Crosstalk between Notch, HIF-1α and GPER in Breast Cancer EMT



Abstract
: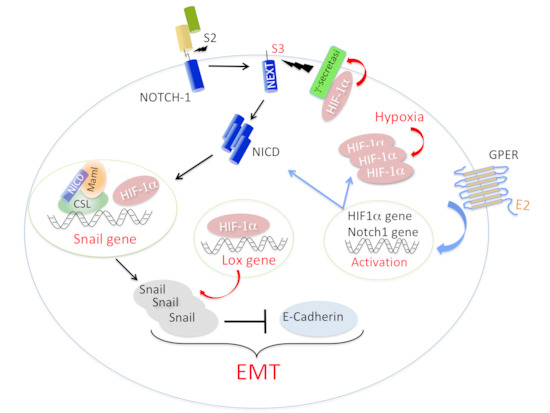
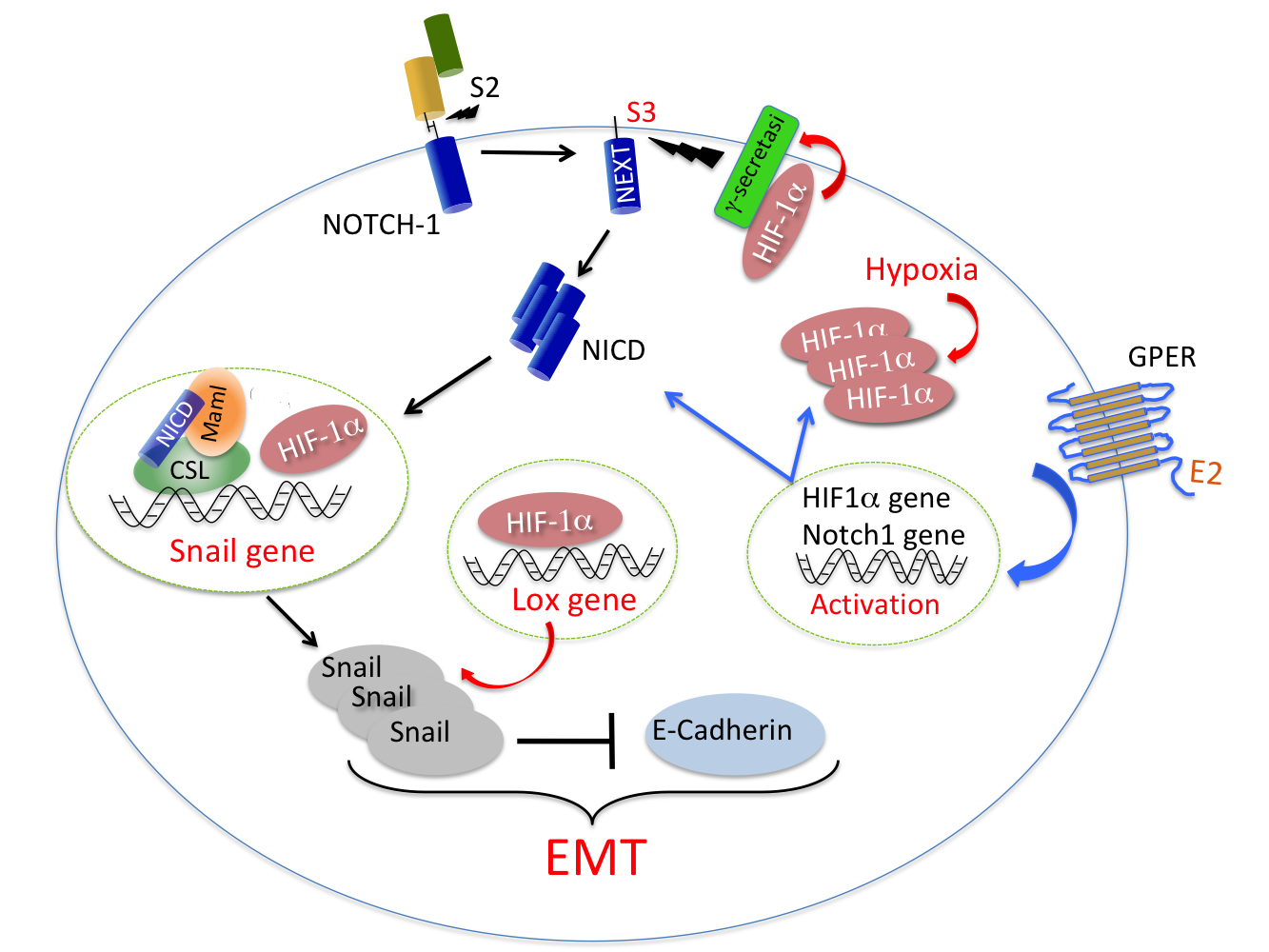{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. The Notch Core Pathway
3. Notch Endosomal Trafficking and Ligand-Independent Activation
4. NICD Transcriptional Strength and Target Selection
5. Notch Signaling in Tumor Angiogenesis and EMT
5.1. Angiogenesis
5.2. EMT
6. Interplay between Notch Signaling and 17β-Estradiol (E2) in Breast Cancer
7. Crosstalk between HIF-1α and Notch in Cancer EMT
8. Hypoxia-Inducible HIF-1α in Breast Cancer Cells and CAFs
8.1. HIF-1α in Breast Cancer Cells
8.2. HIF-1α in CAFs
8.3. Cytokines Regulating HIF-1α and NOTCH Signaling in the Tumor Microenvironment
9. Hypoxia-Independent Activation of HIF-1 in Cancer and Stromal Cells
10. HIF-1α Regulation by E2 (ER e GPER)
11. Does a Signaling Network between Notch, HIF-1α, and GPER Strengthen EMT in Breast Cancer Cells?
12. Therapeutic Approaches Targeting Notch Signaling in Breast Cancer
Author Contributions
Acknowledgments
Conflicts of interest
References
- Siebel, C.; Lendahl, U. Notch Signaling in Development, Tissue Homeostasis, and Disease. Physiol. Rev. 2017, 97, 1235–1294. [Google Scholar] [CrossRef] [PubMed]
- Ranganathan, P.; Weaver, K.L.; Capobianco, A.J. Notch signalling in solid tumours: A little bit of everything but not all the time. Nat. Rev. Cancer 2011, 11, 338–351. [Google Scholar] [CrossRef] [PubMed]
- Ntziachristos, P.; Lim, J.S.; Sage, J.; Aifantis, I. From fly wings to targeted cancer therapies: A centennial for notch signaling. Cancer Cell 2014, 25, 318–334. [Google Scholar] [CrossRef] [PubMed]
- Kopan, R.; Ilagan, M.X. The canonical Notch signaling pathway: Unfolding the activation mechanism. Cell 2009, 137, 216–233. [Google Scholar] [CrossRef] [PubMed]
- Palmer, W.H.; Deng, W.M. Ligand-Independent Mechanisms of Notch Activity. Trends Cell Biol. 2015, 25, 697–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pupo, M.; Pisano, A.; Abonante, S.; Maggiolini, M.; Musti, A.M. GPER activates Notch signaling in breast cancer cells and cancer-associated fibroblasts (CAFs). Int. J. Biochem. Cell Biol. 2014, 46, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Landor, S.K.; Lendahl, U. The interplay between the cellular hypoxic response and Notch signaling. Exp. Cell Res. 2017, 356, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Bray, S.J. Notch signalling in context. Nat. Rev. Mol. Cell Biol. 2016, 17, 722–735. [Google Scholar] [CrossRef] [PubMed]
- Nam, Y.; Weng, A.P.; Aster, J.C.; Blacklow, S.C. Structural requirements for assembly of the CSL.intracellular Notch1.Mastermind-like 1 transcriptional activation complex. J. Biol. Chem. 2003, 278, 21232–21239. [Google Scholar] [CrossRef] [PubMed]
- Bray, S.J.; Gomez-Lamarca, M. Notch after cleavage. Curr. Opin. Cell Biol. 2018, 51, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Giaimo, B.D.; Oswald, F.; Borggrefe, T. Dynamic chromatin regulation at Notch target genes. Transcription 2017, 8, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Lamarca, M.J.; Falo-Sanjuan, J.; Stojnic, R.; Abdul Rehman, S.; Muresan, L.; Jones, M.L.; Pillidge, Z.; Cerda-Moya, G.; Yuan, Z.; Baloul, S.; et al. Activation of the Notch Signaling Pathway In Vivo Elicits Changes in CSL. Nucl. Dyn. Dev. Cell 2018, 44, 611–623.e7. [Google Scholar] [CrossRef] [PubMed]
- Vaccari, T.; Lu, H.; Kanwar, R.; Fortini, M.E.; Bilder, D. Endosomal entry regulates Notch receptor activation in Drosophila melanogaster. J. Cell Biol. 2008, 180, 755–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baron, M. Endocytic routes to Notch activation. Semin. Cell Dev. Biol. 2012, 23, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Gupta-Rossi, N.; Six, E.; LeBail, O.; Logeat, F.; Chastagner, P.; Olry, A.; Israël, A.; Brou, C. Monoubiquitination and endocytosis direct gamma-secretase cleavage of activated Notch receptor. J. Cell Biol. 2004, 166, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Andersson, E.R.; Sandberg, R.; Lendahl, U. Notch signaling: Simplicity in design, versatility in function. Development 2011, 138, 3593–3612. [Google Scholar] [CrossRef] [PubMed]
- Moretti, J.; Chastagner, P.; Gastaldello, S.; Heuss, S.F.; Dirac, A.M.; Bernards, R.; Masucci, M.G.; Israël, A.; Brou, C. The translation initiation factor 3f (eIF3f) exhibits a deubiquitinase activity regulating Notch activation. PLoS Biol. 2010, 8, e1000545. [Google Scholar] [CrossRef] [PubMed]
- Gulino, A.; Di Marcotullio, L.; Screpanti, I. The multiple functions of Numb. Exp. Cell Res. 2010, 316, 900–906. [Google Scholar] [CrossRef] [PubMed]
- Yan, B. Numb—From flies to humans. Brain Dev. 2010, 32, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Chastagner, P.; Israël, A.; Brou, C. AIP4/Itch regulates Notch receptor degradation in the absence of ligand. PLoS ONE 2008, 16, e2735. [Google Scholar] [CrossRef] [PubMed]
- Wilkin, M.B.; Carbery, A.M.; Fostier, M.; Aslam, H.; Mazaleyrat, S.L.; Higgs, J.; Myat, A.; Evans, D.A.; Cornell, M.; Baron, M. Regulation of notch endosomal sorting and signaling by Drosophila Nedd4 family proteins. Curr. Biol. 2004, 14, 2237–2244. [Google Scholar] [CrossRef] [PubMed]
- Vaccari, T.; Bilder, D. The Drosophila tumor suppressor vps25 prevents nonautonomous overproliferation by regulating notch trafficking. Dev. Cell 2005, 9, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Thompson, B.J.; Mathieu, J.; Sung, H.H.; Loeser, E.; Rørth, P.; Cohen, S.M. Tumor suppressor properties of the ESCRT-II complex component Vps25 in Drosophila. Dev. Cell 2005, 9, 711–720. [Google Scholar] [CrossRef] [PubMed]
- Childress, J.L.; Acar, M.; Tao, C.; Halder, G. Lethal giant discs, a novel C2-domain protein, restricts notch activation during endocytosis. Curr. Biol. 2006, 16, 2228–2233. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, T.; Kim, W.S.; Mandal, L.; Banerjee, U. Interaction between Notch and Hif-alpha in development and survival of Drosophila blood cells. Science 2011, 332, 1210–1213. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Roche, O.; Yan, M.S.; Finak, G.; Evans, A.J.; Metcalf, J.L.; Hast, B.E.; Hanna, S.C.; Wondergem, B.; Furge, K.A.; et al. Regulation of endocytosis via the oxygen-sensing pathway. Nat. Med. 2009, 15, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Tagami, S.; Okochi, M.; Yanagida, K.; Ikuta, A.; Fukumori, A.; Matsumoto, N.; Ishizuka-Katsura, Y.; Nakayama, T.; Itoh, N.; Jiang, J.; et al. Regulation of Notch signaling by dynamic changes in the precision of S3 cleavage of Notch-1. Mol. Cell. Biol. 2008, 28, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Kobia, F.; Duchi, S.; Deflorian, G.; Vaccari, T. Pharmacologic inhibition of vacuolar H+ ATPase reduces physiologic and oncogenic Notch signaling. Mol. Oncol. 2014, 8, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.R.; Kalyana-Sundaram, S.; Wu, Y.M.; Shankar, S.; Cao, X.; Ateeq, B.; Asangani, I.A.; Iyer, M.; Maher, C.A.; Grasso, C.S.; et al. Functionally recurrent rearrangements of the MAST kinase and Notch gene families in breast cancer. Nat. Med. 2011, 17, 1646–1651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guruharsha, K.G.; Kankel, M.W.; Artavanis-Tsakonas, S. The Notch signalling system: Recent insights into the complexity of a conserved pathway. Nat. Rev. Genet. 2012, 13, 654–666. [Google Scholar] [CrossRef] [PubMed]
