating toxin of Helicobacter pylori, which becomes acid and pepsin resistant. J. Biol. Chem. 1995, 270, 23937–23940. [Google Scholar] [CrossRef] [PubMed]
- Kao, C.Y.; Sheu, B.S.; Wu, J.J. Helicobacter pylori infection: An overview of bacterial virulence factors and pathogenesis. Biomed. J. 2016, 39, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Henderson, B.; Allan, E.; Coates, A.R. Stress wars: The direct role of host and bacterial molecular chaperones in bacterial infection. Infect. Immun. 2006, 74, 3693–3706. [Google Scholar] [CrossRef] [PubMed]
- Targosz, A.; Pierzchalski, P.; Krawiec, A.; Szczyrk, U.; Brzozowski, T.; Konturek, S.J.; Pawlik, W.W. Helicobacter pylori inhibits expression of heat shock protein 70 (HSP70) in human epithelial cell line. Importance of Cag A protein. J. Physiol. Pharmacol. 2006, 57, 265–278. [Google Scholar] [PubMed]
- Yeo, M.; Park, H.K.; Kim, D.K.; Cho, S.W.; Kim, Y.S.; Cho, S.Y.; Paik, Y.K.; Hahm, K.B. Restoration of heat shock protein 70 suppresses gastric mucosal inducible nitric oxide synthase expression induced by Helicobacter pylori. Proteomics 2004, 4, 3335–3342. [Google Scholar] [CrossRef] [PubMed]
- Konturek, J.W.; Fischer, H.; Konturek, P.C.; Huber, V.; Boknik, P.; Luess, H.; Neumann, J.; Brzozowski, T.; Schmitz, W.; Hahn, E.G.; et al. Heat shock protein 70 (HSP70) in gastric adaptation to aspirin in Helicobacter pylori infection. J. Physiol. Pharmacol. 2001, 52, 153–164. [Google Scholar] [PubMed]
- Axsen, W.S.; Styer, C.M.; Solnick, J.V. Inhibition of heat shock protein expression by Helicobacter pylori. Microb. Pathog. 2009, 47, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Slamti, L.; Livny, J.; Waldor, M.K. Global gene expression and phenotypic analysis of a Vibrio cholerae rpoH deletion mutant. J. Bacteriol. 2007, 189, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Allan, B.; Linseman, M.; MacDonald, L.A.; Lam, J.S.; Kropinski, A.M. Heat shock response of Pseudomonas aeruginosa. J. Bacteriol. 1988, 170, 3668–3674. [Google Scholar] [CrossRef] [PubMed]
- Schumann, W. Regulation of bacterial heat shock stimulons. Cell Stress Chaperones 2016, 21, 959–968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chastanet, A.; Prudhomme, M.; Claverys, J.P.; Msadek, T. Regulation of Streptococcus pneumoniae clp genes and their role in competence development and stress survival. J. Bacteriol. 2001, 183, 7295–7307. [Google Scholar] [CrossRef] [PubMed]
- Musatovova, O.; Dhandayuthapani, S.; Baseman, J.B. Transcriptional heat shock response in the smallest known self-replicating cell, Mycoplasma genitalium. J. Bacteriol. 2006, 188, 2845–2855. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Smyth, D.; O’Hagan, B.M.G.; Heap, J.T.; McMullan, G.; Minton, N.P.; Ternan, N.G. Inactivation of the dnaK gene in Clostridium difficile 630 Δerm yields a temperature-sensitive phenotype and increases biofilm-forming ability. Sci. Rep. 2017, 7, 17522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wetzstein, M.; Völker, U.; Dedio, J.; Löbau, S.; Zuber, U.; Schiesswohl, M.; Herget, C.; Hecker, M.; Schumann, W. Cloning, sequencing, and molecular analysis of the dnaK locus from Bacillus subtilis. J. Bacteriol. 1992, 174, 3300–3310. [Google Scholar] [CrossRef] [PubMed]
