Activating the Anaphase Promoting Complex to Enhance Genomic Stability and Prolong Lifespan

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
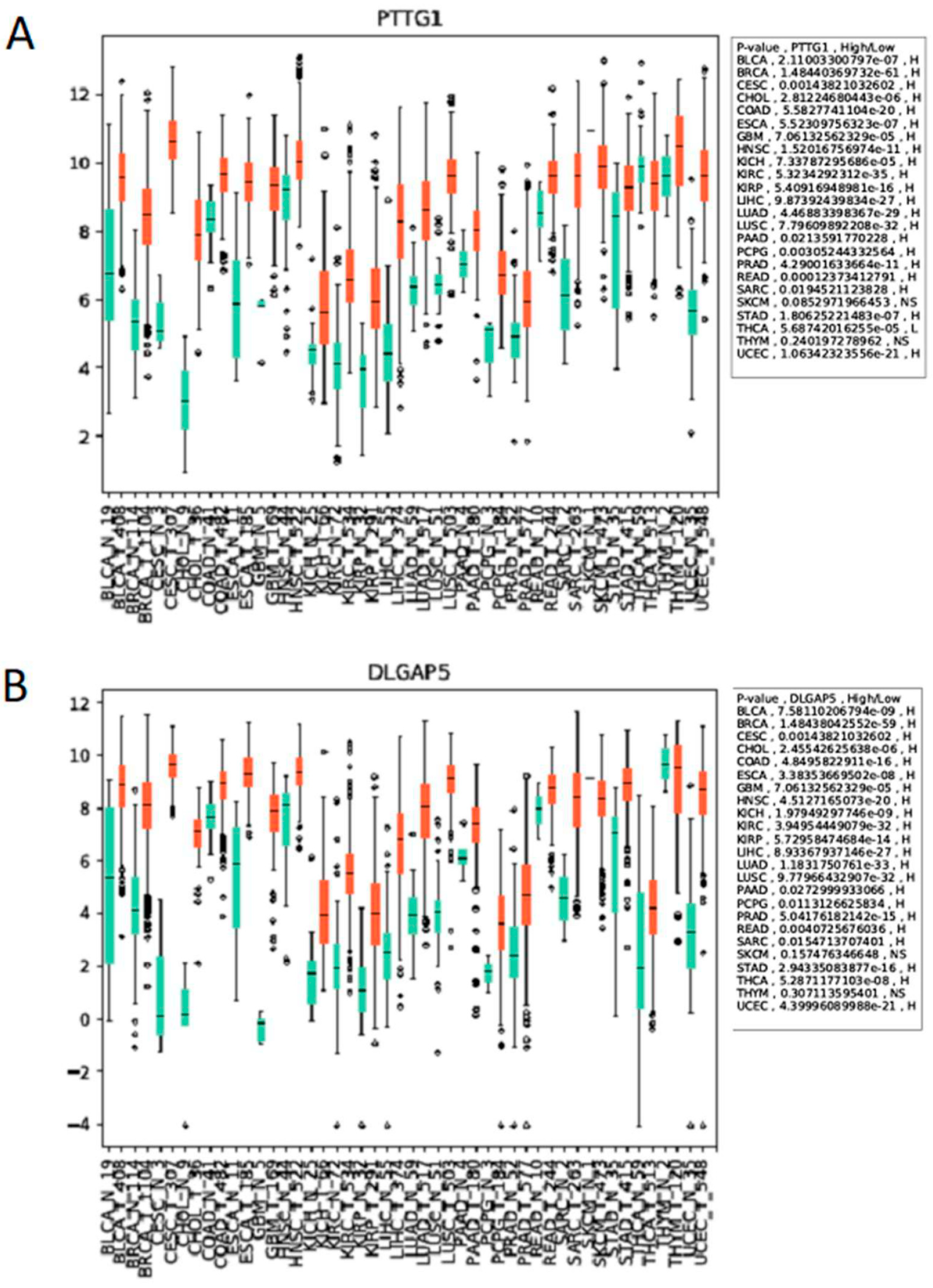{kind=link}
1. Introduction
2. Genetic Control of Longevity
3. Genetic Control of Aging
4. A Ceiling on a Maximum Lifespan?
5. So When Does Aging Begin?
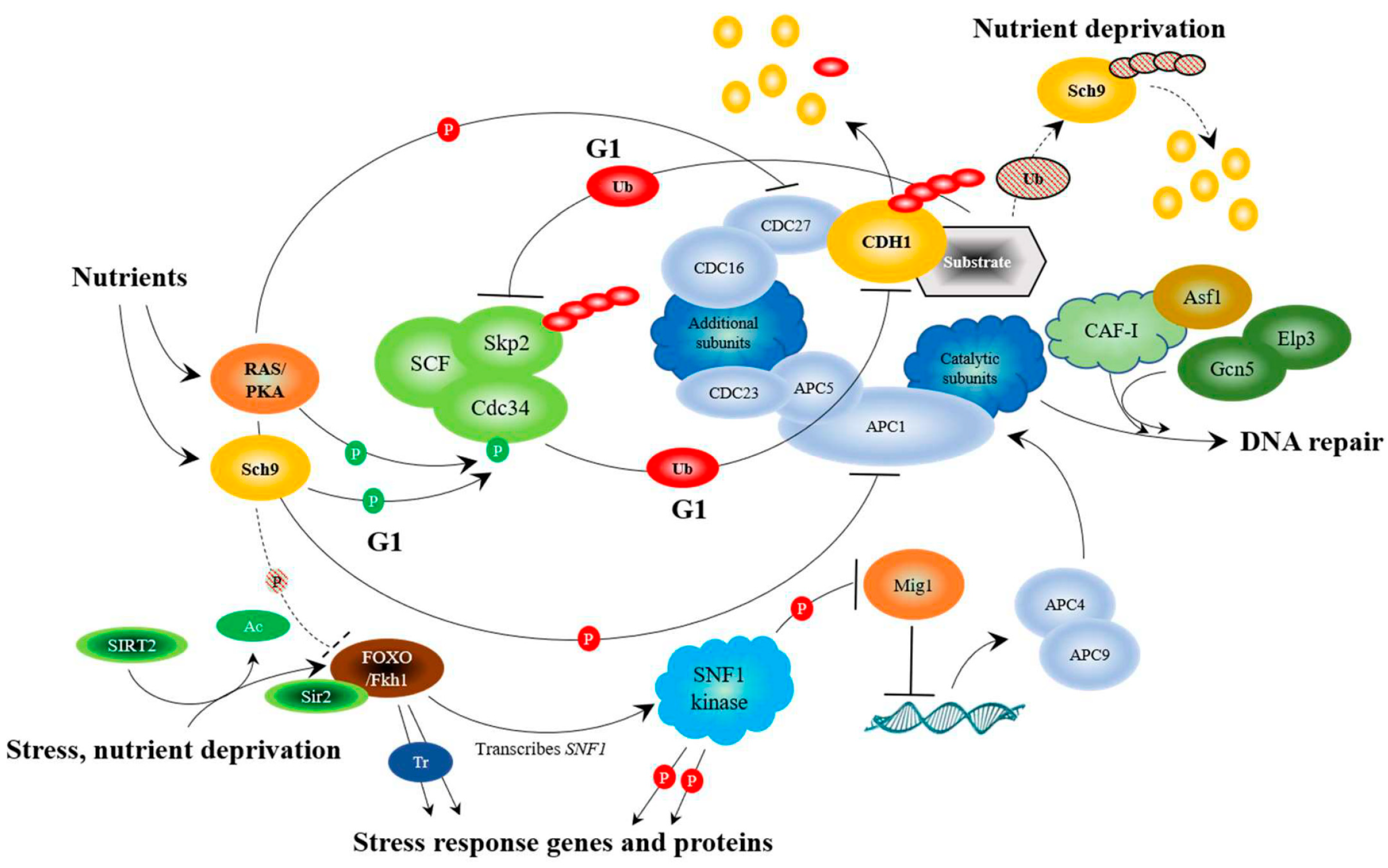
6. Connecting Stress Sensing with Nutrient Sensing
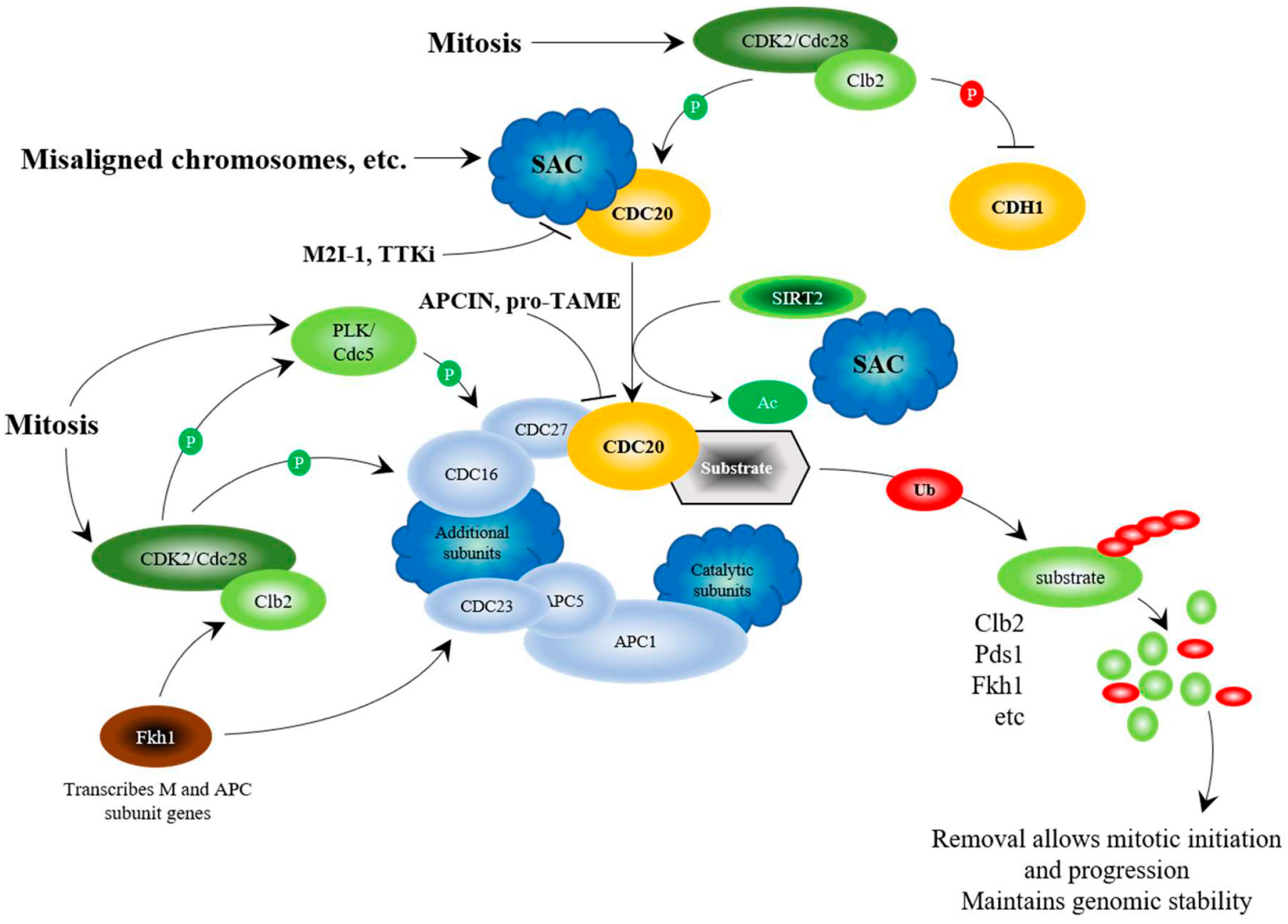
7. The Anaphase Promoting Complex, Using Chromatin Assembly during Mitosis to Maintain Genome Stability
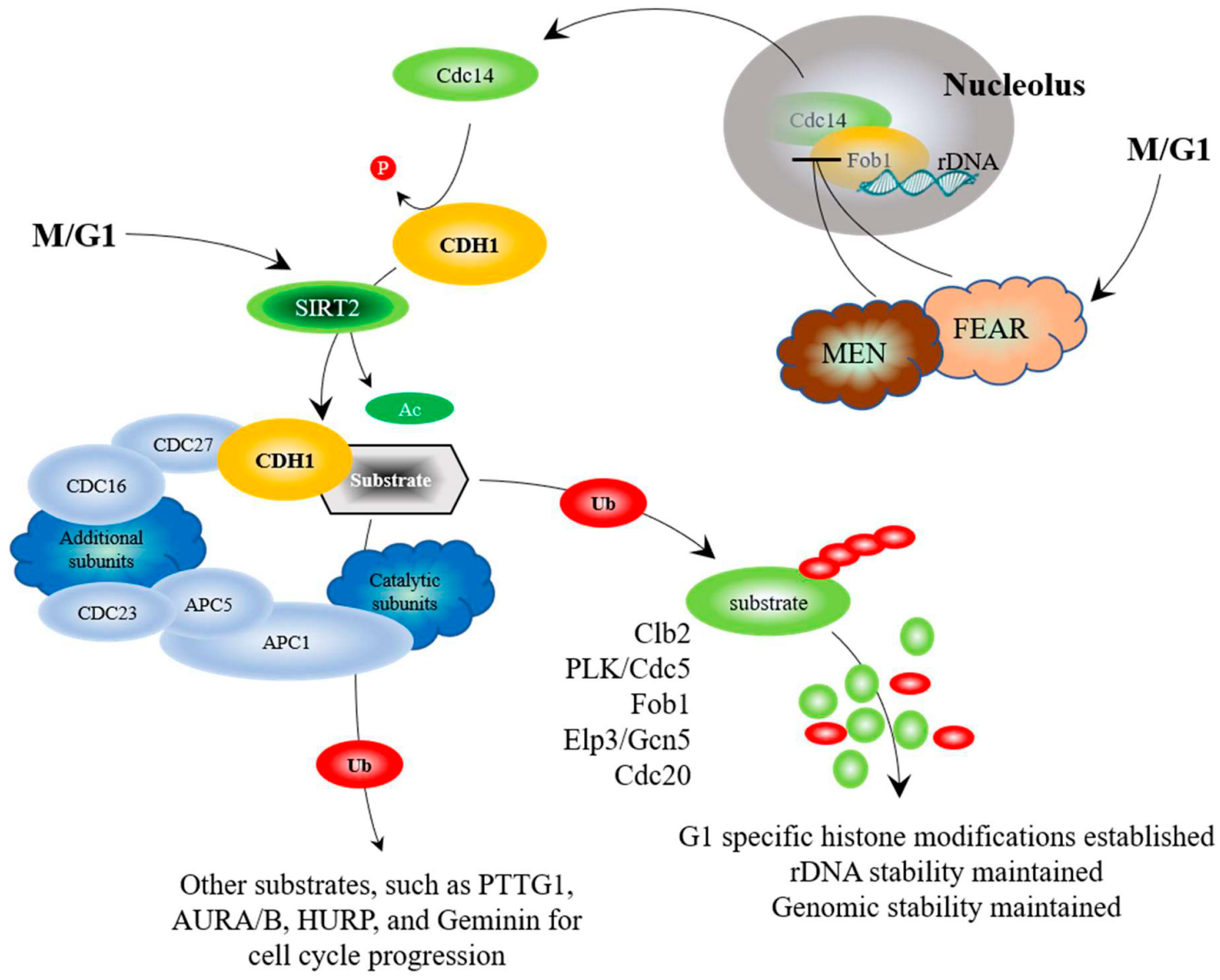
8. Maintaining Genomic Stability via APC-Mediated Histone Modifications
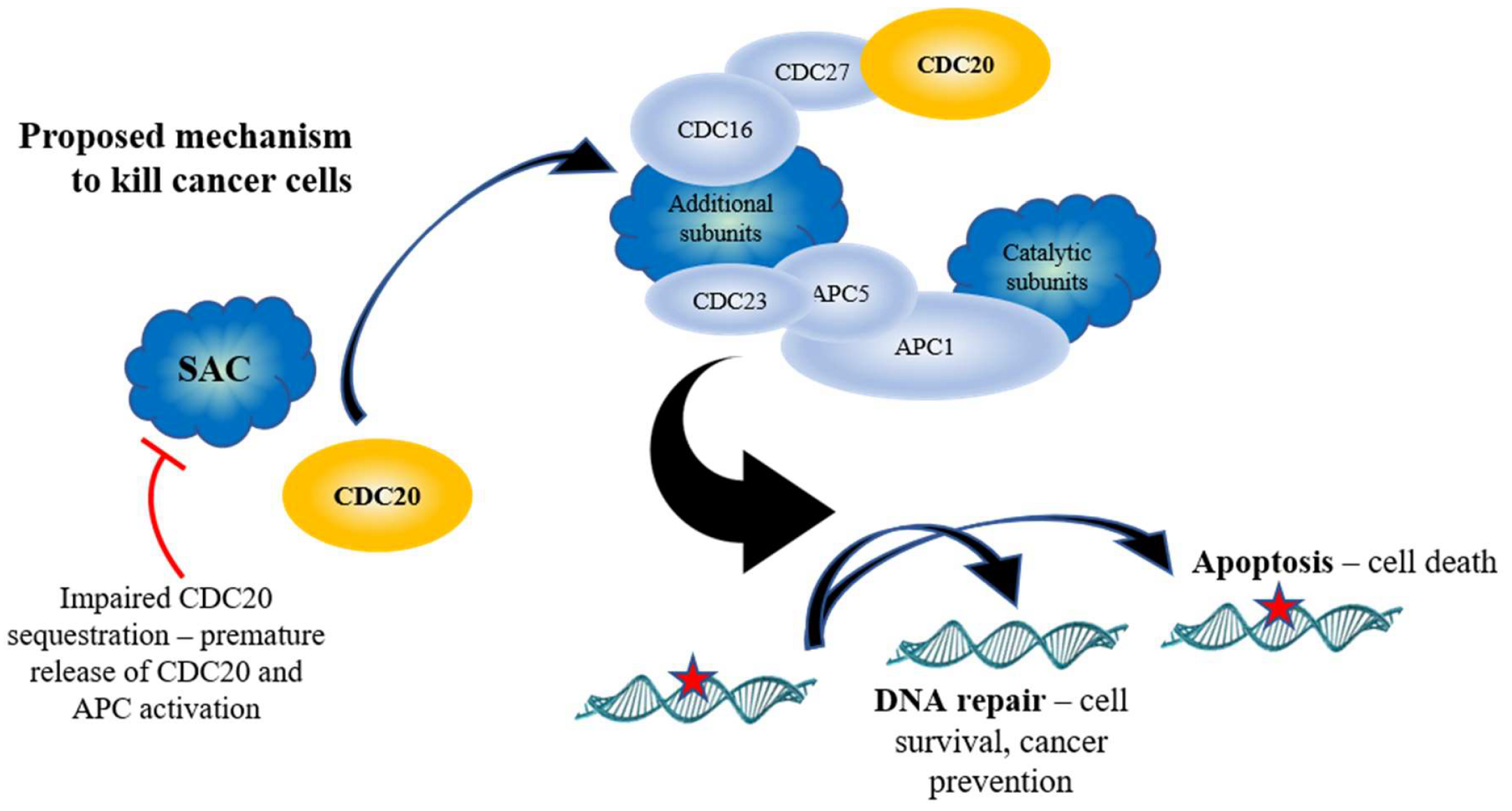
9. Targeting APC Inhibition for Anticancer Therapy
10. Targeting APC Activation for Anticancer Therapy
11. APC Activation Reduces Substrate Levels and Inhibits Cancer Cell Growth
12. APC Activity, via the Fkh/SNF Kinase/Sir2 Pathway, is Required for Prolonged Longevity
13. The APC Triggers the End of Nutrient Signaling in the Presence of Stress
14. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Sobel, H. When does human aging start? Gerontologist 1966, 6, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Lakatta, E.G. So! What’s aging? Is cardiovascular aging a disease? J. Mol. Cell. Cardiol. 2015, 83, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Paashuis-Lew, Y.R.; Heddle, J.A. Spontaneous mutation during fetal development and post-natal growth. Mutagenesis 1998, 13, 613–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Relton, C.L.; Daniel, C.P.; Hammal, D.M.; Parker, L.; Janet Tawn, E.; Burn, J. DNA repair gene polymorphisms, pre-natal factors and the frequency of somatic mutations in the glycophorin-A gene among healthy newborns. Mutat. Res. 2004, 545, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Milne, E.M. When does human ageing begin? Mech. Ageing Dev. 2006, 127, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Kubben, N.; Misteli, T. Shared molecular and cellular mechanisms of premature ageing and ageing-associated diseases. Nat. Rev. Mol. Cell Biol. 2017, 18, 595–609. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, L. When does aging begin? Res. Aging 1984, 6, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, L. The future of ageing. Nature 2000, 408, 267–269. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, L. The not-so-close relationship between biological aging and age-associated pathologies in humans. J. Gerontol. A Biol. Sci. Med. Sci. 2004, 59, B547–B550. [Google Scholar] [CrossRef] [PubMed]
- Kirkwood, T.B.; Austad, S.N. Why do we age? Nature 2000, 408, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Nemoto, S.; Finkel, T. Ageing and the mystery at Arles. Nature 2004, 429, 149–152. [Google Scholar] [CrossRef] [PubMed]
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, L. Entropy explains aging, genetic determination explains longevity, and undefined terminology explains misunderstanding both. PLoS Genet. 2007, 3, e220. [Google Scholar] [CrossRef] [PubMed]
- Labbadia, J.; Morimoto, R.I. Proteostasis and longevity: When does aging really begin? F1000Prime Rep. 2014, 6, 7. [Google Scholar] [CrossRef] [PubMed]
- Kirkwood, T.B.; Holliday, R. The evolution of ageing and longevity. Proc. R. Soc. Lond. B Biol. Sci. 1979, 205, 531–546. [Google Scholar] [CrossRef] [PubMed]
- Kenyon, C.J. The genetics of ageing. Nature 2010, 464, 504–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salminen, A.; Kaarniranta, K. ER stress and hormetic regulation of the aging process. Ageing Res. Rev. 2010, 9, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Martins, I.; Galluzzi, L.; Kroemer, G. Hormesis, cell death and aging. Aging 2011, 3, 821–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, G.C. Pleiotrophy, natural selection and the evolution of senescence. Evolution 1957, 22, 406–421. [Google Scholar]
- Kirkwood, T.B.; Rose, M.R. Evolution of senescence: Late survival sacrificed for reproduction. Philos. Trans. R Soc. Lond. B Biol. Sci. 1991, 332, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Nesse, R.M.; Williams, G.C. Evolution and the origins of disease. Sci. Am. 1998, 279, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Wick, G.; Berger, P.; Jansen-Dürr, P.; Grubeck-Loebenstein, B.A. Darwinian-evolutionary concept of age-related diseases. Exp. Gerontol. 2003, 38, 13–25. [Google Scholar] [CrossRef]
