Implications of Angiogenesis Involvement in Arthritis

 , and
, and
Abstract
:1. Introduction
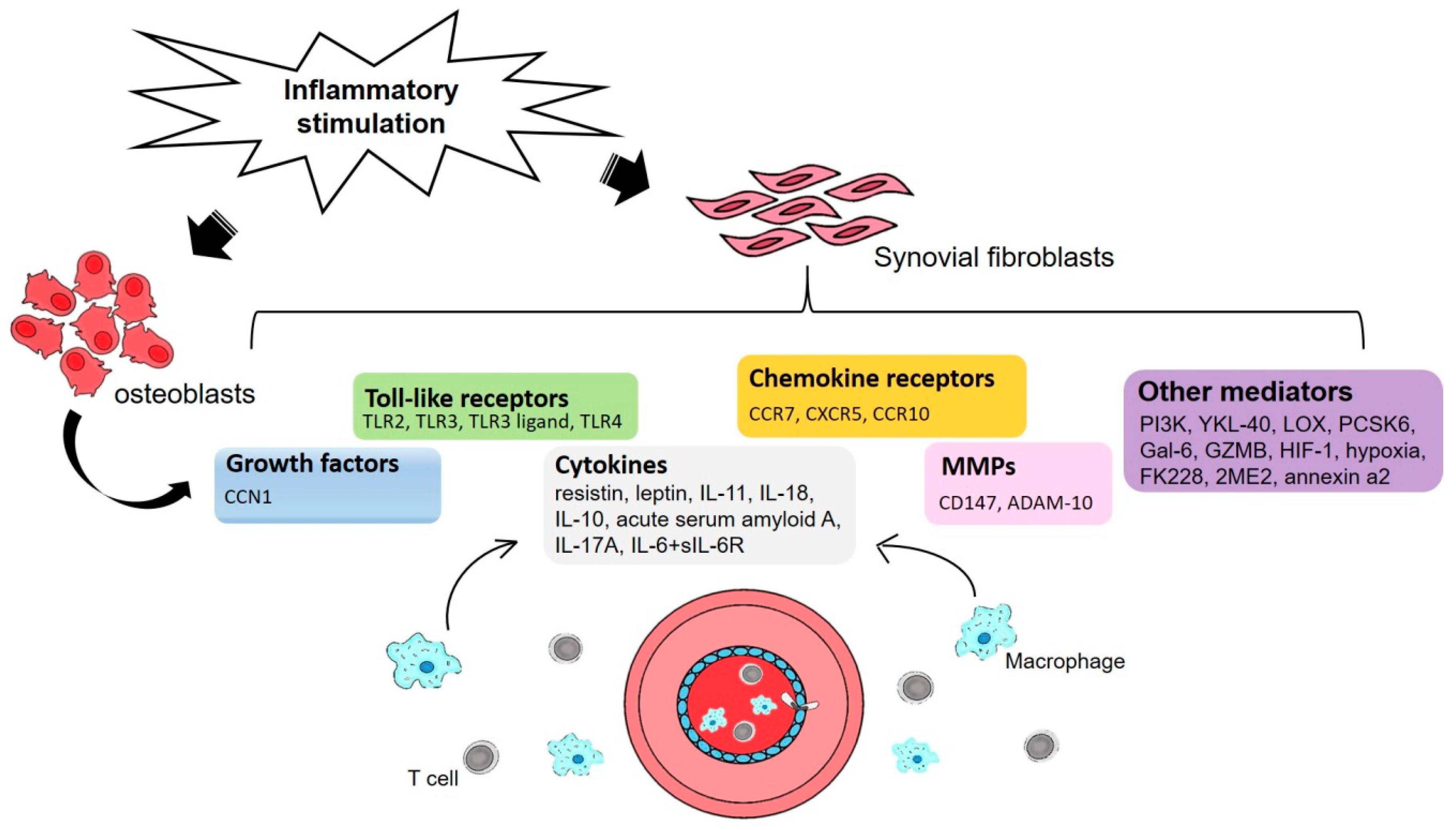
2. Rheumatoid Arthritis
3. The Involvement of Toll-Like Receptors in RA Disease
4. Vasohibin-1 mRNA Expression in RA Synovial Fibroblasts
5. Cytokines Show Angiogenic Activity
TLR2 May Amplify the Effects of Serum Amyloid A
6. Targeting Stromal Cells and Vascular Responses
Possibilities of Stem Cell Therapy and GZMB Gene Silencing
7. Characterizing the Expression and Function of Chemokine Receptors in RA
8. Targeting the MMP Family
9. Chinese Herbal Preparations
10. Other Potentially Targetable Factors That Participate in RA Angiogenesis
Targeting Proinflammatory YKL-40, Cyr61/CCN1, Axna2, and Axna2R
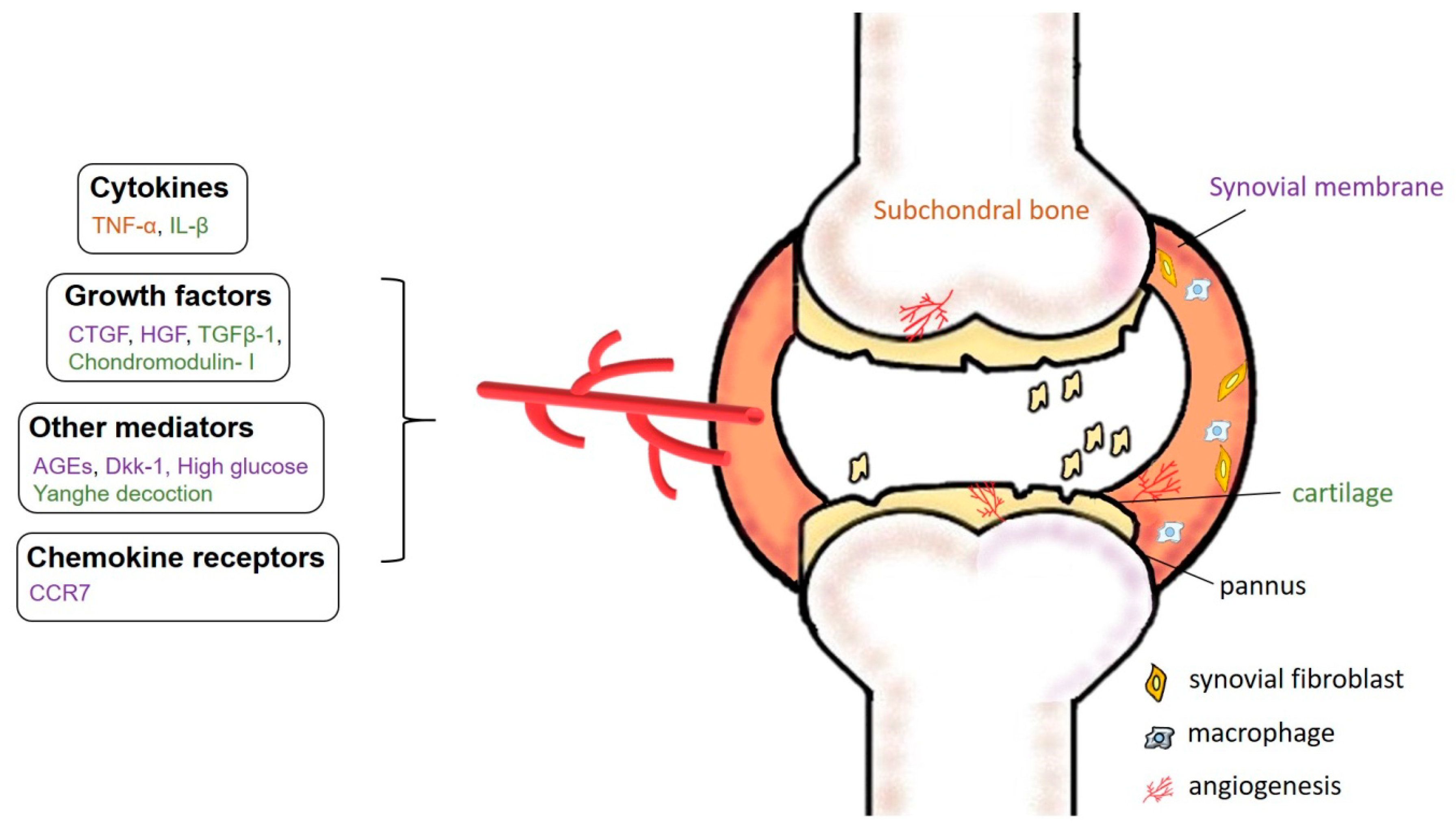
11. Osteoarthritis
12. Angiogenesis in the OA Synovium
13. The Importance of Targeting AGE-Induced Inflammatory Responses
14. Targeting OA Cartilage
15. Subchondral Bone and Articular Cartilage
16. Summary and Future Directions
Funding
Conflicts of Interest
References
- McInnes, I.B.; Schett, G. The pathogenesis of rheumatoid arthritis. N. Engl. J. Med. 2011, 365, 2205–2219. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Ouyang, H.; Dass, C.R.; Xu, J. Current research on pharmacologic and regenerative therapies for osteoarthritis. Bone Res. 2016, 4, 15040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wehling, P.; Evans, C.; Wehling, J.; Maixner, W. Effectiveness of intra-articular therapies in osteoarthritis: A literature review. Ther. Adv. Musculoskelet. Dis. 2017, 9, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Emery, P. Optimizing outcomes in patients with rheumatoid arthritis and an inadequate response to anti-tnf treatment. Rheumatology 2012, 51 (Suppl. 5), v22–v30. [Google Scholar] [CrossRef] [PubMed]
- Kihara, M.; Davies, R.; Kearsley-Fleet, L.; Watson, K.D.; Lunt, M.; Symmons, D.P.; Hyrich, K.L. Use and effectiveness of tocilizumab among patients with rheumatoid arthritis: An observational study from the british society for rheumatology biologics register for rheumatoid arthritis. Clin. Rheumatol. 2017, 36, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.; Nash, P.; Hall, S. Advances in rheumatoid arthritis. Med. J. Aust. 2017, 206, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Wijbrandts, C.A.; Tak, P.P. Prediction of response to targeted treatment in rheumatoid arthritis. Mayo Clin. Proc. 2017, 92, 1129–1143. [Google Scholar] [CrossRef] [PubMed]
- Castaneda, O.M.; Romero, F.J.; Salinas, A.; Citera, G.; Mysler, E.; Rillo, O.; Radominski, S.C.; Cardiel, M.H.; Jaller, J.J.; Alvarez-Moreno, C.; et al. Safety of tofacitinib in the treatment of rheumatoid arthritis in latin america compared with the rest of the world population. J. Clin. Rheumatol. Pract. Rep. Rheum. Musculoskelet. Dis. 2017, 23, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Barclay, N.; Tarallo, M.; Hendrikx, T.; Marett, S. Patient preference for oral versus injectable and intravenous methods of treatment for rheumatoid arthritis. Value Health 2013, 16, A568. [Google Scholar] [CrossRef]
