Mitochondrial Dysfunctions in Type I Endometrial Carcinoma: Exploring Their Role in Oncogenesis and Tumor Progression

 ,
,  , ,
, ,  ,
,  and
and
Abstract
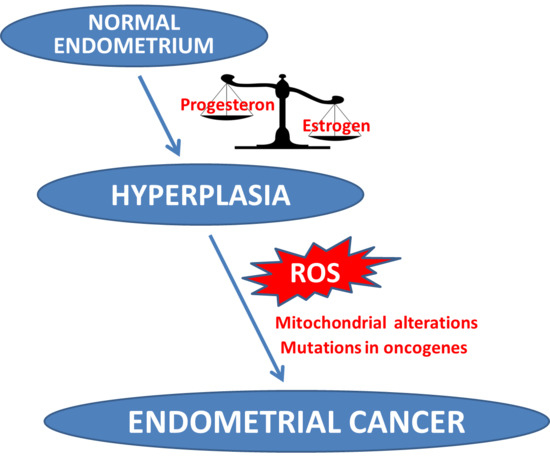
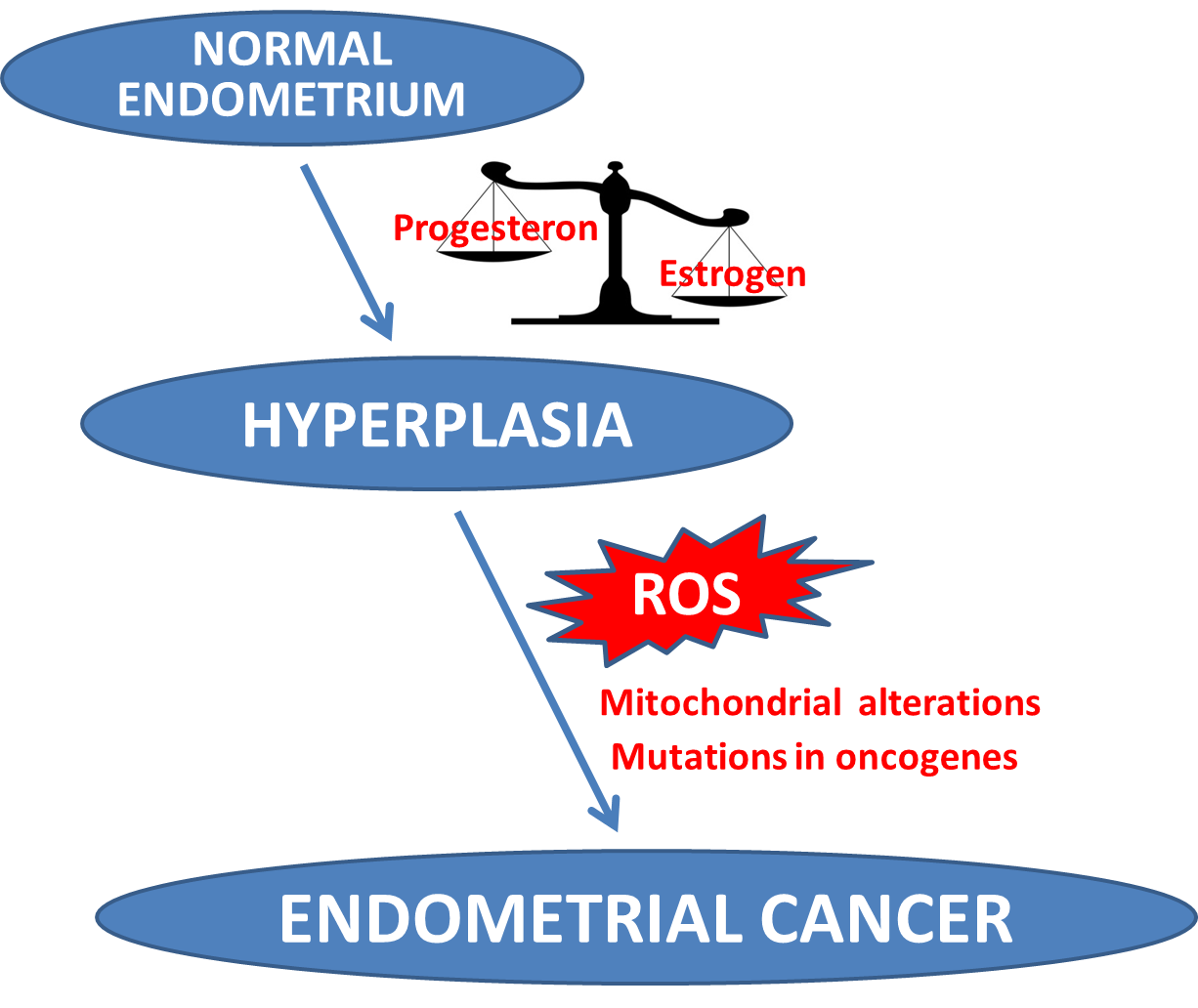
:
1. Introduction
2. MtDNA Mutations in EC Development and Progression
3. Deficit of Respiratory Complex I in Type I EC
4. Mitochondrial Biogenesis Increase in Hyperplasia and Type I EC
5. Activation of Antioxidant Response in Type I EC
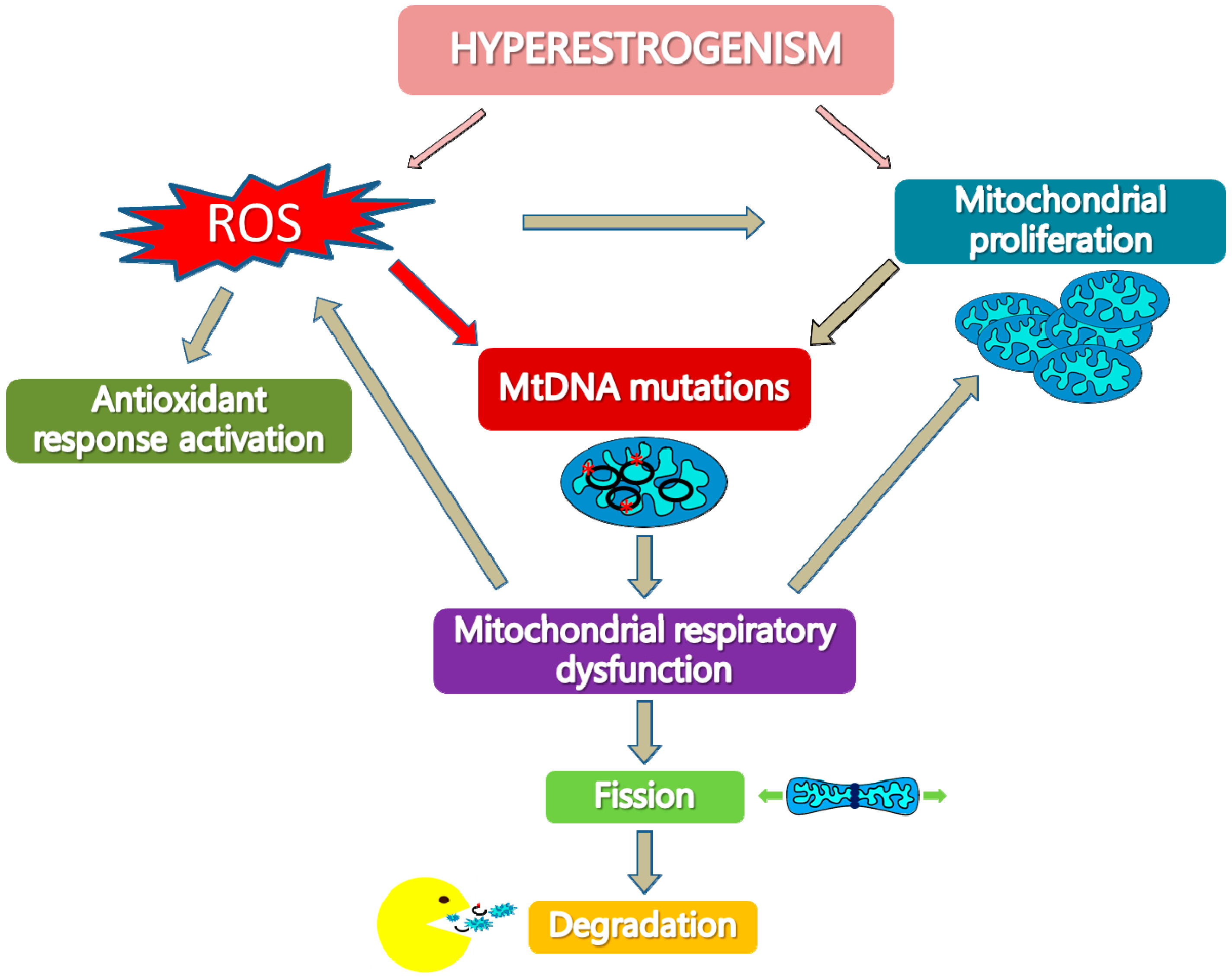
6. Activation of The Mitochondrial Quality Control Systems in Type I EC
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.S. Calcium, ATP and ROS: A mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef] [PubMed]
- Cadenas, E.; Davies, K. Mitochondrial free radical generation, oxidative stress and aging. Free Radic. Biol. Med. 2000, 29, 222–230. [Google Scholar] [CrossRef]
- Attardi, G. Animal mitochondrial DNA: An extreme example of genetic economy. Int. Rev. Cytol. 1985, 93, 93–145. [Google Scholar] [PubMed]
- Lightowlers, R.N.; Chinnery, P.F.; Turnbull, D.M.; Howell, N. Mammalian mitochondrial genetics: Heredity, heteroplasmy and disease. Trends Genet. 1997, 13, 450–455. [Google Scholar] [CrossRef]
- Johns, D.R. Paternal transmission of mitochondrial DNA is (fortunately) rare. Ann. Neurol. 2003, 54, 422–424. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Butow, R.A. The organization and inheritance of the mitochondrial genome. Nat. Rev. Genet. 2005, 6, 815–825. [Google Scholar] [CrossRef] [PubMed]
- DiMauro, S.; Tanji, K.; Bonilla, E.; Pallotti, F.; Schon, E.A. Mitochondrial abnormalities in muscle and other aging cells: Classification, causes and effects. Muscle Nerve 2002, 26, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Moraes, C.T.; Ciacci, F.; Silvestri, G.; Shanske, S.; Sciacco, M.; Hirano, M.; Schon, EA.; Bonilla, E.; DiMauro, S. Atypical clinical presentations associated with the MELAS mutation at position 3243 of human mitochondrial DNA. Neuromuscul. Disord. 1993, 3, 43–50. [Google Scholar] [CrossRef]
- Matthews, P.M.; Hopkin, J.; Brown, R.M.; Stephenson, J.B.; Hilton-Jones, D.; Brown, G.K. Comparison of the relative levels of the 3243 (A-->G) mtDNA mutation in heteroplasmic adult and fetal tissues. J. Med. Genet. 1994, 31, 41–44. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, R.; Faustin, B.; Rocher, C.; Malgat, M.; Mazat, J.P.; Letellier, T. Mitochondrial threshold effects. Biochem. J. 2003, 370, 751–762. [Google Scholar] [CrossRef] [PubMed]