- Artandi, S.E.; DePinho, R.A. A critical role for telomeres in suppressing and facilitating carcinogenesis. Curr. Opin. Genet. Dev. 2000, 10, 39–46. [Google Scholar] [CrossRef]
- Shay, J.W.; Wright, W.E. Ageing and cancer: The telomere and telomerase connection. Novartis Found. Symp. 2001, 235, 116–125. [Google Scholar] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Aunan, J.R.; Watson, M.M.; Hagland, H.R.; Søreide, K. Molecular and biological hallmarks of ageing. Br. J. Surg. 2016, 103, e29–e46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaeberlein, M.; McVey, M.; Guarente, L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 1999, 13, 2570–2580. [Google Scholar] [CrossRef] [PubMed]
- Fabrizio, P.; Pozza, F.; Pletcher, S.D.; Gendron, C.M.; Longo, V.D. Regulation of longevity and stress resistance by Sch9 in yeast. Science 2001, 292, 288–290. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.; Fabrizio, P.; Hu, J.; Ge, H.; Cheng, C.; Li, L.; Longo, V.D. Life span extension by calorie restriction depends on Rim15 and transcription factors downstream of Ras/PKA, Tor, and Sch9. PLoS Genet. 2008, 4, e13. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.Y.; Lin, Y.Y.; Sheu, J.C.; Wu, J.T.; Lee, F.J.; Chen, Y.; Lin, M.I.; Chiang, F.T.; Tai, T.Y.; Berger, S.L.; et al. Acetylation of yeast AMPK controls intrinsic aging independently of caloric restriction. Cell 2011, 146, 969–979. [Google Scholar] [CrossRef] [PubMed]
- Postnikoff, S.D.; Malo, M.M.; Wong, B.; Harkness, T.A. The yeast forkhead transcription factors fkh1 and fkh2 regulate lifespan and stress response together with the anaphase-promoting complex. PLoS Genet. 2012, 8, e1002583. [Google Scholar] [CrossRef] [PubMed]
- Jiao, R.; Postnikoff, S.; Harkness, T.A.; Arnason, T.G. The SNF1 Kinase Ubiquitin-associated Domain Restrains Its Activation, Activity, and the Yeast Life Span. J. Biol. Chem. 2015, 290, 15393–15404. [Google Scholar] [CrossRef] [PubMed]
- Mirisola, M.G.; Taormina, G.; Fabrizio, P.; Wei, M.; Hu, J.; Longo, V.D. Serine- and threonine/valine-dependent activation of PDK and Tor orthologs converge on Sch9 to promote aging. PLoS Genet. 2014, 10, e1004113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, X.; Milholland, B.; Vijg, J. Evidence for a limit to human lifespan. Nature 2016, 538, 257–259. [Google Scholar] [CrossRef] [PubMed]
- Robine, J.M.; Allard, M. The oldest human. Science 1998, 279, 1834–1835. [Google Scholar] [CrossRef] [PubMed]
- Weon, B.M.; Je, J.H. Theoretical estimation of maximum human lifespan. Biogerontology 2009, 10, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Modig, K.; Andersson, T.; Vaupel, J.; Rau, R.; Ahlbom, A. How long do centenarians survive? Life expectancy and maximum lifespan. J. Intern. Med. 2017, 282, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Gerontology Research Group. Available online: http://supercentenarian-research-foundation.org/TableE.aspx (accessed on 30 April 2018).
- De Grey, A. A strategy for postponing aging indefinitely. Stud. Health Technol. Inform. 2005, 118, 209–219. [Google Scholar] [PubMed]
- Lenart, A.; Vaupel, J.W. Questionable evidence for a limit to human lifespan. Nature 2017, 546, E13–E14. [Google Scholar] [CrossRef] [PubMed]
- Hughes, B.G.; Hekimi, S. Many possible maximum lifespan trajectories. Nature 2017, 546, E8–E9. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Kuro-o, M.; Ishikawa, F. Aging mechanisms. Proc. Natl. Acad. Sci. USA 2000, 97, 12407–12408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucke, J.C.; Hall, W. Who wants to live forever? EMBO Rep. 2005, 6, 98–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tacutu, R.; Thornton, D.; Johnson, E.; Budovsky, A.; Barardo, D.; Craig, T.; Diana, E.; Lehmann, G.; Toren, D.; Wang, J.; et al. Human Ageing Genomic Resources: New and updated databases. Nucleic Acids Res. 2018, 46, D1083–D1090. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, T. Instability and decay of the primary structure of DNA. Nature 1993, 362, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Hoeijmakers, J.H. DNA damage, aging, and cancer. N. Engl. J. Med. 2009, 361, 1475–1485. [Google Scholar] [CrossRef] [PubMed]