- Elshabrawy, H.A.; Chen, Z.; Volin, M.V.; Ravella, S.; Virupannavar, S.; Shahrara, S. The pathogenic role of angiogenesis in rheumatoid arthritis. Angiogenesis 2015, 18, 433–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, S.J.; Park, M.K.; Oh, H.J.; Lee, S.Y.; Kwok, S.K.; Cho, M.L.; Ju, J.H.; Park, K.S.; Kim, H.Y.; Park, S.H. Engagement of toll-like receptor 3 induces vascular endothelial growth factor and interleukin-8 in human rheumatoid synovial fibroblasts. Korean J. Intern. Med. 2010, 25, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Saber, T.; Veale, D.J.; Balogh, E.; McCormick, J.; NicAnUltaigh, S.; Connolly, M.; Fearon, U. Toll-like receptor 2 induced angiogenesis and invasion is mediated through the tie2 signalling pathway in rheumatoid arthritis. PLoS ONE 2011, 6, e23540. [Google Scholar] [CrossRef] [PubMed]
- Miyake, K.; Nishida, K.; Kadota, Y.; Yamasaki, H.; Nasu, T.; Saitou, D.; Tanabe, K.; Sonoda, H.; Sato, Y.; Maeshima, Y.; et al. Inflammatory cytokine-induced expression of vasohibin-1 by rheumatoid synovial fibroblasts. Acta Med. Okayama 2009, 63, 349–358. [Google Scholar] [PubMed]
- Manabe, H.; Nasu, Y.; Komiyama, T.; Furumatsu, T.; Kitamura, A.; Miyazawa, S.; Ninomiya, Y.; Ozaki, T.; Asahara, H.; Nishida, K. Inhibition of histone deacetylase down-regulates the expression of hypoxia-induced vascular endothelial growth factor by rheumatoid synovial fibroblasts. Inflamm. Res. Off. J. Eur. Histamine Res. Soc. 2008, 57, 4–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brahn, E.; Banquerigo, M.L.; Lee, J.K.; Park, E.J.; Fogler, W.E.; Plum, S.M. An angiogenesis inhibitor, 2-methoxyestradiol, involutes rat collagen-induced arthritis and suppresses gene expression of synovial vascular endothelial growth factor and basic fibroblast growth factor. J. Rheumatol. 2008, 35, 2119–2128. [Google Scholar] [CrossRef] [PubMed]
- Hashizume, M.; Hayakawa, N.; Suzuki, M.; Mihara, M. Il-6/sil-6r trans-signalling, but not tnf-alpha induced angiogenesis in a huvec and synovial cell co-culture system. Rheumatol. Int. 2009, 29, 1449–1454. [Google Scholar] [CrossRef] [PubMed]
- Moran, E.M.; Connolly, M.; Gao, W.; McCormick, J.; Fearon, U.; Veale, D.J. Interleukin-17a induction of angiogenesis, cell migration, and cytoskeletal rearrangement. Arthritis Rheum. 2011, 63, 3263–3273. [Google Scholar] [CrossRef] [PubMed]
- Amin, M.A.; Rabquer, B.J.; Mansfield, P.J.; Ruth, J.H.; Marotte, H.; Haas, C.S.; Reamer, E.N.; Koch, A.E. Interleukin 18 induces angiogenesis in vitro and in vivo via src and jnk kinases. Ann. Rheum. Dis. 2010, 69, 2204–2212. [Google Scholar] [CrossRef] [PubMed]
- Kobori, T.; Hamasaki, S.; Kitaura, A.; Yamazaki, Y.; Nishinaka, T.; Niwa, A.; Nakao, S.; Wake, H.; Mori, S.; Yoshino, T.; et al. Interleukin-18 amplifies macrophage polarization and morphological alteration, leading to excessive angiogenesis. Front. Immunol. 2018, 9, 334. [Google Scholar] [CrossRef] [PubMed]