- Torroni, A.; Achilli, A.; Macaulay, V.; Richards, M.; Bandelt, H.J. Harvesting the fruit of the human mtDNA tree. Trends Genet. 2006, 22, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Bioenergetics in human evolution and disease: Implications for the origins of biological complexity and the missing genetic variation of common diseases. Philos. Trans. R. Soc. Lond: Biol. Sci. 2013, 368, 2012–2067. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.M.; Jung, Y.K. A Molecular Approach to Mitophagy and Mitochondrial Dynamics. Mol. Cells 2018, 41, 18–26. [Google Scholar] [PubMed]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [PubMed]
- Gentric, G.; Mieulet, V.; Mechta-Grigoriou, F. Heterogeneity in Cancer Metabolism: New Concepts in an Old Field. Antioxid. Redox Signal. 2017, 26, 462–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz-Bermúdez, A.; Vicente-Blanco, R.J.; Gonzalez-Vioque, E.; Provencio, M.; Fernández-Moreno, M.Á.; Garesse, R. Spotlight on the relevance of mtDNA in cancer. Clin. Transl. Oncol. 2017, 19, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.S.; Patel, J.; Wise, D.R.; Abdel-Wahab, O.; Bennett, B.D.; Coller, H.A.; Cross, J.R.; Fantin, V.R.; Hedvat, C.V.; Perl, A.E.; et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010, 17, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Chandra, D.; Singh, K.K. Genetic insights into OXPHOS defect and its role in cancer. Biochim. Biophys. Acta 2011, 1807, 620–625. [Google Scholar] [CrossRef] [PubMed]
- Picaud, S.; Kavanagh, K.L.; Yue, W.W.; Lee, W.H.; Muller-Knapp, S.; Gileadi, O.; Sacchettini, J.; Oppermann, U. Structural basis of fumarate hydratase deficiency. J. Inherit. Metab. Dis. 2011, 34, 671–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bardella, C.; Pollard, P.; Tomlinson, I. SDH mutations in cancer. Biochim. Biophys. Acta 2011, 1807, 1432–1443. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Heredia, J.M.; Carnero, A. Decoding Warburg’s hypothesis: Tumor-related mutations in the mitochondrial respiratory chain. Oncotarget 2015, 6, 41582–41599. [Google Scholar] [CrossRef] [PubMed]