- Cervelli, T.; Borghini, A.; Galli, A.; Andreassi, M.G. DNA damage and repair in atherosclerosis: Current insights and future perspectives. Int. J. Mol. Sci. 2012, 13, 16929–16944. [Google Scholar] [CrossRef] [PubMed]
- Vijg, J.; Suh, Y. Genome instability and aging. Annu. Rev. Physiol. 2013, 75, 645–668. [Google Scholar] [CrossRef] [PubMed]
- Grindel, A.; Brath, H.; Nersesyan, A.; Knasmueller, S.; Wagner, K.H. Association of genomic instability with HbA1c levels and medication in diabetic patients. Sci. Rep. 2017, 7, 41985. [Google Scholar] [CrossRef] [PubMed]
- Yuza, K.; Nagahashi, M.; Watanabe, S.; Takabe, K.; Wakai, T. Hypermutation and microsatellite instability in gastrointestinal cancers. Oncotarget 2017, 8, 112103–112115. [Google Scholar] [CrossRef] [PubMed]
- Barzilai, A.; Schumacher, B.; Shiloh, Y. Genome instability: Linking ageing and brain degeneration. Mech. Ageing Dev. 2017, 161, 4–18. [Google Scholar] [CrossRef] [PubMed]
- Henderson, K.A.; Gottschling, D.E. A mother’s sacrifice: what is she keeping for herself? Curr. Opin. Cell Biol. 2008, 20, 723–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottschling, D.E.; Nyström, T. The Upsides and Downsides of Organelle Interconnectivity. Cell 2017, 169, 24–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.C.; Sun, X.F.; Dyce, P.W.; Shen, W.; Chen, H. The role of germ cell loss during primordial follicle assembly: A review of current advances. Int. J. Biol. Sci. 2017, 13, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Sivakumar, S.; Gorbsky, G.J. Spatiotemporal regulation of the anaphase-promoting complex in mitosis. Nat. Rev. Mol. Cell Biol. 2015, 16, 82–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Chang, L.; Alfieri, C.; Zhang, Z.; Yang, J.; Maslen, S.; Skehel, M.; Barford, D. Molecular mechanism of APC/C activation by mitotic phosphorylation. Nature 2016, 533, 260–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.; He, M.; Shah, A.A.; Wan, Y. Insights into APC/C: From cellular function to diseases and therapeutics. Cell Div. 2016, 11, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukushima, H.; Ogura, K.; Wan, L.; Lu, Y.; Li, V.; Gao, D.; Liu, P.; Lau, A.W.; Wu, T.; Kirschner, M.W.; et al. SCF-mediated Cdh1 degradation defines a negative feedback system that coordinates cell-cycle progression. Cell Rep. 2013, 4, 803–816. [Google Scholar] [CrossRef] [PubMed]
- Simpson-Lavy, K.J.; Sajman, J.; Zenvirth, D.; Brandeis, M. APC/CCdh1 specific degradation of Hsl1 and Clb2 is required for proper stress responses of S. cerevisiae. Cell Cycle 2009, 8, 3003–3009. [Google Scholar] [CrossRef] [PubMed]
- Harkness, T.A.; Davies, G.F.; Ramaswamy, V.; Arnason, T.G. The ubiquitin-dependent targeting pathway in Saccharomyces cerevisiae plays a critical role in multiple chromatin assembly regulatory steps. Genetics 2002, 162, 615–632. [Google Scholar] [PubMed]
- Harkness, T.A.; Shea, K.A.; Legrand, C.; Brahmania, M.; Davies, G.F. A functional analysis reveals dependence on the Anaphase Promoting Complex for prolonged life span in yeast. Genetics 2004, 168, 759–774. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, D.L.; Bonham-Smith, P.C.; Postnikoff, S.; Gray, G.R.; Harkness, T.A. A role for the anaphase promoting complex in hormone regulation. Planta 2011, 233, 1223–1235. [Google Scholar] [CrossRef] [PubMed]
- Postnikoff, S.D.; Harkness, T.A. Mechanistic insights into aging, cell-cycle progression, and stress response. Front. Physiol. 2012, 3, 183. [Google Scholar] [CrossRef] [PubMed]
- Menzel, J.; Malo, M.E.; Chan, C.; Prusinkiewicz, M.; Arnason, T.G.; Harkness, T.A. The anaphase promoting complex regulates yeast lifespan and rDNA stability by targeting Fob1 for degradation. Genetics 2014, 196, 693–709. [Google Scholar] [CrossRef] [PubMed]