- Moretti, J.; Brou, C. Ubiquitinations in the notch signaling pathway. Int. J. Mol. Sci. 2013, 19, 6359–6381. [Google Scholar] [CrossRef] [PubMed]
- Yumimoto, K.; Akiyoshi, S.; Ueo, H.; Sagara, Y.; Onoyama, I.; Ueo, H.; Ohno, S.; Mori, M.; Mimori, K.; Nakayama, K.I. F-box protein FBXW7 inhibits cancer metastasis in a non-cell-autonomous manner. J. Clin. Investig. 2015, 125, 621–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, K.; Nihira, N.T.; Inuzuka, H.; Wei, W. Physiological functions of FBW7 in cancer and metabolism. Cell. Signal. 2018, 46, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Anzi, S.; Finkin, S.; Shaulian, E. Transcriptional repression of c-Jun’s E3 ubiquitin ligases contributes to c-Jun induction by UV. Cell. Signal. 2008, 20, 862–871. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Brunskill, E.; Varnum-Finney, B.; Zhang, C.; Zhang, A.; Jay, P.Y.; Bernstein, I.; Morimoto, M.; Kopan, R. The intracellular domains of Notch1 and Notch2 are functionally equivalent during development and carcinogenesis. Development 2015, 142, 2452–2463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skalska, L.; Stojnic, R.; Li, J.; Fischer, B.; Cerda-Moya, G.; Sakai, H.; Tajbakhsh, S.; Russell, S.; Adryan, B.; Bray, S.J. Chromatin signatures at Notch-regulated enhancers reveal large-scale changes in H3K56ac upon activation. EMBO J. 2015, 34, 1889–1904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Zang, C.; Taing, L.; Arnett, K.L.; Wong, Y.J.; Pear, W.S.; Blacklow, S.C.; Liu, X.S.; Aster, J.C. NOTCH1-RBPJ complexes drive target gene expression through dynamic interactions with superenhancers. Proc. Natl. Acad. Sci. USA 2014, 111, 705–710. [Google Scholar] [CrossRef] [PubMed]
- Sahlgren, C.; Gustafsson, M.V.; Jin, S.; Poellinger, L.; Lendahl, U. Notch signaling mediates hypoxia-induced tumor cell migration and invasion. Proc. Natl. Acad. Sci. USA 2008, 29, 6392–6397. [Google Scholar] [CrossRef] [PubMed]
- Yamamizu, K.; Matsunaga, T.; Uosaki, H.; Fukushima, H.; Katayama, S.; Hiraoka-Kanie, M.; Mitani, K.; Yamashita, J.K. Convergence of Notch and beta-catenin signaling induces arterial fate in vascular progenitors. J. Cell. Biol. 2010, 189, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Geudens, I.; Gerhardt, H. Coordinating cell behaviour during blood vessel formation. Development 2011, 138, 4569–4583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Fan, F.; Wang, A.; Zheng, S.; Lu, Y. Dll4-Notch signaling in regulation of tumor angiogenesis. J. Cancer Res. Clin. Oncol. 2014, 140, 525–536. [Google Scholar] [CrossRef] [PubMed]
- Bridges, E.; Oon, C.E.; Harris, A. Notch regulation of tumor angiogenesis. Future Oncol. 2011, 7, 569–788. [Google Scholar] [CrossRef] [PubMed]
- Boareto, M.; Jolly, M.K.; Ben-Jacob, E.; Onuchic, J.N. Jagged mediates differences in normal and tumor angiogenesis by affecting tip-stalk fate decision. Proc. Natl. Acad. Sci. USA 2015, 112, E3836–E3844. [Google Scholar] [CrossRef] [PubMed]
- Kuhnert, F.; Chen, G.; Coetzee, S.; Thambi, N.; Hickey, C.; Shan, J.; Kovalenko, P.; Noguera-Troise, I.; Smith, E.; Fairhurst, J.; et al. Dll4 Blockade in Stromal Cells Mediates Antitumor Effects in Preclinical Models of Ovarian Cancer. Cancer Res. 2015, 75, 4086–4096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noguera-Troise, I.; Daly, C.; Papadopoulos, N.J.; Coetzee, S.; Boland, P.; Gale, N.W.; Lin, H.C.; Yancopoulos, G.D.; Thurston, G. Blockade of Dll4 inhibits tumour growth by promoting non-productive angiogenesis. Nature 2006, 444, 1032–1037. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Ding, B.S.; Guo, P.; Lee, S.B.; Butler, J.M.; Casey, S.C.; Simons, M.; Tam, W.; Felsher, D.W.; Shido, K.; et al. Angiocrine factors deployed by tumor vascular niche induce B cell lymphoma invasiveness and chemoresistance. Cancer Cell 2014, 25, 350–365. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Ye, X.; Fan, F.; Xia, L.; Bhattacharya, R.; Bellister, S.; Tozzi, F.; Sceusi, E.; Zhou, Y.; Tachibana, I.; et al. Endothelial cells promote the colorectal cancer stem cell phenotype through a soluble form of Jagged-1. Cancer Cell 2013, 23, 171–185. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shi, J.; Chai, K.; Ying, X.; Zhou, B.P. The Role of Snail in EMT and Tumorigenesis. Curr. Cancer Drug Targets 2013, 13, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Jain, S.; Azad, A.K.; Xu, X.; Yu, H.C.; Xu, Z.; Godbout, R.; Fu, Y. Notch and TGFβ form a positive regulatory loop and regulate EMT in epithelial ovarian cancer cells. Cell. Signal. 2016, 28, 838–849. [Google Scholar] [CrossRef] [PubMed]
- Matsuno, Y.; Coelho, A.L.; Jarai, G.; Westwick, J.; Hogaboam, C.M. Notch signaling mediates TGF-β1-induced epithelial-mesenchymal transition through the induction of Snai1. Int. J. Biochem. Cell Biol. 2012, 44, 776–789. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Ballestar, E.; Esteller, M.; Cano, A. Snail mediates E-cadherin repression by the recruitment of the Sin3A/histone deacetylase 1 (HDAC1)/HDAC2 complex. Mol. Cell. Biol. 2004, 24, 306–319. [Google Scholar] [CrossRef] [PubMed]
- Herranz, N.; Pasini, D.; Díaz, V.M.; Francí, C.; Gutierrez, A.; Dave, N.; Escrivà, M.; Hernandez-Muñoz, I.; Di Croce, L.; Helin, K.; et al. Polycomb complex 2 is required for E-cadherin repression by the Snail1 transcription factor. Mol. Cell. Biol. 2008, 28, 4772–4781. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Wu, Y.; Li, J.; Dong, C.; Ye, X.; Chi, Y.I.; Evers, B.M.; Zhou, B.P. The SNAG domain of Snail1 functions as a molecular hook for recruiting lysine-specific demethylase 1. EMBO J. 2010, 29, 1803–1816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Ma, J.; Qian, X.; Wu, Q.; Xia, J.; Miele, L.; Sarkar, F.H.; Wang, Z. Regulation of EMT by Notch signaling pathway in tumor progression. Curr. Cancer Drug Targets 2013, 13, 957–962. [Google Scholar] [CrossRef] [PubMed]
- Fujiki, K.; Inamura, H.; Miyayama, T.; Matsuoka, M. Involvement of Notch1 signaling in malignant progression of A549 cells subjected to prolonged cadmium exposure. J. Biol. Chem. 2017, 292, 7942–7953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reedijk, M.; Odorcic, S.; Chang, L.; Zhang, H.; Miller, N.; McCready, D.R.; Lockwood, G.; Egan, S.E. High-level coexpression of JAG1 and NOTCH1 is observed in human breast cancer and is associated with poor overall survival. Cancer Res. 2005, 65, 8530–8537. [Google Scholar] [CrossRef] [PubMed]