- Elshabrawy, H.A.; Volin, M.V.; Essani, A.B.; Chen, Z.; McInnes, I.B.; Van Raemdonck, K.; Palasiewicz, K.; Arami, S.; Gonzalez, M.; Ashour, H.M.; et al. Il-11 facilitates a novel connection between ra joint fibroblasts and endothelial cells. Angiogenesis 2018, 21, 215–228. [Google Scholar] [CrossRef] [PubMed]
- Connolly, M.; Marrelli, A.; Blades, M.; McCormick, J.; Maderna, P.; Godson, C.; Mullan, R.; FitzGerald, O.; Bresnihan, B.; Pitzalis, C.; et al. Acute serum amyloid a induces migration, angiogenesis, and inflammation in synovial cells in vitro and in a human rheumatoid arthritis/scid mouse chimera model. J. Immunol. 2010, 184, 6427–6437. [Google Scholar] [CrossRef] [PubMed]
- Su, C.M.; Hsu, C.J.; Tsai, C.H.; Huang, C.Y.; Wang, S.W.; Tang, C.H. Resistin promotes angiogenesis in endothelial progenitor cells through inhibition of microrna206: Potential implications for rheumatoid arthritis. Stem Cells 2015, 33, 2243–2255. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Wei, J.; Tang, Y.; Wang, B.; Zhang, Y.; Shi, L.; Guo, J.; Hu, F.; Li, X. Leptin-induced migration and angiogenesis in rheumatoid arthritis is mediated by reactive oxygen species. FEBS Open Bio 2017, 7, 1899–1908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chien, S.Y.; Huang, C.Y.; Tsai, C.H.; Wang, S.W.; Lin, Y.M.; Tang, C.H. Interleukin-1beta induces fibroblast growth factor 2 expression and subsequently promotes endothelial progenitor cell angiogenesis in chondrocytes. Clin. Sci. 2016, 130, 667–681. [Google Scholar] [CrossRef] [PubMed]
- del Rey, M.J.; Izquierdo, E.; Caja, S.; Usategui, A.; Santiago, B.; Galindo, M.; Pablos, J.L. Human inflammatory synovial fibroblasts induce enhanced myeloid cell recruitment and angiogenesis through a hypoxia-inducible transcription factor 1alpha/vascular endothelial growth factor-mediated pathway in immunodeficient mice. Arthritis Rheum. 2009, 60, 2926–2934. [Google Scholar] [CrossRef] [PubMed]
- Ansboro, S.; Roelofs, A.J.; De Bari, C. Mesenchymal stem cells for the management of rheumatoid arthritis: Immune modulation, repair or both? Curr. Opin. Rheumatol. 2017, 29, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Akhavani, M.A.; Madden, L.; Buysschaert, I.; Sivakumar, B.; Kang, N.; Paleolog, E.M. Hypoxia upregulates angiogenesis and synovial cell migration in rheumatoid arthritis. Arthritis Res. Ther. 2009, 11, R64. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, J.; Wan, L.; Sun, Y.; Wang, F.; Qi, Y.; Huang, C. Up-regulated expressions of hif-1alpha, vegf and cd34 promote synovial angiogenesis in rats with adjuvant arthritis. Chin. J. Cell. Mol. Immunol. 2015, 31, 1053–1056. [Google Scholar]
- Nam, Y.; Jung, S.M.; Rim, Y.A.; Jung, H.; Lee, K.; Park, N.; Kim, J.; Jang, Y.; Park, Y.B.; Park, S.H.; et al. Intraperitoneal infusion of mesenchymal stem cell attenuates severity of collagen antibody induced arthritis. PLoS ONE 2018, 13, e0198740. [Google Scholar] [CrossRef] [PubMed]