- Burke, W.M.; Orr, J.; Leitao, M.; Salom, E.; Gehrig, P.; Olawaiye, A.B.; Brewer, M.; Boruta, D.; Villella, J.; Herzog, T.; et al. Endometrial Cancer: A review and current management strategies: Part I. SGO Clinical Practice Endometrial Cancer Working Group. Gynecol. Oncol. 2014, 134, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Hecht, J.L.; Mutter, G.L. Molecular and pathologic aspects of endometrial carcinogenesis. J. Clin. Oncol. 2006, 24, 4783–4791. [Google Scholar] [CrossRef] [PubMed]
- Matias-Guiu, X.; Catasus, L.; Bussaglia, E.; Lagarda, H.; Garcia, A.; Pons, C.; Muñoz, J.; Argüelles, R.; Machin, P.; Prat, J. Molecular pathology of endometrial hyperplasia and carcinoma. Hum. Pathol. 2001, 32, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Cormio, A.; Cormio, G.; Musicco, C.; Sardanelli, A.M.; Gasparre, G.; Gadaleta, M.N. Mitochondrial changes in endometrial carcinoma: Possible role in tumor diagnosis and prognosis. Oncol. Rep. 2015, 33, 1011–1018. [Google Scholar] [CrossRef] [PubMed]
- Klinge, C.M. Estrogens regulate life and death in mitochondria. J. Bioenerg. Biomembr. 2017, 49, 307–324. [Google Scholar] [CrossRef] [PubMed]
- Van Gisbergen, M.W.; Voets, A.M.; Starmans, M.H.; de Coo, I.F.; Yadak, R.; Hoffmann, R.F.; Boutros, P.C.; Smeets, H.J.; Dubois, L.; Lambin, P. How do changes in the mtDNA and mitochondrial dysfunction influence cancer and cancer therapy? Challenges, opportunities and models. Mutat. Res. Rev. 2015, 764, 16–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandon, M.; Baldi, P.; Wallace, D.C. Mitochondrial mutations in cancer. Oncogene 2006, 25, 4647–4662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, V.W.; Wang, Y.; Yang, H.J.; Tsang, P.C.; Ng, T.Y.; Wong, L.C.; Nagley, P.; Ngan, H.Y. Mitochondrial DNA variant 16189T>C is associated with susceptibility to endometrial cancer. Hum. Mutat. 2003, 22, 173–174. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Hu, Y.; Chen, B.; Tang, W.; Han, X.; Yu, H.; Xiao, C. Mitochondrial polymorphisms as risk factors for endometrial cancer in southwest China. Int. J. Gynecol. Cancer 2006, 16, 1661–1667. [Google Scholar] [CrossRef] [PubMed]