- Stylianou, S.; Clarke, R.B.; Brennan, K. Aberrant activation of notch signaling in human breast cancer. Cancer Res. 2006, 66, 1517–1525. [Google Scholar] [CrossRef] [PubMed]
- Heldring, N.; Pike, A.; Andersson, S.; Matthews, J.; Cheng, G.; Hartman, J.; Tujague, M.; Ström, A.; Treuter, E.; Warner, M.; et al. Estrogen receptors: How do they signal and what are their targets. Physiol. Rev. 2007, 87, 905–931. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, P.; Miao, H.; D’Souza, G.; Osipo, C.; Song, L.L.; Yun, J.; Zhao, H.; Mascarenhas, J.; Wyatt, D.; Antico, G.; et al. Cross-talk between notch and the estrogen receptor in breast cancer suggests novel therapeutic approaches. Cancer Res. 2008, 68, 5226–5235. [Google Scholar] [CrossRef] [PubMed]
- Haughian, J.M.; Pinto, M.P.; Harrell, J.C.; Bliesner, B.S.; Joensuu, K.M.; Dye, W.W.; Sartorius, C.A.; Tan, A.C.; Heikkilä, P.; Perou, C.M.; et al. Maintenance of hormone responsiveness in luminal breast cancers by suppression of Notch. Proc. Natl. Acad. Sci. USA 2012, 109, 2742–2747. [Google Scholar] [CrossRef] [PubMed]
- Stinson, S.; Lackner, M.R.; Adai, A.T.; Yu, N.; Kim, H.J.; O’Brien, C.; Spoerke, J.; Jhunjhunwala, S.; Boyd, Z.; Januario, T.; et al. TRPS1 targeting by miR-221/222 promotes the epithelial-to-mesenchymal transition in breast cancer. Sci. Signal. 2011, 4, ra41. [Google Scholar] [CrossRef] [PubMed]
- Momeni, P.; Glöckner, G.; Schmidt, O.; von Holtum, D.; Albrecht, B.; Gillessen-Kaesbach, G.; Hennekam, R.; Meinecke, P.; Zabel, B.; Rosenthal, A.; et al. Mutations in a new gene, encoding a zinc-finger protein, cause tricho-rhino-phalangeal syndrome type I. Nat. Genet. 2000, 24, 71–74. [Google Scholar] [CrossRef] [PubMed]
- Golan, T.; Messer, A.R.; Amitai-Lange, A.; Melamed, Z.; Ohana, R.; Bell, R.E.; Kapitansky, O.; Lerman, G.; Greenberger, S.; Khaled, M.; et al. Interactions of Melanoma Cells with Distal Keratinocytes Trigger Metastasis via Notch Signaling Inhibition of MITF. Mol. Cell 2015, 59, 664–676. [Google Scholar] [CrossRef] [PubMed]
- Han, S.H.; Kim, H.J.; Gwak, J.M.; Kim, M.; Chung, Y.R.; Park, S.Y. MicroRNA-222 Expression as a Predictive Marker for Tumor Progression in Hormone Receptor-Positive Breast Cancer. J. Breast Cancer 2017, 20, 35–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, N.; Jaye, D.L.; Kajita, M.; Geigerman, C.; Moreno, C.S.; Wade, P.A. MTA3, a Mi-2/NuRD complex subunit, regulates an invasive growth pathway in breast cancer. Cell 2003, 113, 207–219. [Google Scholar] [CrossRef]
- Dhasarathy, A.; Kajita, M.; Wade, P.A. The transcription factor snail mediates epithelial to mesenchymal transitions by repression of estrogen receptor-alpha. Mol. Endocrinol. 2007, 21, 2907–2918. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.W.; Chen, M.N.; Wei, X.L.; Li, Y.C.; Lin, H.Y.; Chen, M.; Li, J.W.; Du, C.W.; Man, K.; Zhang, G.J. The zinc-finger transcriptional factor Slug transcriptionally downregulates ERα by recruiting lysine-specific demethylase 1 in human breast cancer. Oncogenesis 2017, 6, e330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barton, M.; Filardo, E.J.; Lolait, S.J.; Thomas, P.; Maggiolini, M.; Prossnitz, E.R. Twenty years of the G protein-coupled estrogen receptor GPER: Historical and personal perspectives. J. Steroid. Biochem. Mol. Biol. 2018, 176, 14–15. [Google Scholar] [CrossRef] [PubMed]
- Filardo, E.J.; Graeber, C.T.; Quinn, J.A.; Resnick, M.B.; Giri, D.; DeLellis, R.A.; Steinhoff, M.M.; Sabo, E. Distribution of GPR30, a seven membrane-spanning estrogen receptor, in primary breast cancer and its association with clinicopathologic determinants of tumor progression. Clin. Cancer Res. 2006, 12, 6359–6366. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.O.; Leslie, K.K.; Singh, M.; Qualls, C.R.; Revankar, M.; Joste, N.E.; Prossnitz, E.R. GPR30: A novel indicator of poor survival for endometrial carcinoma. Am. J. Obstet. Gynecol. 2007, 196, 386.e1–386.e11. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.O.; Arias-Pulido, H.; Kuo, D.Y.; Howard, T.; Qualls, C.R.; Lee, S.J.; Verschraegen, C.F.; Hathaway, H.J.; Joste, N.E.; Prossnitz, E.R. GPR30 predicts poor survival for ovarian cancer. Gynecol. Oncol. 2009, 114, 465–471. [Google Scholar] [Green Version]
- Sjöström, M.; Hartman, L.; Grabau, D.; Fornander, T.; Malmström, P.; Nordenskjöld, B.; Sgroi, D.C.; Skoog, L.; Stål, O.; Leeb-Lundberg, L.M.; et al. Lack of G protein-coupled estrogen receptor (GPER) in the plasma membrane is associated with excellent long-term prognosis in breast cancer. Breast Cancer Res. Treat. 2014, 145, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Zhong, Q.; Rhodes, L.V.; Townley, I.; Bratton, M.R.; Zhang, Q.; Martin, E.C.; Elliott, S.; Collins-Burow, B.M.; Burow, M.E.; et al. Proteomic analysis of acquired tamoxifen resistance in MCF-7 cells reveals expression signatures associated with enhanced migration. Breast Cancer Res. 2012, 14, R45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lappano, R.; Pisano, A.; Maggiolini, M. GPER Function in Breast Cancer: An Overview. Front. Endocrinol. 2014, 5, 66. [Google Scholar] [CrossRef] [PubMed]
- Lappano, R.; Maggiolini, M. GPER is involved in the functional liaison between breast tumor cells and cancer-associated fibroblasts (CAFs). J. Steroid Biochem. Mol. Biol. 2018, 176, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Madeo, A.; Maggiolini, M. Nuclear alternate estrogen receptor GPR30 mediates 17beta-estradiol-induced gene expression and migration in breast cancer-associated fibroblasts. Cancer Res. 2010, 70, 6036–6046. [Google Scholar] [CrossRef] [PubMed]
- Pandey, D.P.; Lappano, R.; Albanito, L.; Madeo, A.; Maggiolini, M.; Picard, D. Estrogenic GPR30 signalling induces proliferation and migration of breast cancer cells through CTGF. EMBO J. 2009, 28, 523–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Q.F.; Wu, T.T.; Yang, J.Y.; Dong, C.R.; Wang, N.; Liu, X.H.; Liu, Z.M. 17β-estradiol promotes the invasion and migration of nuclear estrogen receptor-negative breast cancer cells through cross-talk between GPER1 and CXCR1. J. Steroid Biochem. Mol. Biol. 2013, 138, 314–324. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Wang, S.; Wang, Z.; Feng, X.; Liu, P.; Lv, X.B.; Li, F.; Yu, F.X.; Sun, Y.; Yuan, H.; et al. Estrogen regulates Hippo signaling via GPER in breast cancer. J. Clin. Investig. 2015, 125, 2123–2135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Marco, P.; Lappano, R.; De Francesco, E.M.; Cirillo, F.; Pupo, M.; Avino, S.; Vivacqua, A.; Abonante, S.; Picard, D.; Maggiolini, M. GPER signalling in both cancer-associated fibroblasts and breast cancer cells mediates a feedforward IL1β/IL1R1 response. Sci. Rep. 2016, 6, 24354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenza, G.L. Angiogenesis in ischemic and neoplastic disorders. Annu. Rev. Med. 2003, 54, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L.; Roth, P.H.; Fang, H.M.; Wang, G.L. Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J. Biol. Chem. 1994, 269, 23757–23763. [Google Scholar] [PubMed]