- Bao, C.X.; Chen, H.X.; Mou, X.J.; Zhu, X.K.; Zhao, Q.; Wang, X.G. Gzmb gene silencing confers protection against synovial tissue hyperplasia and articular cartilage tissue injury in rheumatoid arthritis through the mapk signaling pathway. Biomed. Pharmacother. 2018, 103, 346–354. [Google Scholar] [CrossRef] [PubMed]
- O'Brien, M.J.; Shu, Q.; Stinson, W.A.; Tsou, P.S.; Ruth, J.H.; Isozaki, T.; Campbell, P.L.; Ohara, R.A.; Koch, A.E.; Fox, D.A.; et al. A unique role for galectin-9 in angiogenesis and inflammatory arthritis. Arthritis Res. Ther. 2018, 20, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruhl, H.; Mack, M.; Niedermeier, M.; Lochbaum, D.; Scholmerich, J.; Straub, R.H. Functional expression of the chemokine receptor ccr7 on fibroblast-like synoviocytes. Rheumatology 2008, 47, 1771–1774. [Google Scholar] [CrossRef] [PubMed]
- Wengner, A.M.; Hopken, U.E.; Petrow, P.K.; Hartmann, S.; Schurigt, U.; Brauer, R.; Lipp, M. Cxcr5- and ccr7-dependent lymphoid neogenesis in a murine model of chronic antigen-induced arthritis. Arthritis Rheum. 2007, 56, 3271–3283. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Kim, S.J.; Essani, A.B.; Volin, M.V.; Vila, O.M.; Swedler, W.; Arami, S.; Volkov, S.; Sardin, L.V.; Sweiss, N.; et al. Characterising the expression and function of ccl28 and its corresponding receptor, ccr10, in ra pathogenesis. Ann. Rheum. Dis. 2015, 74, 1898–1906. [Google Scholar] [CrossRef] [PubMed]
- Pickens, S.R.; Chamberlain, N.D.; Volin, M.V.; Pope, R.M.; Mandelin, A.M., 2nd; Shahrara, S. Characterization of ccl19 and ccl21 in rheumatoid arthritis. Arthritis Rheum. 2011, 63, 914–922. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.H.; Yao, H.; Chen, L.N.; Jia, J.F.; Wang, L.; Dai, J.Y.; Zheng, Z.H.; Chen, Z.N.; Zhu, P. Cd147 induces angiogenesis through a vascular endothelial growth factor and hypoxia-inducible transcription factor 1alpha-mediated pathway in rheumatoid arthritis. Arthritis Rheum. 2012, 64, 1818–1827. [Google Scholar] [CrossRef] [PubMed]
- Zong, J.; Li, Y.; Du, D.; Liu, Y.; Yin, Y. Cd147 induces up-regulation of vascular endothelial growth factor in u937-derived foam cells through pi3k/akt pathway. Arch. Biochem. Biophys. 2016, 609, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Isozaki, T.; Rabquer, B.J.; Ruth, J.H.; Haines, G.K., 3rd; Koch, A.E. Adam-10 is overexpressed in rheumatoid arthritis synovial tissue and mediates angiogenesis. Arthritis Rheum. 2013, 65, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Pan, R.; Dai, Y.; Gao, X.; Xia, Y. Scopolin isolated from erycibe obtusifolia benth stems suppresses adjuvant-induced rat arthritis by inhibiting inflammation and angiogenesis. Int. Immunopharmacol. 2009, 9, 859–869. [Google Scholar] [CrossRef] [PubMed]