- Czarnecka, A.M.; Klemba, A.; Semczuk, A.; Plak, K.; Marzec, B.; Krawczyk, T.; Kofler, B.; Golik, P.; Bartnik, E. Common mitochondrial polymorphisms as risk factor for endometrial cancer. Int. Arch. Med. 2009, 2, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, V.W.; Yang, H.J.; Wang, Y.; Tsang, P.C.; Cheung, A.N.; Chiu, P.M.; Ng, T.Y.; Wong, L.C.; Nagley, P.; Ngan, H.Y. High frequency of mitochondrial genome instability in human endometrial carcinomas. Br. J. Cancer 2003, 89, 697–701. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xue, W.C.; Liu, V.W.; Ngan, H.Y. Detection of mosaic pattern of mitochondrial DNA alterations in different populations of cells from the same endometrial tumor. Mitochondrion 2007, 7, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, V.W.; Tsang, P.C.; Chiu, P.M.; Cheung, A.N.; Khoo, U.S.; Nagley, P.; Ngan, H.Y. Microsatellite instability in mitochondrial genome of common female cancers. Int. J. Gynecol. Cancer 2006, 16, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Parrella, P.; Seripa, D.; Matera, M.G.; Rabitti, C.; Rinaldi, M.; Mazzarelli, P.; Gravina, C.; Gallucci, M.; Altomare, V.; Flammia, G.; et al. Mutations of the D310 mitochondrial mononucleotide repeat in primary tumors and cytological specimens. Cancer Lett. 2003, 190, 73–77. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, V.W.; Ngan, H.Y.; Nagley, P. Frequent occurrence of mitochondrial microsatellite instability in the D-loop region of human cancers. Ann. N. Y. Acad. Sci. 2005, 1042, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Guerra, F.; Girolimetti, G.; Perrone, A.M.; Procaccini, M.; Kurelac, I.; Ceccarelli, C.; de Biase, D.; Caprara, G.; Zamagni, C.; de Iaco, P.; et al. Mitochondrial DNA genotyping efficiently reveals clonality of synchronous endometrial and ovarian cancers. Mod. Pathol. 2014, 27, 1412–1420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerra, F.; Kurelac, I.; Cormio, A.; Zuntini, R.; Amato, L.B.; Ceccarelli, C.; Santini, D.; Cormio, G.; Fracasso, F.; Selvaggi, L.; et al. Placing mitochondrial DNA mutations within the progression model of type I endometrial carcinoma. Hum. Mol. Genet. 2011, 20, 2394–2405. [Google Scholar] [Green Version]
- Semczuk, A.; Lorenc, A.; Putowski, L.; Futyma, K.; Bryk, J.; Miotla, P.; Bartnik, E. Clinicoprognostical features of endometrial cancer patients with somatic mtDNA mutations. Oncol. Rep. 2006, 16, 1041–1045. [Google Scholar] [CrossRef] [PubMed]
- Roy, D.; Cai, Q.; Felty, Q.; Narayan, S. Estrogen-induced generation of reactive oxygen and nitrogen species, gene damage, and estrogen-dependent cancers. J. Toxicol. Environ. Health B Crit. Rev. 2007, 10, 235–257. [Google Scholar] [CrossRef] [PubMed]