- Simon, M.C.; Keith, B. The role of oxygen availability in embryonic development and stem cell function. Nat. Rev. Mol. Cell Biol. 2008, 9, 285–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, G.; Liu, Y. Hypoxia-inducible factors in cancer stem cells and inflammation. Trends Pharmacol. Sci. 2015, 36, 374–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansson, E.M.; Lendahl, U.; Chapman, G. Notch signaling in development and disease. Semin. Cancer Biol. 2004, 14, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, M.V.; Zheng, X.; Pereira, T.; Gradin, K.; Jin, S.; Lundkvist, J.; Ruas, J.L.; Poellinger, L.; Lendahl, U.; Bondesson, M. Hypoxia requires notch signaling to maintain the undifferentiated cell state. Dev. Cell 2005, 9, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Jögi, A.; Øra, I.; Nilsson, H.; Lindeheim, A.; Makino, Y.; Poellinger, L.; Axelson, H.; Påhlman, S. Hypoxia alters gene expression in human neuroblastoma cells toward an immature and neural crest-like phenotype. Proc. Natl. Acad. Sci. USA 2002, 99, 7021–7026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irshad, K.; Mohapatra, S.K.; Srivastava, C.; Garg, H.; Mishra, S.; Dikshit, B.; Sarkar, C.; Gupta, D.; Chandra, P.S.; Chattopadhyay, P.; et al. A combined gene signature of hypoxia and notch pathway in human glioblastoma and its prognostic relevance. PLoS ONE 2015, 10, e0118201. [Google Scholar] [CrossRef] [PubMed]
- Bao, B.; Azmi, A.S.; Ali, S.; Ahmad, A.; Li, Y.; Banerjee, S.; Kong, D.; Sarkar, F.H. The biological kinship of hypoxia with CSC and EMT and their relationship with deregulated expression of miRNAs and tumor aggressiveness. Biochim. Biophys. Acta 2012, 1826, 272–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imai, T.; Horiuchi, A.; Wang, C.; Oka, K.; Ohira, S.; Nikaido, T.; Konishi, I. Hypoxia attenuates the expression of E-cadherin via up-regulation of SNAIL in ovarian carcinoma cells. Am. J. Pathol. 2003, 163, 1437–1447. [Google Scholar] [CrossRef]
- Choi, B.J.; Park, S.A.; Lee, S.Y.; Cha, Y.N.; Surh, Y.J. Hypoxia induces epithelial-mesenchymal transition in colorectal cancer cells through ubiquitin-specific protease 47-mediated stabilization of Snail: A potential role of Sox9. Sci. Rep. 2017, 7, 15918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Imanaka, N.; Chen, J.; Griffin, J.D. Hypoxia potentiates Notch signaling in breast cancer leading to decreased E-cadherin expression and increased cell migration and invasion. Br. J. Cancer 2010, 102, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Ishida, T.; Hijioka, H.; Kume, K.; Miyawaki, A.; Nakamura, N. Notch signaling induces EMT in OSCC cell lines in a hypoxic environment. Oncol. Lett. 2013, 6, 1201–1206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Craene, B.; Berx, G. Regulatory networks defining EMT during cancer initiation and progression. Nat. Rev. Cancer 2013, 13, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.Z.; Tsai, Y.P.; Yang, M.H.; Huang, C.H.; Chang, S.Y.; Chang, C.C.; Teng, S.C.; Wu, K.J. Interplay between HDAC3 and WDR5 is essential for hypoxia-induced epithelial-mesenchymal transition. Mol. Cell 2011, 43, 811–822. [Google Scholar] [CrossRef] [PubMed]
- Villa, J.C.; Chiu, D.; Brandes, A.H.; Escorcia, F.E.; Villa, C.H.; Maguire, W.F.; Hu, C.J.; de Stanchina, E.; Simon, M.C.; Sisodia, S.S.; et al. Nontranscriptional role of Hif-1α in activation of γ-secretase and notch signaling in breast cancer. Cell. Rep. 2014, 8, 1077–1092. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.M.; Giaccia, A.J. The unique physiology of solid tumors: Opportunities (and problems) for cancer therapy. Cancer Res. 1998, 58, 1408–1416. [Google Scholar] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 4, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Paolicchi, E.; Gemignani, F.; Krstic-Demonacos, M.; Dedhar, S.; Mutti, L.; Landi, S. Targeting hypoxic response for cancer therapy. Oncotarget 2016, 7, 13464–13478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wigerup, C.; Påhlman, S.; Bexell, D. Therapeutic targeting of hypoxia and hypoxia-inducible factors in cancer. Pharmacol. Ther. 2016, 164, 152–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rundqvist, H.; Johnson, R.S. Tumour oxygenation: Implications for breast cancer prognosis. J. Intern. Med. 2013, 274, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Masoud, G.N.; Li, W. HIF-1α pathway: Role, regulation and intervention for cancer therapy. Acta Pharm. Sin. B 2015, 5, 378–389. [Google Scholar] [CrossRef] [PubMed]
- Gilkes, D.M.; Semenza, G.L. Role of hypoxia-inducible factors in breast cancer metastasis. Future Oncol. 2013, 9, 1623–1636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schito, L.; Semenza, G.L. Hypoxia-Inducible Factors: Master Regulators of Cancer Progression. Trends Cancer 2016, 2, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-inducible factors in physiology and medicine. Cell 2012, 3, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.L.; Semenza, G.L. Purification and characterization of hypoxia-inducible factor 1. J. Biol. Chem. 1995, 20, 1230–1237. [Google Scholar] [CrossRef]
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [PubMed]
- Majmundar, A.J.; Wong, W.J.; Simon, M.C. Hypoxia-inducible factors and the response to hypoxic stress. Mol. Cell 2010, 40, 294–309. [Google Scholar] [CrossRef] [PubMed]
- Bracken, C.P.; Fedele, A.O.; Linke, S.; Balrak, W.; Lisy, K.; Whitelaw, M.L.; Peet, D.J. Cell-specific regulation of hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha stabilization and transactivation in a graded oxygen environment. J. Biol. Chem. 2006, 281, 22575–22585. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hydroxylation of HIF-1: Oxygen sensing at the molecular level. Physiology 2004, 19, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Hirota, K.; Semenza, G.L. Regulation of hypoxia-inducible factor 1 by prolyl and asparaginyl hydroxylases. Biochem. Biophys. Res. Commun. 2005, 338, 610–616. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Zhao, Q.; Mooney, S.M.; Lee, F.S. Sequence determinants in hypoxia-inducible factor-1alpha for hydroxylation by the prolyl hydroxylases PHD1, PHD2, and PHD3. J. Biol. Chem. 2002, 277, 39792–39800. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, M.A.; Dunning, S.P.; Bunn, H.F. Regulation of the erythropoietin gene: Evidence that the oxygen sensor is a heme protein. Science 1988, 242, 1412–1415. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L.; Nejfelt, M.K.; Chi, S.M.; Antonarakis, S.E. Hypoxia-inducible nuclear factors bind to an enhancer element located 3′ to the human erythropoietin gene. Proc. Natl. Acad. Sci. USA 1991, 88, 5680–5684. [Google Scholar] [CrossRef] [PubMed]