- Deng, Q.; Bai, S.; Gao, W.; Tong, L. Pristimerin inhibits angiogenesis in adjuvant-induced arthritic rats by suppressing vegfr2 signaling pathways. Int. Immunopharmacol. 2015, 29, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Shankar, J.; Thippegowda, P.B.; Kanum, S.A. Inhibition of hif-1alpha activity by bp-1 ameliorates adjuvant induced arthritis in rats. Biochem. Biophys. Res. Commun. 2009, 387, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Wang, L.; Wang, F.; Pan, J. Proprotein convertase subtilisin/kexin type 6 promotes in vitro proliferation, migration and inflammatory cytokine secretion of synovial fibroblastlike cells from rheumatoid arthritis via nuclearkappab, signal transducer and activator of transcription 3 and extracellular signal regulated 1/2 pathways. Mol. Med. Rep. 2017, 16, 8477–8484. [Google Scholar] [PubMed]
- Wang, F.; Wan, J.; Li, Q.; Zhang, M.; Wan, Q.; Ji, C.; Li, H.; Liu, R.; Han, M. Lysyl oxidase is involved in synovial hyperplasia and angiogenesis in rats with collageninduced arthritis. Mol. Med. Rep. 2017, 16, 6736–6742. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Hong, C.; Wang, Y.; Liu, J.; Zhang, N.; Shen, C.; Wei, W.; Zheng, F. Calreticulin promotes angiogenesis via activating nitric oxide signalling pathway in rheumatoid arthritis. Clin. Exp. Immunol. 2014, 178, 236–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.M.; Liu, S.C.; Huang, Y.H.; Huang, C.C.; Hsu, C.J.; Tsai, C.H.; Wang, S.W.; Tang, C.H. Ykl-40-induced inhibition of mir-590-3p promotes interleukin-18 expression and angiogenesis of endothelial progenitor cells. Int. J. Mol. Sci. 2017, 18, 920. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Y.; Su, C.M.; Hsu, C.J.; Huang, C.C.; Wang, S.W.; Liu, S.C.; Chen, W.C.; Fuh, L.J.; Tang, C.H. Ccn1 promotes vegf production in osteoblasts and induces endothelial progenitor cell angiogenesis by inhibiting mir-126 expression in rheumatoid arthritis. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2017, 32, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.; Zhu, Y.; Jia, Y.; Jiang, H.; Zheng, X.; Liu, D.; Gao, S.; Sun, M.; Hu, B.; Jiao, B.; et al. The annexin a2 promotes development in arthritis through neovascularization by amplification hedgehog pathway. PLoS ONE 2016, 11, e0150363. [Google Scholar] [CrossRef] [PubMed]
- Robinson, W.H.; Lepus, C.M.; Wang, Q.; Raghu, H.; Mao, R.; Lindstrom, T.M.; Sokolove, J. Low-grade inflammation as a key mediator of the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol. 2016, 12, 580–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazzerini, P.E.; Capecchi, P.L.; Laghi-Pasini, F. Systemic inflammation and arrhythmic risk: Lessons from rheumatoid arthritis. Eur. Heart J. 2017, 38, 1717–1727. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.C.; Chuang, S.M.; Hsu, C.J.; Tsai, C.H.; Wang, S.W.; Tang, C.H. Ctgf increases vascular endothelial growth factor-dependent angiogenesis in human synovial fibroblasts by increasing mir-210 expression. Cell Death Dis. 2014, 5, e1485. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.M.; Huang, Y.L.; Fong, Y.C.; Tsai, C.H.; Chou, M.C.; Tang, C.H. Hepatocyte growth factor increases vascular endothelial growth factor-a production in human synovial fibroblasts through c-met receptor pathway. PLoS ONE 2012, 7, e50924. [Google Scholar] [CrossRef] [PubMed]