- Malo, M.E.; Postnikoff, S.D.; Arnason, T.G.; Harkness, T.A. Mitotic degradation of yeast Fkh1 by the Anaphase Promoting Complex is required for normal longevity, genomic stability and stress resistance. Aging 2016, 8, 810–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganem, N.J.; Pellman, D. Linking abnormal mitosis to the acquisition of DNA damage. J. Cell Biol. 2012, 199, 871–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harkness, T.A.; Arnason, T.G.; Legrand, C.; Pisclevich, M.G.; Davies, G.F.; Turner, E.L. Contribution of CAF-I to anaphase-promoting-complex-mediated mitotic chromatin assembly in Saccharomyces cerevisiae. Eukaryot Cell 2005, 4, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Arnason, T.G.; Pisclevich, M.G.; Dash, M.D.; Davies, G.F.; Harkness, T.A. Novel interaction between Apc5p and Rsp5p in an intracellular signaling pathway in Saccharomyces cerevisiae. Eukaryot Cell 2005, 4, 134–146. [Google Scholar] [CrossRef] [PubMed]
- Turner, E.L.; Malo, M.E.; Pisclevich, M.G.; Dash, M.D.; Davies, G.F.; Arnason, T.G.; Harkness, T.A. The Saccharomyces cerevisiae Anaphase Promoting Complex interacts with multiple histone-modifying enzymes to regulate cell cycle progression. Eukaryot Cell 2010, 9, 1418–1431. [Google Scholar] [CrossRef] [PubMed]
- Islam, A.; Turner, E.L.; Menzel, J.; Malo, M.E.; Harkness, T.A. Antagonistic Gcn5-Hda1 interactions revealed by mutations to the Anaphase Promoting Complex in yeast. Cell Div. 2011, 6, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linger, J.G.; Tyler, J.K. Chromatin Disassembly and Reassembly during DNA Repair. Mutat. Res. 2007, 618, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Tyler, J.K. Nucleosome disassembly during human non-homologous end joining followed by concerted HIRA- and CAF-1-dependent reassembly. eLife 2016, 5, e15129. [Google Scholar] [CrossRef] [PubMed]
- Mello, J.A.; Silljé, H.H.W.; Roche, D.M.J.; Kirschner, D.B.; Nigg, E.A.; Almouzni, G. Human Asf1 and CAF-1 interact and synergize in a repair-coupled nucleosome assembly pathway. EMBO Rep. 2002, 3, 329–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.C.; Tyler, J. Chromatin reassembly signals the end of DNA repair. Cell Cycle 2008, 7, 3792–3797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.A.; Haber, J.E. Chromatin assembly factors Asf1 and CAF-1 have overlapping roles in deactivating the DNA damage checkpoint when DNA repair is complete. Proc. Natl. Acad. Sci. USA 2009, 106, 1151–1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriges Blanko, E.; Kadyrova, L.Y.; Kadyrov, F.A. DNA Mismatch Repair Interacts with CAF-1- and ASF1A-H3-H4-dependent Histone (H3-H4)2 Tetramer Deposition. J. Biol. Chem. 2016, 291, 9203–9217. [Google Scholar] [CrossRef] [PubMed]
- Izawa, D.; Pines, J. The mitotic checkpoint complex binds a second CDC20 to inhibit active APC/C. Nature 2015, 517, 631–634. [Google Scholar] [CrossRef] [PubMed]
- Alfieri, C.; Chang, L.; Zhang, Z.; Yang, J.; Maslen, S.; Skehel, M.; Barford, D. Molecular basis of APC/C regulation by the spindle assembly checkpoint. Nature 2016, 536, 431–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, G.; Spellman, P.T.; Volpe, T.; Brown, P.O.; Botstein, D.; Davis, T.N.; Futcher, B. Two yeast forkhead genes regulate the cell cycle and pseudohyphal growth. Nature 2000, 406, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Rudner, A.D.; Murray, A.W. Phosphorylation by Cdc28 activates the Cdc20-dependent activity of the anaphase-promoting complex. J. Cell Biol. 2000, 149, 1377–1390. [Google Scholar] [CrossRef] [PubMed]
- Kotani, S.; Tugendreich, S.; Fujii, M.; Jorgensen, P.M.; Watanabe, N.; Hoog, C.; Hieter, P.; Todokoro, K. PKA and MPF-activated polo-like kinase regulate anaphase-promoting complex activity and mitosis progression. Mol. Cell 1998, 1, 371–380. [Google Scholar] [CrossRef]
- Crasta, K.; Lim, H.H.; Giddings, T.H., Jr.; Winey, M.; Surana, U. Inactivation of Cdh1 by synergistic action of Cdk1 and polo kinase is necessary for proper assembly of the mitotic spindle. Nat. Cell Biol. 2008, 10, 665–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.S.; Vassilopoulos, A.; Wang, R.H.; Lahusen, T.; Xiao, Z. SIRT2 maintains genome integrity and suppresses tumorigenesis through regulating APC/C activity. Cancer Cell 2011, 20, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wan, L.; Dai, X.; Sun, Y.; Wei, W. Functional characterization of Anaphase Promoting Complex/Cyclosome (APC/C) E3 ubiquitin ligases in tumorigenesis. Biochim. Biophys. Acta 2014, 1845, 277–293. [Google Scholar] [PubMed] [Green Version]