- Snell, C.E.; Turley, H.; McIntyre, A.; Li, D.; Masiero, M.; Schofield, C.J.; Gatter, K.C.; Harris, A.L.; Pezzella, F. Proline-hydroxylated hypoxia-inducible factor 1α (HIF-1α) upregulation in human tumours. PLoS ONE 2014, 9, e88955. [Google Scholar] [CrossRef] [PubMed]
- Zia, M.K.; Rmali, K.A.; Watkins, G.; Mansel, R.E.; Jiang, W.G. The expression of the von Hippel-Lindau gene product and its impact on invasiveness of human breast cancer cells. Int. J. Mol. Med. 2007, 20, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Fox, S.B.; Generali, D.; Berruti, A.; Brizzi, M.P.; Campo, L.; Bonardi, S.; Bersiga, A.; Allevi, G.; Milani, M.; Aguggini, S.; et al. The prolyl hydroxylase enzymes are positively associated with hypoxia-inducible factor-1α and vascular endothelial growth factor in human breast cancer and alter in response to primary systemic treatment with epirubicin and tamoxifen. Breast Cancer Res. 2011, 13, R16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baek, S.H.; Kim, K.I. Regulation of HIF-1α stability by lysine methylation. BMB Rep. 2016, 49, 245–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soysal, S.D.; Tzankov, A.; Muenst, S.E. Role of the Tumor Microenvironment in Breast Cancer. Pathobiology 2015, 82, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 23, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Polyak, K.; Kalluri, R. The role of the microenvironment in mammary gland development and cancer. Cold Spring Harb. Perspect. Biol. 2010, 2, a003244. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Keller, E.T.; Garfield, D.H.; Shen, K.; Wang, J. Stromal cells in tumor microenvironment and breast cancer. Cancer Metastasis Rev. 2013, 32, 303–315. [Google Scholar] [CrossRef] [PubMed]
- Polyak, K.; Weinberg, R.A. Transitions between epithelial and mesenchymal states: Acquisition of malignant and stem cell traits. Nat. Rev. Cancer 2009, 9, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, E.M.; Potenta, S.; Xie, L.; Zeisberg, M.; Kalluri, R. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res. 2007, 67, 10123–10128. [Google Scholar] [CrossRef] [PubMed]
- Qiao, A.; Gu, F.; Guo, X.; Zhang, X.; Fu, L. Breast cancer-associated fibroblasts: Their roles in tumor initiation, progression and clinical applications. Front. Med. 2016, 10, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Comito, G.; Giannoni, E.; Di Gennaro, P.; Segura, C.P.; Gerlini, G.; Chiarugi, P. Stromal fibroblasts synergize with hypoxic oxidative stress to enhance melanoma aggressiveness. Cancer Lett. 2012, 324, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Toullec, A.; Gerald, D.; Despouy, G.; Bourachot, B.; Cardon, M.; Lefort, S.; Richardson, M.; Rigaill, G.; Parrini, M.C.; Lucchesi, C.; et al. Oxidative stress promotes myofibroblast differentiation and tumour spreading. EMBO Mol. Med. 2010, 2, 211–230. [Google Scholar] [CrossRef] [PubMed]
- Waghray, M.; Cui, Z.; Horowitz, J.C.; Subramanian, I.M.; Martinez, F.J.; Toews, G.B.; Thannickal, V.J. Hydrogen peroxide is a diffusible paracrine signal for the induction of epithelial cell death by activated myofibroblasts. FASEB J. 2005, 19, 854–856. [Google Scholar] [CrossRef] [PubMed]
- Cirri, P.; Chiarugi, P. Cancer-associated-fibroblasts and tumour cells: A diabolic liaison driving cancer progression. Cancer Metastasis Rev. 2012, 31, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Wottawa, M.; Leisering, P.; Ahlen, M.V.; Schnelle, M.; Vogel, S.; Malz, C.; Bordoli, M.R.; Camenisch, G.; Hesse, A.; Napp, J.; et al. Knockdown of prolyl-4-hydroxylase domain 2 inhibits tumor growth of human breast cancer MDA-MB-231 cells by affecting TGF-β1 processing. Int. J. Cancer 2013, 132, 2787–2798. [Google Scholar] [CrossRef] [PubMed]
- Kuchnio, A.; Dewerchin, M.; Carmeliet, P. The PHD2 oxygen sensor paves the way to metastasis. Oncotarget 2015, 6, 35149–35150. [Google Scholar] [CrossRef] [PubMed]
- Madsen, C.D.; Pedersen, J.T.; Venning, F.A.; Singh, L.B.; Moeendarbary, E.; Charras, G.; Cox, T.R.; Sahai, E.; Erler, J.T. Hypoxia and loss of PHD2 inactivate stromal fibroblasts to decrease tumour stiffness and metastasis. EMBO Rep. 2015, 16, 1394–1408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiavarina, B.; Martinez-Outschoorn, U.E.; Whitaker-Menezes, D.; Howell, A.; Tanowitz, H.B.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Metabolic reprogramming and two-compartment tumor metabolism: Opposing role(s) of HIF1α and HIF2α in tumor-associated fibroblasts and human breast cancer cells. Cell Cycle 2012, 11, 3280–3289. [Google Scholar] [CrossRef] [PubMed]
- Migneco, G.; Whitaker-Menezes, D.; Chiavarina, B.; Castello-Cros, R.; Pavlides, S.; Pestell, R.G.; Fatatis, A.; Flomenberg, N.; Tsirigos, A.; Howell, A.; et al. Glycolytic cancer associated fibroblasts promote breast cancer tumor growth, without a measurable increase in angiogenesis: Evidence for stromal-epithelial metabolic coupling. Cell Cycle 2010, 9, 2412–2422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, A.; Scholer-Dahirel, A.; Mechta-Grigoriou, F. The role of reactive oxygen species and metabolism on cancer cells and their microenvironment. Semin. Cancer Biol. 2014, 25, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Lisanti, M.P.; Sotgia, F. Catabolic cancer-associated fibroblasts transfer energy and biomass to anabolic cancer cells, fueling tumor growth. Semin. Cancer Biol. 2014, 25, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed]
- Haddad, J.J.; Harb, H.L. Cytokines and the regulation of hypoxia-inducible factor (HIF)-1alpha. Int. Immunopharmacol. 2005, 5, 461–483. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Brüne, B. Cytokines and hormones in the regulation of hypoxia inducible factor-1alpha (HIF-1alpha). Cardiovasc. Hematol. Agents Med. Chem. 2006, 4, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Balamurugan, K. HIF-1 at the crossroads of hypoxia, inflammation, and cancer. Int. J. Cancer 2016, 1, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, R.; Singh, V.; Asif, M.; Negi, M.P.S.; Bhadauria, S. Oncostatin M upregulates HIF-1α in breast tumor associated macrophages independent of intracellular oxygen concentration. Life Sci. 2018, 194, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 2005, 6, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Andreou, K.; Rajendran, R.; Krstic-Demonacos, M.; Demonacos, C. Regulation of CXCR4 gene expression in breast cancer cells under diverse stress conditions. Int. J. Oncol. 2012, 41, 2253–2259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curran, C.S.; Keely, P.J. Breast tumor and stromal cell responses to TGF-β and hypoxia in matrix deposition. Matrix Biol. 2013, 32, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Dunn, L.K.; Mohammad, K.S.; Fournier, P.G.; McKenna, C.R.; Davis, H.W.; Niewolna, M.; Peng, X.H.; Chirgwin, J.M.; Guise, T.A. Hypoxia and TGF-beta drive breast cancer bone metastases through parallel signaling pathways in tumor cells and the bone microenvironment. PLoS ONE 2009, 4, e6896. [Google Scholar] [CrossRef] [PubMed]