- Weng, L.H.; Ko, J.Y.; Wang, C.J.; Sun, Y.C.; Wang, F.S. Dkk-1 promotes angiogenic responses and cartilage matrix proteinase secretion in synovial fibroblasts from osteoarthritic joints. Arthritis Rheum. 2012, 64, 3267–3277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.J.; Chan, D.C.; Chiang, C.K.; Wang, C.C.; Yang, T.H.; Lan, K.C.; Chao, S.C.; Tsai, K.S.; Yang, R.S.; Liu, S.H. Advanced glycation end-products induced vegf production and inflammatory responses in human synoviocytes via rage-nf-kappab pathway activation. J. Orthop. Res. Off. Publ. Orthop. Res. Soc. 2016, 34, 791–800. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.H.; Chiang, Y.C.; Chen, H.T.; Huang, P.H.; Hsu, H.C.; Tang, C.H. High glucose induces vascular endothelial growth factor production in human synovial fibroblasts through reactive oxygen species generation. Biochim. Biophys. Acta 2013, 1830, 2649–2658. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.L.; Zou, C.; Chen, Y.; Zhu, W.; Liu, W.; Huang, J.; Liu, Q.; Wang, D.; Duan, L.; Xiong, J.; et al. Tgfbeta1 induces hypertrophic change and expression of angiogenic factors in human chondrocytes. Oncotarget 2017, 8, 91316–91327. [Google Scholar] [PubMed]
- Deng, B.; Chen, C.; Gong, X.; Guo, L.; Chen, H.; Yin, L.; Yang, L.; Wang, F. Chondromodulini expression and correlation with angiogenesis in human osteoarthritic cartilage. Mol. Med. Rep. 2017, 16, 2142–2148. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.A.; McWilliams, D.F.; Turley, M.J.; Dixon, M.R.; Franses, R.E.; Mapp, P.I.; Wilson, D. Angiogenesis and nerve growth factor at the osteochondral junction in rheumatoid arthritis and osteoarthritis. Rheumatology 2010, 49, 1852–1861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashraf, S.; Wibberley, H.; Mapp, P.I.; Hill, R.; Wilson, D.; Walsh, D.A. Increased vascular penetration and nerve growth in the meniscus: A potential source of pain in osteoarthritis. Ann. Rheum. Dis. 2011, 70, 523–529. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, J.; Zhang, X.; Wang, C.; Huang, Y.; Dai, K. Tnf-alpha-induced lrg1 promotes angiogenesis and mesenchymal stem cell migration in the subchondral bone during osteoarthritis. Cell Death Dis. 2017, 8, e2715. [Google Scholar] [CrossRef] [PubMed]
- Kuyinu, E.L.; Narayanan, G.; Nair, L.S.; Laurencin, C.T. Animal models of osteoarthritis: Classification, update, and measurement of outcomes. J. Orthop. Surg. Res. 2016, 11, 19. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.W.; Chen, Y.Q. Effects of yanghe decoction on vascular endothelial growth factor in cartilage cells of osteoarthritis rabbits. J. Chin. Integr. Med. 2008, 6, 372–375. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G.; Schlossman, R.L.; Weller, E.; Hideshima, T.; Mitsiades, C.; Davies, F.; LeBlanc, R.; Catley, L.P.; Doss, D.; Kelly, K.; et al. Immunomodulatory drug cc-5013 overcomes drug resistance and is well tolerated in patients with relapsed multiple myeloma. Blood 2002, 100, 3063–3067. [Google Scholar] [CrossRef] [PubMed]