- Peters, J.M. The anaphase promoting complex/cyclosome: A machine designed to destroy. Nat. Rev. Mol. Cell Biol. 2006, 7, 644–656. [Google Scholar] [CrossRef] [PubMed]
- Sackton, K.L.; Dimova, N.; Zeng, X.; Tian, W.; Zhang, M.; Sackton, T.B.; Meaders, J.; Pfaff, K.L.; Sigoillot, F.; Yu, H.; et al. Synergistic blockade of mitotic exit by two chemical inhibitors of the APC/C. Nature 2014, 514, 646–649. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, J.; Wan, L.; Zhou, X.; Wang, Z.; Wei, W. Targeting Cdc20 as a novel cancer therapeutic strategy. Pharmacol. Ther. 2015, 151, 141–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sansregret, L.; Patterson, J.O.; Dewhurst, S.; López-García, C.; Koch, A.; McGranahan, N.; Chao, W.C.H.; Barry, D.J.; Rowan, A.; Instrell, R.; et al. APC/C Dysfunction Limits Excessive Cancer Chromosomal Instability. Cancer Discov. 2017, 7, 218–233. [Google Scholar] [CrossRef] [PubMed]
- Thu, K.L.; Silvester, J.; Elliott, M.J.; Ba-Alawi, W.; Duncan, M.H.; Elia, A.C.; Mer, A.S.; Smirnov, P.; Safikhani, Z.; Haibe-Kains, B.; et al. Disruption of the anaphase-promoting complex confers resistance to TTK inhibitors in triple-negative breast cancer. Proc. Natl. Acad. Sci. USA 2018, 115, E1570–E1577. [Google Scholar] [CrossRef] [PubMed]
- Kastl, J.; Braun, J.; Prestel, A.; Möller, H.M.; Huhn, T.; Mayer, T.U. Mad2 Inhibitor-1 (M2I-1): A Small Molecule Protein-Protein Interaction Inhibitor Targeting the Mitotic Spindle Assembly Checkpoint. ACS Chem. Biol. 2015, 10, 1661–1666. [Google Scholar] [CrossRef] [PubMed]
- Stegmeier, F.; Huang, J.; Rahal, R.; Zmolik, J.; Moazed, D.; Amon, A. The replication fork block protein Fob1 functions as a negative regulator of the FEAR network. Curr. Biol. 2004, 14, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Waples, W.G.; Chahwan, C.; Ciechonska, M.; Lavoie, B.D. Putting the brake on FEAR: Tof2 promotes the biphasic release of Cdc14 phosphatase during mitotic exit. Mol. Biol. Cell 2009, 20, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Visintin, R.; Craig, K.; Hwang, E.S.; Prinz, S.; Tyers, M.; Amon, A. The phosphatase Cdc14 triggers mitotic exit by reversal of Cdk-dependent phosphorylation. Mol. Cell 1998, 2, 709–718. [Google Scholar] [CrossRef]
- Karra, H.; Repo, H.; Ahonen, I.; Löyttyniemi, E.; Pitkänen, R.; Lintunen, M.; Kuopio, T.; Söderström, M.; Kronqvist, P. Cdc20 and securin overexpression predict short-term breast cancer survival. Br. J. Cancer 2014, 110, 2905–2913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmit, T.L.; Ledesma, M.C.; Ahmad, N. Modulating polo-like kinase 1 as a means for cancer chemoprevention. Pharm. Res. 2010, 27, 989–998. [Google Scholar] [CrossRef] [PubMed]
- Staff, S.; Isola, J.; Jumppanen, M.; Tanner, M. Aurora-A gene is frequently amplified in basal-like breast cancer. Oncol. Rep. 2010, 23, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Heredia, F.F.; de Sousa, J.C.; Ribeiro Junior, H.L.; Carvalho, A.F.; Magalhaes, S.M.; Pinheiro, R.F. Proteins related to the spindle and checkpoint mitotic emphasize the different pathogenesis of hypoplastic MDS. Leuk. Res. 2014, 38, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Kuo, T.C.; Lu, H.P.; Chao, C.C. The tyrosine kinase inhibitor sorafenib sensitizes hepatocellular carcinoma cells to taxol by suppressing the HURP protein. Biochem. Pharmacol. 2011, 82, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Xiang, W.; Wu, X.; Huang, C.; Wang, M.; Zhao, X.; Luo, G.; Li, Y.; Jiang, G.; Xiao, X.; Zeng, F. PTTG1 regulated by miR-146a-3p promotes bladder cancer migration, invasion, metastasis and growth. Oncotarget 2017, 8, 664–678. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Cai, M.; Gong, Z.; Zhang, B.; Li, Y.; Guan, L.; Hou, X.; Li, Q.; Liu, G.; Xue, Z.; et al. Geminin facilitates FoxO3 deacetylation to promote breast cancer cell metastasis. J. Clin. Investig. 2017, 127, 2159–2175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irniger, S.; Bäumer, M.; Braus, G.H. Glucose and ras activity influence the ubiquitin ligases APC/C and SCF in Saccharomyces cerevisiae. Genetics 2000, 154, 1509–1521. [Google Scholar] [PubMed]