- Hung, S.P.; Yang, M.H.; Tseng, K.F.; Lee, O.K. Hypoxia-induced secretion of TGF-β1 in mesenchymal stem cell promotes breast cancer cell progression. Cell Transplant. 2013, 22, 1869–1882. [Google Scholar] [CrossRef] [PubMed]
- Stantic, M.; Sakil, H.A.; Zirath, H.; Fang, T.; Sanz, G.; Fernandez-Woodbridge, A.; Marin, A.; Susanto, E.; Mak, T.W.; Henriksson, M.A.; et al. TAp73 suppresses tumor angiogenesis through repression of proangiogenic cytokines and HIF-1α activity. Proc. Natl. Acad. Sci. USA 2015, 112, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Sanguinetti, A.; Santini, D.; Bonafè, M.; Taffurelli, M.; Avenia, N. Interleukin-6 and pro inflammatory status in the breast tumor microenvironment. World J. Surg. Oncol. 2015, 13, 129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Studebaker, A.W.; Storci, G.; Werbeck, J.L.; Sansone, P.; Sasser, A.K.; Tavolari, S.; Huang, T.; Chan, M.W.; Marini, F.C.; Rosol, T.J.; et al. Fibroblasts isolated from common sites of breast cancer metastasis enhance cancer cell growth rates and invasiveness in an interleukin-6-dependent manner. Cancer Res. 2008, 68, 9087–9095. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Xu, J.; Liu, B.; He, X.; Zhou, L.; Hu, X.; Qiao, F.; Zhang, A.; Xu, X.; Zhang, H.; et al. IL6 blockade potentiates the anti-tumor effects of γ-secretase inhibitors in Notch3-expressing breast cancer. Cell Death Differ. 2018, 25, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Kong, W.; He, L.; Richards, E.J.; Challa, S.; Xu, C.X.; Permuth-Wey, J.; Lancaster, J.M.; Coppola, D.; Sellers, T.A.; Djeu, J.Y.; et al. Upregulation of miRNA-155 promotes tumour angiogenesis by targeting VHL and is associated with poor prognosis and triple-negative breast cancer. Oncogene 2014, 33, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Zhou, X.; Liu, X.; Jia, H.H.; Zhao, X.H.; Wang, Q.X.; Han, L.; Song, X.; Zhu, Z.Y.; Sun, T.; et al. Reprogramming carcinoma associated fibroblasts by AC1MMYR2 impedes tumor metastasis and improves chemotherapy efficacy. Cancer Lett. 2016, 374, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.V.; Baek, J.H.; Zhang, H.; Diez, R.; Cole, R.N.; Semenza, G.L. RACK1 competes with HSP90 for binding to HIF-1alpha and is required for O(2)-independent and HSP90 inhibitor-induced degradation of HIF-1alpha. Mol. Cell 2007, 25, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Dewhirst, M.W.; Cao, Y.; Moeller, B. Cycling hypoxia and free radicals regulate angiogenesis and radiotherapy response. Nat. Rev. Cancer 2008, 8, 425–437. [Google Scholar] [CrossRef] [PubMed]
- Iommarini, L.; Porcelli, A.M.; Gasparre, G.; Kurelac, I. Non-Canonical Mechanisms Regulating Hypoxia-Inducible Factor 1 Alpha in Cancer. Front. Oncol. 2017, 7, 286. [Google Scholar] [CrossRef] [PubMed]
- Kazi, A.A.; Gilani, R.A.; Schech, A.J.; Chumsri, S.; Sabnis, G.; Shah, P.; Goloubeva, O.; Kronsberg, S.; Brodie, A.H. Nonhypoxic regulation and role of hypoxia-inducible factor 1 in aromatase inhibitor resistant breast cancer. Breast Cancer Res. 2014, 16, R15. [Google Scholar] [CrossRef] [PubMed]
- Carroll, V.A.; Ashcroft, M. Role of hypoxia-inducible factor (HIF)-1alpha versus HIF-2alpha in the regulation of HIF target genes in response to hypoxia, insulin-like growth factor-I, or loss of von Hippel-Lindau function: Implications for targeting the HIF pathway. Cancer Res. 2006, 66, 6264–6270. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.D.; Zhou, X.; Zhou, K.Y. Dauricine inhibits insulin-like growth factor-I-induced hypoxia inducible factor 1alpha protein accumulation and vascular endothelial growth factor expression in human breast cancer cells. Acta Pharmacol. Sin. 2009, 30, 605–616. [Google Scholar] [CrossRef] [PubMed]
- De Francesco, E.M.; Sims, A.H.; Maggiolini, M.; Sotgia, F.; Lisanti, M.P.; Clarke, R.B. GPER mediates the angiocrine actions induced by IGF1 through the HIF-1α/VEGF pathway in the breast tumor microenvironment. Breast Cancer Res. 2017, 19, 129. [Google Scholar] [CrossRef] [PubMed]
- Samanta, D.; Gilkes, D.M.; Chaturvedi, P.; Xiang, L.; Semenza, G.L. Hypoxia-inducible factors are required for chemotherapy resistance of breast cancer stem cells. Proc. Natl. Acad. Sci. USA 2014, 111, E5429–5438. [Google Scholar] [CrossRef] [PubMed]
- Rigiracciolo, D.C.; Scarpelli, A.; Lappano, R.; Pisano, A.; Santolla, M.F.; De Marco, P.; Cirillo, F.; Cappello, A.R.; Dolce, V.; Belfiore, A.; et al. Copper activates HIF-1α/GPER/VEGF signalling in cancer cells. Oncotarget 2015, 6, 34158–34177. [Google Scholar] [CrossRef] [PubMed]
- De Francesco, E.M.; Pellegrino, M.; Santolla, M.F.; Lappano, R.; Ricchio, E.; Abonante, S.; Maggiolini, M. GPER mediates activation of HIF1α/VEGF signaling by estrogens. Cancer Res. 2014, 74, 4053–4064. [Google Scholar] [CrossRef] [PubMed]
- Bartella, V.; De Francesco, E.M.; Perri, M.G.; Curcio, R.; Dolce, V.; Maggiolini, M.; Vivacqua, A. The G protein estrogen receptor (GPER) is regulated by endothelin-1 mediated signaling in cancer cells. Cell. Signal. 2016, 28, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Liu, Z.; Zandi, E. AP-1 function and regulation. Curr. Opin. Cell Biol. 1997, 9, 240–246. [Google Scholar] [CrossRef]
- Kazi, A.A.; Jones, J.M.; Koos, R.D. Chromatin immunoprecipitation analysis of gene expression in the rat uterus in vivo: Estrogen-induced recruitment of both estrogen receptor alpha and hypoxia-inducible factor 1 to the vascular endothelial growth factor promoter. Mol. Endocrinol. 2005, 19, 2006–2019. [Google Scholar] [CrossRef] [PubMed]
- Hua, K.; Din, J.; Cao, Q.; Feng, W.; Zhang, Y.; Yao, L.; Huang, Y.; Zhao, Y.; Feng, Y. Estrogen and progestin regulate HIF-1alpha expression in ovarian cancer cell lines via the activation of Akt signaling transduction pathway. Oncol. Rep. 2009, 21, 893–898. [Google Scholar] [PubMed]
- George, A.L.; Rajoria, S.; Suriano, R.; Mittleman, A.; Tiwari, R.K. Hypoxia and estrogen are functionally equivalent in breast cancer-endothelial cell interdependence. Mol. Cancer 2012, 11, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazi, A.A.; Koos, R.D. Estrogen-induced activation of hypoxia-inducible factor-1alpha, vascular endothelial growth factor expression, and edema in the uterus are mediated by the phosphatidylinositol 3-kinase/Akt pathway. Endocrinology 2007, 148, 2363–2374. [Google Scholar] [CrossRef] [PubMed]
- Padró, M.; Louie, R.J.; Lananna, B.V.; Krieg, A.J.; Timmerman, L.A.; Chan, D.A. Genome-independent hypoxic repression of estrogen receptor alpha in breast cancer cells. BMC Cancer 2017, 17, 203. [Google Scholar] [CrossRef] [PubMed]
- Frump, A.L.; Selej, M.; Wood, J.A.; Albrecht, M.; Yakubov, B.; Petrache, I.; Lahm, T. Hypoxia Up-regulates Estrogen Receptor β in Pulmonary Artery Endothelial Cells in a HIF-1α Dependent Manner. Am. J. Respir. Cell. Mol. Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Recchia, A.G.; De Francesco, E.M.; Vivacqua, A.; Sisci, D.; Panno, M.L.; Andò, S.; Maggiolini, M. The G protein-coupled receptor 30 is up-regulated by hypoxia-inducible factor-1alpha (HIF-1alpha) in breast cancer cells and cardiomyocytes. J. Biol. Chem. 2011, 286, 10773–10782. [Google Scholar] [CrossRef] [PubMed]