- Bucca, G.; Ferina, G.; Puglia, A.M.; Smith, C.P. The dnaK operon of Streptomyces coelicolor encodes a novel heat-shock protein which binds to the promoter region of the operon. Mol. Microbiol. 1995, 17, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Tomb, J.F.; White, O.; Kerlavage, A.R.; Clayton, R.A.; Sutton, G.G.; Fleischmann, R.D.; Ketchum, K.A.; Klenk, H.P.; Gill, S.; Dougherty, B.A.; et al. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature 1997, 388, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Spohn, G.; Scarlato, V. The autoregulatory HspR repressor protein governs chaperone gene transcription in Helicobacter pylori. Mol. Microbiol. 1999, 34, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Spohn, G.; Danielli, A.; Roncarati, D.; Delany, I.; Rappuoli, R.; Scarlato, V. Dual control of Helicobacter pylori heat shock gene transcription by HspR and HrcA. J. Bacteriol. 2004, 186, 2956–2965. [Google Scholar] [CrossRef] [PubMed]
- Roncarati, D.; Danielli, A.; Spohn, G.; Delany, I.; Scarlato, V. Transcriptional regulation of stress response and motility functions in Helicobacter pylori is mediated by HspR and HrcA. J. Bacteriol. 2007, 189, 7234–7243. [Google Scholar] [CrossRef] [PubMed]
- Roncarati, D.; Spohn, G.; Tango, N.; Danielli, A.; Delany, I.; Scarlato, V. Expression, purification and characterization of the membrane-associated HrcA repressor protein of Helicobacter pylori. Protein Expr. Purif. 2007, 51, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Homuth, G.; Domm, S.; Kleiner, D.; Schumann, W. Transcriptional analysis of major heat shock genes of Helicobacter pylori. J. Bacteriol. 2000, 182, 4257–4263. [Google Scholar] [CrossRef] [PubMed]
- Spohn, G.; Delany, I.; Rappuoli, R.; Scarlato, V. Characterization of the HspR-mediated stress response in Helicobacter pylori. J. Bacteriol. 2002, 184, 2925–2930. [Google Scholar] [CrossRef] [PubMed]
- Huesca, M.; Borgia, S.; Hoffman, P.; Lingwood, C.A. Acidic pH changes receptor binding specificity of Helicobacter pylori: A binary adhesion model in which surface heat shock (stress) proteins mediate sulfatide recognition in gastric colonization. Infect. Immun. 1996, 64, 2643–2648. [Google Scholar] [PubMed]
- Huesca, M.; Goodwin, A.; Bhagwansingh, A.; Hoffman, P.; Lingwood, C.A. Characterization of an acidic-pH-inducible stress protein (hsp70), a putative sulfatide binding adhesin, from Helicobacter pylori. Infect. Immun. 1998, 66, 4061–4067. [Google Scholar] [PubMed]
- De la Cruz, M.A.; Ares, M.A.; von Bargen, K.; Panunzi, L.G.; Martínez-Cruz, J.; Valdez-Salazar, H.A.; Jiménez-Galicia, C.; Torres, J. Gene expression profiling of transcription factors of Helicobacter pylori under different environmental conditions. Front. Microbiol. 2017, 8, 615. [Google Scholar] [CrossRef] [PubMed]
- Lim, B.; Miyazaki, R.; Neher, S.; Siegele, D.A.; Ito, K.; Walter, P.; Akiyama, Y.; Yura, T.; Gross, C.A. Heat shock transcription factor σ32 co-opts the signal recognition particle to regulate protein homeostasis in E. coli. PLoS Biol. 2013, 11, e1001735. [Google Scholar] [CrossRef] [PubMed]
- Mogk, A.; Völker, A.; Engelmann, S.; Hecker, M.; Schumann, W.; Völker, U. Nonnative proteins induce expression of the Bacillus subtilis CIRCE regulon. J. Bacteriol. 1998, 180, 2895–2900. [Google Scholar] [PubMed]
- Wilson, A.C.; Wu, C.C.; Yates, J.R., 3rd; Tan, M. Chlamydial GroEL autoregulates its own expression through direct interactions with the HrcA repressor protein. J. Bacteriol. 2005, 187, 7535–7542. [Google Scholar] [CrossRef] [PubMed]