- Bolte, M.; Dieckhoff, P.; Krause, C.; Braus, G.H.; Irniger, S. Synergistic inhibition of APC/C by glucose and activated Ras proteins can be mediated by each of the Tpk1-3 proteins in Saccharomyces cerevisiae. Microbiology 2003, 149, 1205–1216. [Google Scholar] [CrossRef] [PubMed]
- Searle, J.S.; Schollaert, K.L.; Wilkins, B.J.; Sanchez, Y. The DNA damage checkpoint and PKA pathways converge on APC substrates and Cdc20 to regulate mitotic progression. Nat. Cell Biol. 2004, 6, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Cocklin, R.; Goebl, M. Nutrient sensing kinases PKA and Sch9 phosphorylate the catalytic domain of the ubiquitin-conjugating enzyme Cdc34. PLoS ONE 2011, 6, e27099. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, R.; Bonacci, T.; Arceci, A.; Lahiri, D.; Mills, C.A.; Kernan, J.L.; Branigan, T.B.; DeCaprio, J.A.; Burke, D.J.; Emanuele, M.J. APC/C and SCF(cyclin F) constitute a reciprocal feedback circuit controlling S-phase entry. Cell Rep. 2016, 16, 3359–3372. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Nguyen, M.; Qin, F.X.; Tong, Q. SIRT2 deacetylates FOXO3a in response to oxidative stress and caloric restriction. Aging Cell 2007, 6, 505–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linke, C.; Klipp, E.; Lehrach, H.; Barberis, M.; Krobitsch, S. Fkh1 and Fkh2 associate with Sir2 to control CLB2 transcription under normal and oxidative stress conditions. Front. Physiol. 2013, 4, 173. [Google Scholar] [CrossRef] [PubMed]
- Jiao, R.; Lobanova, L.; Waldner, A.; Fu, A.; Xiao, L.; Harkness, T.A.; Arnason, T.G. The ubiquitin-conjugating enzyme, Ubc1, indirectly regulates SNF1 kinase activity via Forkhead-dependent transcription. Microb. Cell 2016, 3, 540–553. [Google Scholar] [CrossRef] [PubMed]
- Chiacchiera, F.; Simone, C. The AMPK-FoxO3A axis as a target for cancer treatment. Cell Cycle 2010, 9, 1091–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salminen, A.; Kaarniranta, K. AMP-activated protein kinase (AMPK) controls the aging process via an integrated signaling network. Ageing Res. Rev. 2012, 11, 230–241. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Ayad, N.G.; Wan, Y.; Zhang, G.J.; Kirschner, M.W.; Kaelin, W.G., Jr. Degradation of the SCF component Skp2 in cell-cycle phase G1 by the anaphase-promoting complex. Nature 2004, 428, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Bashir, T.; Dorrello, N.V.; Amador, V.; Guardavaccaro, D.; Pagano, M. Control of the SCF(Skp2-Cks1) ubiquitin ligase by the APC/C(Cdh1) ubiquitin ligase. Nature 2004, 428, 190–193. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Higgins, J.M. Histone modifications and mitosis: countermarks, landmarks, and bookmarks. Trends Cell Biol. 2013, 23, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Guppy, B.J.; McManus, K.J. Mitotic accumulation of dimethylated lysine 79 of histone H3 is important for maintaining genome integrity during mitosis in human cells. Genetics 2015, 199, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Recht, J.; Tsubota, T.; Tanny, J.C.; Diaz, R.L.; Berger, J.M.; Zhang, X.; Garcia, B.A.; Shabanowitz, J.; Burlingame, A.L.; Hunt, D.F.; et al. Histone chaperone Asf1 is required for histone H3 lysine 56 acetylation, a modification associated with S phase in mitosis and meiosis. Proc. Natl. Acad. Sci. USA 2006, 103, 6988–6993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, Y.S.; Cho, S.; Park, J.S.; Ko, Y.; Kang, Y.K. Phosphorylation of serine-10 of histone H3 shields modified lysine-9 selectively during mitosis. Genes Cells 2010, 15, 181–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karmodiya, K.; Krebs, A.R.; Oulad-Abdelghani, M.; Kimura, H.; Tora, L.H. 3K9 and H3K14 acetylation co-occur at many gene regulatory elements, while H3K14ac marks a subset of inactive inducible promoters in mouse embryonic stem cells. BMC Genom. 2012, 13, 424. [Google Scholar] [CrossRef] [PubMed]
- Gates, L.A.; Shi, J.; Rohira, A.D.; Feng, Q.; Zhu, B.; Bedford, M.T.; Sagum, C.A.; Jung, S.Y.; Qin, J.; Tsai, M.J.; et al. Acetylation on histone H3 lysine 9 mediates a switch from transcription initiation to elongation. J. Biol. Chem. 2017, 292, 14456–14472. [Google Scholar] [CrossRef] [PubMed]