- Bos, R.; Zhong, H.; Hanrahan, C.F.; Mommers, E.C.; Semenza, G.L.; Pinedo, H.M.; Abeloff, M.D.; Simons, J.W.; van Diest, P.J.; van der Wall, E. Levels of hypoxia-inducible factor-1 alpha during breast carcinogenesis. J. Natl. Cancer Inst. 2001, 93, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Brunnberg, S.; Pettersson, K.; Rydin, E.; Matthews, J.; Hanberg, A.; Pongratz, I. The basic helix-loop-helix-PAS protein ARNT functions as a potent coactivator of estrogen receptor-dependent transcription. Proc. Natl. Acad. Sci. USA 2003, 100, 6517–6522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, K.; Park, C.; Lee, Y. Hypoxia-inducible factor 1 alpha represses the transcription of the estrogen receptor alpha gene in human breast cancer cells. Biochem. Biophys. Res. Commun. 2011, 407, 831–836. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.; Kim, D.; Lee, S.; Lee, Y. Cobalt chloride-induced estrogen receptor alpha down-regulation involves hypoxia-inducible factor-1alpha in MCF-7 human breast cancer cells. Mol. Endocrinol. 2005, 19, 1191–1199. [Google Scholar] [CrossRef] [PubMed]
- Kronblad, A.; Helczynska, K.; Nielsen, N.H.; Påhlman, E.; Emdin, S.; Påhlman, S.; Landberg, G. Regional cyclin D1 overexpression or hypoxia correlate inversely with heterogeneous oestrogen receptor-alpha expression in human breast cancer. In Vivo 2003, 17, 311–318. [Google Scholar] [PubMed]
- Kronblad, A.; Hedenfalk, I.; Nilsson, E.; Påhlman, S.; Landberg, G. ERK1/2 inhibition increases antiestrogen treatment efficacy by interfering with hypoxia-induced downregulation of ERalpha: A combination therapy potentially targeting hypoxic and dormant tumor cells. Oncogene. 2005, 24, 6835–6841. [Google Scholar] [CrossRef] [PubMed]
- Ogba, N.; Doughman, Y.Q.; Chaplin, L.J.; Hu, Y.; Gargesha, M.; Watanabe, M.; Montano, M.M. HEXIM1 modulates vascular endothelial growth factor expression and function in breast epithelial cells and mammary gland. Oncogene 2010, 29, 3639–3649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sudhagar, S.; Sathya, S.; Lakshmi, B.S. Rapid non-genomic signalling by 17β-oestradiol through c-Src involves mTOR-dependent expression of HIF-1α in breast cancer cells. Br. J. Cancer 2011, 105, 953–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, H.; Rogerson, L.; Gregson, H.J.; Brennan, K.R.; Clarke, R.B.; Landberg, G. Contrasting hypoxic effects on breast cancer stem cell hierarchy is dependent on ER-α status. Cancer Res. 2013, 73, 1420–1433. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; AlTahan, A.; Jones, D.T.; Buffa, F.M.; Bridges, E.; Interiano, R.B.; Qu, C.; Vogt, N.; Li, J.L.; Baban, D.; et al. Estrogen receptor-α directly regulates the hypoxia-inducible factor 1 pathway associated with antiestrogen response in breast cancer. Proc. Natl. Acad. Sci. USA 2015, 112, 15172–15177. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Jubb, A.M.; Pike, L.; Buffa, F.M.; Turley, H.; Baban, D.; Leek, R.; Gatter, K.C.; Ragoussis, J.; Harris, A.L. The histone demethylase JMJD2B is regulated by estrogen receptor alpha and hypoxia, and is a key mediator of estrogen induced growth. Cancer Res. 2010, 70, 6456–6466. [Google Scholar] [CrossRef] [PubMed]
- Fuady, J.H.; Gutsche, K.; Santambrogio, S.; Varga, Z.; Hoogewijs, D.; Wenger, R.H. Estrogen-dependent downregulation of hypoxia-inducible factor (HIF)-2α in invasive breast cancer cells. Oncotarget 2016, 7, 31153–31165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, W.; Park, Y.; Cho, J.; Park, C.; Park, J.; Park, Y.K.; Park, H.; Lee, Y. Estrogen receptor beta inhibits transcriptional activity of hypoxia inducible factor-1 through the downregulation of arylhydrocarbon receptor nuclear translocator. Breast Cancer Res. 2011, 13, R32. [Google Scholar] [CrossRef] [PubMed]
- Park, C.; Lee, Y. Overexpression of ERβ is sufficient to inhibit hypoxia-inducible factor-1 transactivation. Biochem. Biophys. Res. Commun. 2014, 450, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Koda, M.; Kanczuga-Koda, L.; Sulkowska, M.; Surmacz, E.; Sulkowski, S. Relationships between hypoxia markers and the leptin system, estrogen receptors in human primary and metastatic breast cancer: Effects of preoperative chemotherapy. BMC Cancer 2010, 10, 320. [Google Scholar] [CrossRef] [PubMed]
- Bialesova, L.; Xu, L.; Gustafsson, J.Å.; Haldosen, L.A.; Zhao, C.; Dahlman-Wright, K. Estrogen receptor β2 induces proliferation and invasiveness of triple negative breast cancer cells: Association with regulation of PHD3 and HIF-1α. Oncotarget 2017, 8, 76622–76633. [Google Scholar] [CrossRef] [PubMed]
- De Francesco, E.M.; Lappano, R.; Santolla, M.F.; Marsico, S.; Caruso, A.; Maggiolini, M. HIF-1α/GPER signaling mediates the expression of VEGF induced by hypoxia in breast cancer associated fibroblasts (CAFs). Breast Cancer Res. 2013, 15, R64. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Guo, H.; Wu, H.; Tian, T.; Dong, D.; Zhang, Y.; Sui, Y.; Zhang, Y.; Zhao, D.; Wang, S.; et al. GPER in CAFs regulates hypoxia-driven breast cancer invasion in a CTGF-dependent manner. Oncol. Rep. 2015, 33, 1929–1937. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Yang, G.; Hou, Y.; Tang, X.; Wu, C.; Wu, X.A.; Guo, L.; Zhu, Q.; Luo, H.; Du, Y.E. Cytoplasmic GPER translocation in cancer-associated fibroblasts mediates cAMP/PKA/CREB/glycolytic axis to confer tumor cells with multidrug resistance. Oncogene 2017, 36, 2131–2145. [Google Scholar] [CrossRef] [PubMed]
- Sonveaux, P.; Copetti, T.; De Saedeleer, C.J.; Végran, F.; Verrax, J.; Kennedy, K.M.; Moon, E.J.; Dhup, S.; Danhier, P.; Frérart, F.; et al. Targeting the lactate transporter MCT1 in endothelial cells inhibits lactate-induced HIF-1 activation and tumor angiogenesis. PLoS ONE 2012, 7, e33418. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Forbes, R.A.; Verma, A. Hypoxia-inducible factor 1 activation by aerobic glycolysis implicates the Warburg effect in carcinogenesis. J. Biol. Chem. 2002, 277, 23111–23115. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.Y.; Song, H.S.; Park, S.Y.; Chung, S.H.; Kim, Y.J. Pyruvate promotes tumor angiogenesis through HIF-1-dependent PAI-1 expression. Int. J. Oncol. 2011, 38, 571–576. [Google Scholar] [PubMed]
- Zhang, L.; Xiong, W.; Li, N.; Liu, H.; He, H.; Du, Y.; Zhang, Z.; Liu, Y. Estrogen stabilizes hypoxia-inducible factor 1α through G protein-coupled estrogen receptor 1 in eutopic endometrium of endometriosis. Fertil. Steril. 2017, 107, 439–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, F.; Wang, X.; Wu, N.; He, S.; Yi, W.; Xiang, S.; Zhang, P.; Xie, X.; Ying, C. Bisphenol A induces proliferative effects on both breast cancer cells and vascular endothelial cells through a shared GPER-dependent pathway in hypoxia. Environ. Pollut. 2017, 231, 1609–1620. [Google Scholar] [CrossRef] [PubMed]