- Davies, F.E.; Raje, N.; Hideshima, T.; Lentzsch, S.; Young, G.; Tai, Y.T.; Lin, B.; Podar, K.; Gupta, D.; Chauhan, D.; et al. Thalidomide and immunomodulatory derivatives augment natural killer cell cytotoxicity in multiple myeloma. Blood 2001, 98, 210–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, B.; Yu, R.L.; Chi, X.H.; Lu, X.C. Lenalidomide treatment for multiple myeloma: Systematic review and meta-analysis of randomized controlled trials. PLoS ONE 2013, 8, e64354. [Google Scholar] [CrossRef] [PubMed]
- Bridoux, F.; Chen, N.; Moreau, S.; Arnulf, B.; Moumas, E.; Abraham, J.; Desport, E.; Jaccard, A.; Fermand, J.P. Pharmacokinetics, safety, and efficacy of lenalidomide plus dexamethasone in patients with multiple myeloma and renal impairment. Cancer Chemother. Pharmacol. 2016, 78, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Leuci, V.; Maione, F.; Rotolo, R.; Giraudo, E.; Sassi, F.; Migliardi, G.; Todorovic, M.; Gammaitoni, L.; Mesiano, G.; Giraudo, L.; et al. Lenalidomide normalizes tumor vessels in colorectal cancer improving chemotherapy activity. J. Transl. Med. 2016, 14, 119. [Google Scholar] [CrossRef] [PubMed]
- Luz-Crawford, P.; Ipseiz, N.; Espinosa-Carrasco, G.; Caicedo, A.; Tejedor, G.; Toupet, K.; Loriau, J.; Scholtysek, C.; Stoll, C.; Khoury, M.; et al. Pparbeta/delta directs the therapeutic potential of mesenchymal stem cells in arthritis. Ann. Rheum. Dis. 2016, 75, 2166–2174. [Google Scholar] [CrossRef] [PubMed]
- Dold, A.P.; Zywiel, M.G.; Taylor, D.W.; Dwyer, T.; Theodoropoulos, J. Platelet-rich plasma in the management of articular cartilage pathology: A systematic review. Clin. J. Sport Med. Off. J. Can. Acad. Sport Med. 2014, 24, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Tong, S.; Zhang, C.; Liu, J. Platelet-rich plasma exhibits beneficial effects for rheumatoid arthritis mice by suppressing inflammatory factors. Mol. Med. Rep. 2017, 16, 4082–4088. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Stimulation | Target Factors | Effects in Tissue | Known Pathways | References | |
|---|---|---|---|---|---|
| Toll-like receptors | |||||
| Toll-like receptor 3 | VEGF, IL-8 | ↑ | Synovium | NF-κB | [11] |
| Toll-like receptor 2 | Ang2/Tie2 | ↑ | HMVEC | Ang2/Tie2 | [12] |
| Cytokines | |||||
| Resistin | VEGF | ↑ | EPC | PKC AMPK/miR-206 | [22] |
| Leptin | VEGF, IL-8 | ↑ | Synovium | ROS/HIF-1 | [23] |
| IL-11 | VEGF, IL-8 | ↑ | Synovium | N/A | [20] |
| IL-18, IL-10 | OPN | ↑ | M2 macrophage (Mφ) | N/A | [19] |
| Acute serum amyloid A | Synovium | N/A | [21] | ||
| IL-18 | IL-18 | ↑ | HMVEC | Src/JNK | [18] |
| IL-17A | IL-17A | ↑ | HDECs | N/A | [17] |
| IL-6/SL-IL-6R | VEGF | ↑ | Synovium | IL-6/SL-IL-6R | [16] |
| IL-1β | bFGF | ↑ | Cartilage | ROS/AMPK/p38/NF-κB | [24] |
| Chemokine receptors | |||||
| CCR7 | VEGF | ↑ | Synovium | N/A | [32] |
| CCL28 | CCR10 | ↑ | Synovium and EPC | ERK1/2 | [34] |
| CXCR5 | ↑ | CIA model | N/A | [33] | |
| MMPs | |||||
| CD147 | VEGF, HIF-1α | ↑ | Synovium | PI3K/AKT/HIF-1α | [36] |