- Downs, J.A. Histone H3 K56 acetylation, chromatin assembly, and the DNA damage checkpoint. DNA Repair 2008, 7, 2020–2024. [Google Scholar] [CrossRef] [PubMed]
- Farooq, Z.; Banday, S.; Pandita, T.K.; Altaf, M. The many faces of histone H3K79 methylation. Mutat. Res. Rev. Mutat. Res. 2016, 768, 46–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, K.; Tellier, M.; Murphy, S. DOT1L and H3K79 Methylation in Transcription and Genomic Stability. Biomolecules 2018, 8, E11. [Google Scholar] [CrossRef] [PubMed]
- Cea, M.; Cagnetta, A.; Adamia, S.; Acharya, C.; Tai, Y.T.; Fulciniti, M.; Ohguchi, H.; Munshi, A.; Acharya, P.; Bhasin, M.K.; et al. Evidence for a role of the histone deacetylase SIRT6 in DNA damage response of multiple myeloma cells. Blood 2016, 127, 1138–1150. [Google Scholar] [CrossRef] [PubMed]
- Ercilla, A.; Llopis, A.; Feu, S.; Aranda, S.; Ernfors, P. New origin firing is inhibited by APC/CCdh1 activation in S-phase after severe replication stress. Nucleic Acids Res. 2016, 44, 4745–4762. [Google Scholar] [CrossRef] [PubMed]
- Garzón, J.; Rodríguez, R.; Kong, Z.; Chabes, A.; Rodríguez-Acebes, S.; Méndez, J.; Moreno, S.; García-Higuera, I. Shortage of dNTPs underlies altered replication dynamics and DNA breakage in the absence of the APC/C cofactor Cdh1. Oncogene 2017, 36, 5808–5818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wäsch, R.; Engelbert, D. Anaphase-promoting complex-dependent proteolysis of cell cycle regulators and genomic instability of cancer cells. Oncogene 2005, 24, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardozo, T.; Pagano, M. Wrenches in the works: Drug discovery targeting the SCF ubiquitin ligase and APC/C complexes. BMC Biochem. 2007, 8 (Suppl. 1), S9. [Google Scholar] [CrossRef] [PubMed]
- Bolanos-Garcia, V.M. Assessment of the mitotic spindle assembly checkpoint (SAC) as the target of anticancer therapies. Curr. Cancer Drug Targets 2009, 9, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Momiyama, N.; Ueda, M.; Matsuyama, R.; Mori, R.; Fujii, Y.; Ichikawa, Y.; Endo, I.; Togo, S.; Shimada, H. Targeting of CDC20 via Small Interfering RNA Causes Enhancement of the Cytotoxicity of Chemoradiation. Anticancer Res. 2008, 28, 1559–1563. [Google Scholar] [PubMed]
- Chang, D.Z.; Ma, Y.; Ji, B.; Liu, Y.; Hwu, P.; Abbruzzese, J.L.; Logsdon, C.; Wang, H. Increased CDC20 expression is associated with pancreatic ductal adenocarcinoma differentiation and progression. J. Hematol. Oncol. 2012, 5, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lub, S.; Maes, A.; Maes, K.; De Veirman, K.; De Bruyne, E.; Menu, E.; Fostier, K.; Kassambara, A.; Moreaux, J.; Hose, D.; et al. Inhibiting the anaphase promoting complex/cyclosome induces a metaphase arrest and cell death in multiple myeloma cells. Oncotarget 2016, 7, 4062–4076. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, E.M.; Haas, W.; Gygi, M.; Gygi, S.P.; Kellogg, D.R. Cdc28-dependent regulation of the Cdc5/Polo kinase. Curr. Biol. 2005, 15, 2033–2037. [Google Scholar] [CrossRef] [PubMed]
- Visintin, C.; Tomson, B.N.; Rahal, R.; Paulson, J.; Cohen, M.; Taunton, J.; Amon, A.; Visintin, R. APC/C-Cdh1-mediated degradation of the Polo kinase Cdc5 promotes the return of Cdc14 into the nucleolus. Genes Dev. 2008, 22, 79–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pramila, T.; Wu, W.; Miles, S.; Noble, W.S.; Breeden, L.L. The Forkhead transcription factor Hcm1 regulates chromosome segregation genes and fills the S-phase gap in the transcriptional circuitry of the cell cycle. Genes Dev. 2006, 20, 2266–2278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamb, J.R.; Michaud, W.A.; Sikorski, R.S.; Hieter, P.A. Cdc16p, Cdc23p and Cdc27p form a complex essential for mitosis. EMBO J. 1994, 13, 4321–4328. [Google Scholar] [PubMed]
- Irniger, S.; Nasmyth, K. The anaphase-promoting complex is required in G1 arrested yeast cells to inhibit B-type cyclin accumulation and to prevent uncontrolled entry into S-phase. J. Cell Sci. 1997, 110, 1523–1531. [Google Scholar] [PubMed]
- Li, M.; York, J.P.; Zhang, P. Loss of Cdc20 causes a securin-dependent metaphase arrest in two-cell mouse embryos. Mol. Cell. Biol. 2007, 27, 3481–3488. [Google Scholar] [CrossRef] [PubMed]
- TCGA. Available online: https://portal.gdc.cancer.gov (accessed on 1 January 2018).