- Weißenborn, C.; Ignatov, T.; Ochel, H.J.; Costa, S.D.; Zenclussen, A.C.; Ignatova, Z.; Ignatov, A. GPER functions as a tumor suppressor in triple-negative breast cancer cells. J. Cancer Res. Clin. Oncol. 2014, 140, 713–723. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.J.; Wei, W.; Jiang, G.M.; Liu, H.; Wei, W.D.; Yang, X.; Wu, Y.M.; Liu, H.; Wong, C.K.; Du, J.; et al. Activation of GPER suppresses epithelial mesenchymal transition of triple negative breast cancer cells via NF-κB signals. Mol. Oncol. 2016, 10, 775–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, S.; Chen, Z.; Jiang, G.; Zhou, Y.; Liu, Q.; Su, Q.; Wei, W.; Du, J.; Wang, H. Activation of GPER suppresses migration and angiogenesis of triple negative breast cancer via inhibition of NF-κB/IL-6 signals. Cancer Lett. 2017, 386, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Hong, Q.; Lei, L.; Li, D.; Li, J.; Mo, M.; Wang, Y.; Shao, Z.; Shen, Z.; Cheng, J.; et al. Basal and therapy-driven hypoxia-inducible factor-1α confers resistance to endocrine therapy in estrogen receptor-positive breast cancer. Oncotarget 2015, 6, 8648–8662. [Google Scholar] [CrossRef] [PubMed]
- Theys, J.; Jutten, B.; Habets, R.; Paesmans, K.; Groot, A.J.; Lambin, P.; Wouters, B.G.; Lammering, G.; Vooijs, M. E-Cadherin loss associated with EMT promotes radioresistance in human tumor cells. Radiother. Oncol. 2011, 99, 392–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borggrefe, T.; Lauth, M.; Zwijsen, A.; Huylebroeck, D.; Oswald, F.; Giaimo, B.D. The Notch intracellular domain integrates signals from Wnt, Hedgehog, TGFβ/BMP and hypoxia pathways. Biochim. Biophys. Acta 2016, 1863, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Pupo, M.; Vivacqua, A.; Perrotta, I.; Pisano, A.; Aquila, S.; Abonante, S.; Gasperi-Campani, A.; Pezzi, V.; Maggiolini, M. The nuclear localization signal is required for nuclear GPER translocation and function in breast Cancer-Associated Fibroblasts (CAFs). Mol. Cell. Endocrinol. 2013, 376, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Wu, H.; Xu, H.; Xiong, H.; Chu, Q.; Yu, S.; Wu, G.S.; Wu, K. Notch signaling: An emerging therapeutic target for cancer treatment. Cancer Lett. 2015, 369, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Li, Y.; Wu, W.; Gordon, W.R.; Chang, D.W.; Lu, M.; Scoggin, S.; Fu, T.; Vien, L.; Histen, G.; et al. Modulation of Notch signaling by antibodies specific for the extracellular negative regulatory region of NOTCH3. J. Biol. Chem. 2008, 283, 8046–8054. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Cain-Hom, C.; Choy, L.; Hagenbeek, T.J.; de Leon, G.P.; Chen, Y.; Finkle, D.; Venook, R.; Wu, X.; Ridgway, J.; et al. Therapeutic antibody targeting of individual Notch receptors. Nature 2010, 464, 1052–1057. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, D.W.; Ross, S.; Veldman-Jones, M.; Foltz, I.N.; Clavette, B.C.; Manchulenko, K.; Eberlein, C.; Kendrew, J.; Petteruti, P.; Cho, S.; et al. MEDI0639: A novel therapeutic antibody targeting Dll4 modulates endothelial cell function and angiogenesis in vivo. Mol. Cancer Ther. 2012, 11, 1650–1660. [Google Scholar] [CrossRef] [PubMed]
- Funahashi, Y.; Hernandez, S.L.; Das, I.; Ahn, A.; Huang, J.; Vorontchikhina, M.; Sharma, A.; Kanamaru, E.; Borisenko, V.; Desilva, D.M.; et al. A notch1 ectodomain construct inhibits endothelial notch signaling, tumor growth, and angiogenesis. Cancer Res. 2008, 68, 4727–4735. [Google Scholar] [CrossRef] [PubMed]
- Kangsamaksin, T.; Murtomaki, A.; Kofler, N.M.; Cuervo, H.; Chaudhri, R.A.; Tattersall, I.W.; Rosenstiel, P.E.; Shawber, C.J.; Kitajewski, J. NOTCH decoys that selectively block DLL/NOTCH or JAG/NOTCH disrupt angiogenesis by unique mechanisms to inhibit tumor growth. Cancer Discov. 2015, 5, 182–197. [Google Scholar] [CrossRef] [PubMed]
- Moellering, R.E.; Cornejo, M.; Davis, T.N.; Del Bianco, C.; Aster, J.C.; Blacklow, S.C.; Kung, A.L.; Gilliland, D.G.; Verdine, G.L.; Bradner, J.E. Direct inhibition of the NOTCH transcription factor complex. Nature 2009, 462, 182–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Astudillo, L.; Da Silva, T.G.; Wang, Z.; Han, X.; Jin, K.; VanWye, J.; Zhu, X.; Weaver, K.; Oashi, T.; Lopes, P.E.; et al. The Small Molecule IMR-1 Inhibits the Notch Transcriptional Activation Complex to Suppress Tumorigenesis. Cancer Res. 2016, 76, 3593–3603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krämer, A.; Mentrup, T.; Kleizen, B.; Rivera-Milla, E.; Reichenbach, D.; Enzensperger, C.; Nohl, R.; Täuscher, E.; Görls, H.; Ploubidou, A.; et al. Small molecules intercept Notch signaling and the early secretory pathway. Nat. Chem. Biol. 2013, 9, 731–738. [Google Scholar] [CrossRef] [PubMed]
- Roti, G.; Carlton, A.; Ross, K.N.; Markstein, M.; Pajcini, K.; Su, A.H.; Perrimon, N.; Pear, W.S.; Kung, A.L.; Blacklow, S.C.; et al. Complementary genomic screens identify SERCA as a therapeutic target in NOTCH1 mutated cancer. Cancer Cell 2013, 23, 390–405. [Google Scholar] [CrossRef] [PubMed]
- Pandya, K.; Meeke, K.; Clementz, A.G.; Rogowski, A.; Roberts, J.; Miele, L.; Albain, K.S.; Osipo, C. Targeting both Notch and ErbB-2 signalling pathways is required for prevention of ErbB-2-positive breast tumour recurrence. Br. J. Cancer 2011, 105, 796–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schott, A.F.; Landis, M.D.; Dontu, G.; Griffith, K.A.; Layman, R.M.; Krop, I.; Paskett, L.A.; Wong, H.; Dobrolecki, L.E.; Lewis, M.T.; et al. Preclinical and clinical studies of gamma secretase inhibitors with docetaxel on human breast tumors. Clin. Cancer Res. 2013, 19, 1512–1524. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.C.; Yan, Z.; Zong, Q.; Fang, D.D.; Painter, C.; Zhang, Q.; Chen, E.; Lira, M.E.; John-Baptiste, A.; Christensen, J.G. Synergistic effect of the γ-secretase inhibitor PF-03084014 and docetaxel in breast cancer models. Stem Cells Transl. Med. 2013, 2, 233–242. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Francesco, E.M.; Maggiolini, M.; Musti, A.M. Crosstalk between Notch, HIF-1α and GPER in Breast Cancer EMT. Int. J. Mol. Sci. 2018, 19, 2011. https://doi.org/10.3390/ijms19072011
De Francesco EM, Maggiolini M, Musti AM. Crosstalk between Notch, HIF-1α and GPER in Breast Cancer EMT. International Journal of Molecular Sciences. 2018; 19(7):2011. https://doi.org/10.3390/ijms19072011
Chicago/Turabian StyleDe Francesco, Ernestina M., Marcello Maggiolini, and Anna Maria Musti. 2018. "Crosstalk between Notch, HIF-1α and GPER in Breast Cancer EMT" International Journal of Molecular Sciences 19, no. 7: 2011. https://doi.org/10.3390/ijms19072011
APA StyleDe Francesco, E. M., Maggiolini, M., & Musti, A. M. (2018). Crosstalk between Notch, HIF-1α and GPER in Breast Cancer EMT. International Journal of Molecular Sciences, 19(7), 2011. https://doi.org/10.3390/ijms19072011