- Bucca, G.; Brassington, A.M.; Schönfeld, H.J.; Smith, C.P. The HspR regulon of Streptomyces coelicolor: A role for the DnaK chaperone as a transcriptional co-repressordagger. Mol. Microbiol. 2000, 38, 1093–1103. [Google Scholar] [CrossRef] [PubMed]
- Parijat, P.; Batra, J.K. Role of DnaK in HspR-HAIR interaction of Mycobacterium tuberculosis. IUBMB Life 2015, 67, 816–827. [Google Scholar] [CrossRef] [PubMed]
- Roncarati, D.; Danielli, A.; Scarlato, V. The HrcA repressor is the thermosensor of the heat-shock regulatory circuit in the human pathogen Helicobacter pylori. Mol. Microbiol. 2014, 92, 910–920. [Google Scholar] [CrossRef] [PubMed]
- Roncarati, D.; Danielli, A.; Scarlato, V. CbpA acts as a modulator of HspR repressor DNA binding activity in Helicobacter pylori. J. Bacteriol. 2011, 193, 5629–5636. [Google Scholar] [CrossRef] [PubMed]
- Chae, C.; Sharma, S.; Hoskins, J.R.; Wickner, S. CbpA, a DnaJ homolog, is a DnaK co-chaperone, and its activity is modulated by CbpM. J. Biol. Chem. 2004, 279, 33147–33153. [Google Scholar] [CrossRef] [PubMed]
- Bird, J.G.; Sharma, S.; Roshwalb, S.C.; Hoskins, J.R.; Wickner, S. Functional analysis of CbpA, a DnaJ homolog and nucleoid-associated DNA-binding protein. J. Biol. Chem. 2006, 281, 34349–34356. [Google Scholar] [CrossRef] [PubMed]
- Roncarati, D.; Department of Pharmacy and Biotechnology, University of Bologna, Bologna, Italy. Preliminary Experimental Evidences on Helicobacter pylori CbpA Roles as a DnaK-co-Chaperone and as a DNA-Binding Protein. 2018. [Google Scholar]
- Kortmann, J.; Narberhaus, F. Bacterial RNA thermometers: Molecular zippers and switches. Nat. Rev. Microbiol. 2012, 10, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Ignatov, D.; Johansson, J. RNA-mediated signal perception in pathogenic bacteria. Wiley Interdiscip. Rev. RNA 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Prosseda, G.; Falconi, M.; Giangrossi, M.; Gualerzi, C.O.; Micheli, G.; Colonna, B. The virF promoter in Shigella: More than just a curved DNA stretch. Mol. Microbiol. 2004, 51, 523–537. [Google Scholar] [CrossRef] [PubMed]
- Nickerson, C.A.; Achberger, E.C. Role of curved DNA in binding of Escherichia coli RNA polymerase to promoters. J. Bacteriol. 1995, 177, 5756–5761. [Google Scholar] [CrossRef] [PubMed]
- Herbst, K.; Bujara, M.; Heroven, A.K.; Opitz, W.; Weichert, M.; Zimmermann, A.; Dersch, P. Intrinsic thermal sensing controls proteolysis of Yersinia virulence regulator RovA. PLoS Pathog. 2009, 5, e1000435. [Google Scholar] [CrossRef] [PubMed]
- Hurme, R.; Berndt, K.D.; Normark, S.J.; Rhen, M. A proteinaceous gene regulatory thermometer in Salmonella. Cell 1997, 90, 55–64. [Google Scholar] [CrossRef]
- Elsholz, A.K.; Michalik, S.; Zühlke, D.; Hecker, M.; Gerth, U. CtsR, the Gram-positive master regulator of protein quality control, feels the heat. EMBO J. 2010, 29, 3621–3629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Servant, P.; Grandvalet, C.; Mazodier, P. The RheA repressor is the thermosensor of the HSP18 heat shock response in Streptomyces albus. Proc. Natl. Acad. Sci. USA 2000, 97, 3538–3543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, G.R.; Wernisch, L.; Stabler, R.; Mangan, J.A.; Hinds, J.; Laing, G.; Young, D.B.; Butcher, P.D. Dissection of the heat-shock response in Mycobacterium tuberculosis using mutants and microarrays. Microbiology 2002, 148, 3129–3138. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Oliver, H.F.; Raengpradub, S.; Palmer, M.E.; Orsi, R.H.; Wiedmann, M.; Boor, K.J. Transcriptomic and phenotypic analyses suggest a network between the transcriptional regulators HrcA and σB in Listeria monocytogenes. Appl. Environ. Microbiol. 2007, 73, 7981–7991. [Google Scholar] [CrossRef] [PubMed]