- Ju, Y.S.; Alexandrov, L.B.; Gerstung, M.; Martincorena, I.; Nik-Zainal, S.; Ramakrishna, M.; Davies, H.R.; Papaemmanuil, E.; Gundem, G.; Shlien, A.; et al. Origins and functional consequences of somatic mitochondrial DNA mutations in human cancer. eLife 2014, 3, e02935. [Google Scholar] [CrossRef] [PubMed]
- Leone, G.; Abla, H.; Gasparre, G.; Porcelli, A.M.; Iommarini, L. The Oncojanus Paradigm of Respiratory Complex I. Genes 2018, 9, 243. [Google Scholar] [CrossRef] [PubMed]
- Guerra, F.; Kurelac, I.; Magini, P.; Cormio, A.; Santini, D.; Ceccarelli, C.; Gasparre, G. Mitochondrial DNA genotyping reveals synchronous nature of simultaneously detected endometrial and ovarian cancers. Gynecol. Oncol. 2011, 122, 457–458. [Google Scholar] [CrossRef] [PubMed]
- Mayr, J.A.; Meierhofer, D.; Zimmermann, F.; Feichtinger, R.; Kögler, C.; Ratschek, M.; Schmeller, N.; Sperl, W.; Kofler, B. Loss of complex I due to mitochondrial DNA mutations in renal oncocytoma. Clin. Cancer Res. 2008, 14, 2270–2275. [Google Scholar] [CrossRef] [PubMed]
- Gasparre, G.; Hervouet, E.; de Laplanche, E.; Demont, J.; Pennisi, L.F.; Colombel, M.; Mège-Lechevallier, F.; Scoazec, J.Y.; Bonora, E.; Smeets, R.; et al. Clonal expansion of mutated mitochondrial DNA is associated with tumor formation and complex I deficiency in the benign renal oncocytoma. Hum. Mol. Genet. 2008, 17, 986–995. [Google Scholar] [CrossRef] [PubMed]
- Gasparre, G.; Iommarini, L.; Porcelli, A.M.; Lang, M.; Ferri, G.G.; Kurelac, I.; Zuntini, R.; Mariani, E.; Pennisi, L.F.; Pasquini, E.; et al. An inherited mitochondrial DNA disruptive mutation shifts to homoplasmy in oncocytic tumor cells. Hum. Mutat. 2009, 30, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Cormio, A.; Musicco, C.; Gasparre, G.; Cormio, G.; Pesce, V.; Sardanelli, A.M.; Gadaleta, M.N. Increase in proteins involved in mitochondrial fission, mitophagy, proteolysis and antioxidant response in type I endometrial cancer as an adaptive response to respiratory complex I deficiency. Biochem. Biophys. Res. Commun. 2017, 491, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Reznik, E.; Wang, Q.; La, K.; Schultz, N.; Sander, C. Mitochondrial respiratory gene expression is suppressed in many cancers. eLife 2017, 6, E21592. [Google Scholar] [CrossRef] [PubMed]