| ADAM-10 | ADAM-10 | ↑ | Synovium | N/A | [38] |
| Chinese Herbs | |||||
| Pristimerin | VEGF-A/VEGFR2 | ↓ | Synovium | PI3K/AKT/mTOR and MAPK | [40] |
| Scopolin | IL-6, VEGF and FGF-2 | ↓ | Synovium | N/A | [39] |
| Growth factors | |||||
| CCN1 | VEGF-A | ↑ | Osteoblast | PKC/miR-126 | [46] |
| VEGF | vasohibin-1 | ↓ | Synovium | N/A | [13] |
| Other mediators | |||||
| YKL-40 | IL-18 | ↑ | Osteoblast | FAK/PI3K/AKT | [45] |
| Lysyl oxidase (LOX) | MMP-2, MMP-9 | ↑ | Synovium | N/A | [43] |
| PCSK6 | IL-1, IL-1, IL-6 | ↑ | Synovium | NF-B | [42] |
| Galectin-9 | Galectin-9 | ↑ | HMVEC | N/A | [31] |
| GZMB | VEGF and bFGF | ↑ | CIA model | MEK/ERK | [30] |
| Annexin A2 | VEGF, Ang-2, MMP-2 | ↑ | HUVEC | HH signaling | [47] |
| HIF-1α | HIF-1, VEGF, CD34 | ↑ | Synovium | HIF-1α | [28] |
| Hypoxia | VEGF and MMP-2, -8, -9 | ↑ | Synovium | N/A | [27] |
| FK228 (inhibitor) | HIF-1α and VEGF | ↓ | Synovium | HIF-1α | [14] |
| 2ME2 (inhibitor) | VEGF and bFGF | ↓ | Synovium | N/A | [15] |
| BP-1 (inhibitor) | HIF-1α and VEGF | ↓ | Synovium | HIF-1α | [41] |
| Stimulation | Target Factors | Effect in Tissue | Known Pathways | References | |
|---|---|---|---|---|---|
| Growth factors | |||||
| CTGF | VEGF-A | ↑ | Synovium | PI3K/AKT/ERK and NF-B/ELK1 | [50] |
| HGF | VEGF-A | ↑ | Synovium | c-Met/PI3K/Akt and mTORC1 | [51] |
| TGF-β1 | VEGF-A | ↑ | Cartilage | N/A | [55] |
| Chondromodulin-I | Chondromodulin-I | ↑ | Cartilage | [56] | |
| Chemokine receptors | |||||
| CCR7 | VEGF | ↑ | Synovium | N/A | [32] |
| Other mediators | |||||
| High glucose | VEGF-A | ↑ | Synovium | ROS, PI3K, Akt, c-Jun and AP-1 | [54] |
| Dkk-1 | Dkk-1 | ↑ | Synovium | β-catenin– and ERK-dependent | [52] |
| AGEs | VEGF-A | ↑ | Synovium | RAGE-NF-κB pathway | [53] |
| Cytokines | |||||
| TNF-α | LRG1 | ↑ | Subchondral bone | p38/ NF-κB | [59] |
| IL-1β | bFGF | ↑ | Cartilage | ROS/AMPK/p38/NF-κB | [24] |
| Chinese Herbs | |||||
| Yanghe Decoction | VEGF-A | ↓ | Cartilage | N/A | [61] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MacDonald, I.J.; Liu, S.-C.; Su, C.-M.; Wang, Y.-H.; Tsai, C.-H.; Tang, C.-H. Implications of Angiogenesis Involvement in Arthritis. Int. J. Mol. Sci. 2018, 19, 2012. https://doi.org/10.3390/ijms19072012
MacDonald IJ, Liu S-C, Su C-M, Wang Y-H, Tsai C-H, Tang C-H. Implications of Angiogenesis Involvement in Arthritis. International Journal of Molecular Sciences. 2018; 19(7):2012. https://doi.org/10.3390/ijms19072012
Chicago/Turabian StyleMacDonald, Iona J., Shan-Chi Liu, Chen-Ming Su, Yu-Han Wang, Chun-Hao Tsai, and Chih-Hsin Tang. 2018. "Implications of Angiogenesis Involvement in Arthritis" International Journal of Molecular Sciences 19, no. 7: 2012. https://doi.org/10.3390/ijms19072012
APA StyleMacDonald, I. J., Liu, S. -C., Su, C. -M., Wang, Y. -H., Tsai, C. -H., & Tang, C. -H. (2018). Implications of Angiogenesis Involvement in Arthritis. International Journal of Molecular Sciences, 19(7), 2012. https://doi.org/10.3390/ijms19072012