- Bucca, G.; Laing, E.; Mersinias, V.; Allenby, N.; Hurd, D.; Holdstock, J.; Brenner, V.; Harrison, M.; Smith, C.P. Development and application of versatile high density microarrays for genome-wide analysis of Streptomyces coelicolor: Characterization of the HspR regulon. Genome Biol. 2009, 10, R5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmes, C.W.; Penn, C.W.; Lund, P.A. The hrcA and hspR regulons of Campylobacter jejuni. Microbiology 2010, 156, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Van Bokhorst-van de Veen, H.; Bongers, R.S.; Wels, M.; Bron, P.A.; Kleerebezem, M. Transcriptome signatures of class I and III stress response deregulation in Lactobacillus plantarum reveal pleiotropic adaptation. Microb. Cell Fact. 2013, 12, 112. [Google Scholar] [CrossRef] [PubMed]
- Andersen, M.T.; Brøndsted, L.; Pearson, B.M.; Mulholland, F.; Parker, M.; Pin, C.; Wells, J.M.; Ingmer, H. Diverse roles for HspR in Campylobacter jejuni revealed by the proteome, transcriptome and phenotypic characterization of an hspR mutant. Microbiology 2005, 151, 905–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pepe, S.; Pinatel, E.; Fiore, E.; Puccio, S.; Peano, C.; Brignoli, T.; Vannini, A.; Danielli, A.; Scarlato, V.; Roncarati, D. The Helicobacter pylori heat-shock repressor HspR: Definition of its direct regulon and characterization of the cooperative DNA-Binding mechanism on its own promoter. Front Microbiol. 2018. Under review. [Google Scholar]
- Pierzchalski, P.; Krawiec, A.; Ptak-Belowska, A.; Barańska, A.; Konturek, S.J.; Pawlik, W.W. The mechanism of heat-shock protein 70 gene expression abolition in gastric epithelium caused by Helicobacter pylori infection. Helicobacter 2006, 11, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Targosz, A.; Brzozowski, T.; Pierzchalski, P.; Szczyrk, U.; Ptak-Belowska, A.; Konturek, S.J.; Pawlik, W. Helicobacter pylori promotes apoptosis, activates cyclooxygenase (COX)-2 and inhibits heat shock protein HSP70 in gastric cancer epithelial cells. Inflamm. Res. 2012, 61, 955–966. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roncarati, D.; Scarlato, V. The Interplay between Two Transcriptional Repressors and Chaperones Orchestrates Helicobacter pylori Heat-Shock Response. Int. J. Mol. Sci. 2018, 19, 1702. https://doi.org/10.3390/ijms19061702
Roncarati D, Scarlato V. The Interplay between Two Transcriptional Repressors and Chaperones Orchestrates Helicobacter pylori Heat-Shock Response. International Journal of Molecular Sciences. 2018; 19(6):1702. https://doi.org/10.3390/ijms19061702
Chicago/Turabian StyleRoncarati, Davide, and Vincenzo Scarlato. 2018. "The Interplay between Two Transcriptional Repressors and Chaperones Orchestrates Helicobacter pylori Heat-Shock Response" International Journal of Molecular Sciences 19, no. 6: 1702. https://doi.org/10.3390/ijms19061702
APA StyleRoncarati, D., & Scarlato, V. (2018). The Interplay between Two Transcriptional Repressors and Chaperones Orchestrates Helicobacter pylori Heat-Shock Response. International Journal of Molecular Sciences, 19(6), 1702. https://doi.org/10.3390/ijms19061702