- Ishizawa, J.; Sugihara, E.; Kuninaka, S.; Mogushi, K.; Kojima, K.; Benton, C.B.; Zhao, R.; Chachad, D.; Hashimoto, N.; Jacamo, R.O.; et al. FZR1 loss increases sensitivity to DNA damage and consequently promotes murine and human B-cell acute leukemia. Blood 2017, 129, 1958–1968. [Google Scholar] [CrossRef] [PubMed]
- Villa-Hernández, S.; Bueno, A.; Bermejo, R. The Multiple Roles of Ubiquitylation in Regulating Challenged DNA Replication. Adv. Exp. Med. Biol. 2017, 1042, 395–419. [Google Scholar] [PubMed]
- Kitao, H.; Iimori, M.; Kataoka, Y.; Wakasa, T.; Tokunaga, E. DNA replication stress and cancer chemotherapy. Cancer Sci. 2018, 109, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Moyret-Lalle, C.; Couzon, F.; Surbiguet-Clippe, C.; Saurin, J.C.; Lorca, T.; Navarro, C.; Puisieux, A. Alterations of anaphase-promoting complex genes in human colon cancer cells. Oncogene 2003, 22, 1486–1490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, K.H.; Choi, S.E.; Eom, M.; Kang, Y. Downregulation of the anaphase-promoting complex (APC)7 in invasive ductal carcinomas of the breast and its clinicopathologic relationships. Breast Cancer Res. 2005, 7, R238–R247. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.Y.; Kwon, H.Y.; Park, K.H.; Kim, D.S. Anaphase-Promoting Complex 7 is a Prognostic Factor in Human Colorectal Cancer. Ann. Coloproctol. 2017, 33, 139–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuda, Y.; Iimori, M.; Nakashima, Y.; Nakanishi, R.; Ando, K.; Ohgaki, K.; Kitao, H.; Saeki, H.; Oki, E.; Maehara, Y. Mitotic slippage and the subsequent cell fates after inhibition of Aurora B during tubulin-binding agent-induced mitotic arrest. Sci. Rep. 2017, 7, 16762. [Google Scholar] [CrossRef] [PubMed]
- Haschka, M.; Karbon, G.; Fava, L.L.; Villunger, A. Perturbing mitosis for anti-cancer therapy: Is cell death the only answer? EMBO Rep. 2018, 19, e45440. [Google Scholar] [CrossRef] [PubMed]
- Kops, G.J.; Foltz, D.R.; Cleveland, D.W. Lethality to human cancer cells through massive chromosome loss by inhibition of the mitotic checkpoint. Proc. Natl. Acad. Sci. USA 2004, 101, 8699–8704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bharadwaj, R.; Yu, H. The spindle checkpoint, aneuploidy, and cancer. Oncogene 2004, 23, 2016–2027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.; Rivera-Rivera, Y.; Moreno, C.S.; Saavedra, H.I. The E2F activators control multiple mitotic regulators and maintain genomic integrity through Sgo1 and BubR1. Oncotarget 2017, 8, 77649–77672. [Google Scholar] [CrossRef] [PubMed]
- Hollenhorst, P.C.; Bose, M.E.; Mielke, M.R.; Müller, U.; Fox, C.A. Forkhead genes in transcriptional silencing, cell morphology and the cell cycle. Overlapping and distinct functions for FKH1 and FKH2 in Saccharomyces cerevisiae. Genetics 2000, 154, 1533–1548. [Google Scholar] [PubMed]
- Rodríguez-Colman, M.J.; Sorolla, M.A.; Vall-Llaura, N.; Tamarit, J.; Ros, J.; Cabiscol, E. The FOX transcription factor Hcm1 regulates oxidative metabolism in response to early nutrient limitation in yeast. Role of Snf1 and Tor1/Sch9 kinases. Biochim. Biophys. Acta 2013, 1833, 2004–2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigo-Brenni, M.C.; Morgan, D.O. Sequential E2s drive polyubiquitin chain assembly on APC targets. Cell 2007, 130, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Horiuchi, T. A yeast gene product, Fob1 protein, required for both replication fork blocking and recombinational hotspot activities. Genes Cells 1996, 1, 465–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, T.; Heck, D.J.; Nomura, M.; Horiuchi, T. Expansion and contraction of ribosomal DNA repeats in Saccharomyces cerevisiae: Requirement of replication fork blocking (Fob1) protein and the role of RNA polymerase I. Genes Dev. 1998, 12, 3821–3830. [Google Scholar] [CrossRef] [PubMed]
- Defossez, P.A.; Prusty, R.; Kaeberlein, M.; Lin, S.J.; Ferrigno, P.; Silver, P.A.; Keil, R.L.; Guarente, L. Elimination of replication block protein Fob1 extends the life span of yeast mother cells. Mol. Cell 1999, 3, 447–455. [Google Scholar] [CrossRef]
- Yamashita, Y.M.; Nakaseko, Y.; Samejima, I.; Kumada, K.; Yamada, H.; Michaelson, D.; Yanagida, M. 20S cyclosome complex formation and proteolytic activity inhibited by the cAMP/PKA pathway. Nature 1996, 384, 276–279. [Google Scholar] [CrossRef] [PubMed]
- Madia, F.; Gattazzo, C.; Wei, M.; Fabrizio, P.; Burhans, W.C.; Weinberger, M.; Galbani, A.; Smith, J.R.; Nguyen, C.; Huey, S.; et al. Longevity mutation in SCH9 prevents recombination errors and premature genomic instability in a Werner/Bloom model system. J. Cell Biol. 2008, 180, 67–81. [Google Scholar] [CrossRef] [PubMed]
- Qie, B.; Lyu, Z.; Lyu, L.; Liu, J.; Gao, X.; Liu, Y.; Duan, W.; Zhang, N.; Du, L.; Liu, K. Sch9 regulates intracellular protein ubiquitination by controlling stress responses. Redox Biol. 2015, 5, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Kaeberlein, M.; Powers, R.W., 3rd; Steffen, K.K.; Westman, E.A.; Hu, D.; Dang, N.; Kerr, E.O.; Kirkland, K.T.; Fields, S.; Kennedy, B.K. Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science 2005, 310, 1193–1196. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harkness, T.A.A. Activating the Anaphase Promoting Complex to Enhance Genomic Stability and Prolong Lifespan. Int. J. Mol. Sci. 2018, 19, 1888. https://doi.org/10.3390/ijms19071888
Harkness TAA. Activating the Anaphase Promoting Complex to Enhance Genomic Stability and Prolong Lifespan. International Journal of Molecular Sciences. 2018; 19(7):1888. https://doi.org/10.3390/ijms19071888
Chicago/Turabian StyleHarkness, Troy A. A. 2018. "Activating the Anaphase Promoting Complex to Enhance Genomic Stability and Prolong Lifespan" International Journal of Molecular Sciences 19, no. 7: 1888. https://doi.org/10.3390/ijms19071888
APA StyleHarkness, T. A. A. (2018). Activating the Anaphase Promoting Complex to Enhance Genomic Stability and Prolong Lifespan. International Journal of Molecular Sciences, 19(7), 1888. https://doi.org/10.3390/ijms19071888