- Scarpulla, R.C. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol. Rev. 2008, 88, 611–638. [Google Scholar] [CrossRef] [PubMed]
- Cormio, A.; Guerra, F.; Cormio, G.; Pesce, V.; Fracasso, F.; Loizzi, V.; Cantatore, P.; Selvaggi, L.; Gadaleta, M.N. The PGC-1alpha-dependent pathway of mitochondrial biogenesis is upregulated in type I endometrial cancer. Biochem. Biophys. Res. Commun. 2009, 390, 1182–1185. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, V.W.; Xue, W.C.; Tsang, P.C.; Cheung, A.N.; Ngan, H.Y. The increase of mitochondrial DNA content in endometrial adenocarcinoma cells: A quantitative study using laser-captured microdissected tissues. Gynecol. Oncol. 2005, 98, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Cormio, A.; Guerra, F.; Cormio, G.; Pesce, V.; Fracasso, F.; Loizzi, V.; Resta, L.; Putignano, G.; Cantatore, P.; Selvaggi, L.E.; et al. Mitochondrial DNA content and mass increase in progression from normal to hyperplastic to cancer endometrium. BMC Res. Notes 2012, 5, 279. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.Q.; Russo, J. Mitochondrial oestrogen receptors and their potential implications in oestrogen carcinogenesis in human breast cancer. J. Nutr. Environ. Med. 2008, 17, 76–89. [Google Scholar] [CrossRef]
- Chen, J.Q.; Cammarata, P.R.; Baines, C.P.; Yager, J.D. Regulation of mitochondrial respiratory chain biogenesis by estrogens/estrogen receptors and physiological, pathological and pharmacological implications. Biochim. Biophys. Acta 2009, 1793, 1540–1570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, A.; Philip, J.; Vinnakota, K.C.; Van den Bergh, F.; Tabima, D.M.; Hacker, T.; Beard, D.A.; Chesler, N.C. Estrogen maintains mitochondrial content and function in the right ventricle of rats with pulmonary hypertension. Physiol. Rep. 2017, 5, e13157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattingly, K.A.; Ivanova, M.M.; Riggs, K.A.; Wickramasinghe, N.S.; Barch, M.J.; Klinge, C.M. Estradiol stimulates transcription of nuclear respiratory factor-1 and increases mitochondrial biogenesis. Mol. Endocrinol. 2008, 22, 609–622. [Google Scholar] [CrossRef] [PubMed]
- Felty, Q.; Xiong, W.C.; Sun, D.; Sarkar, S.; Singh, K.P.; Parkash, J.; Roy, D. Estrogen induced mitochondrial reactive oxygen species as signal-transducing messengers. Biochemistry 2005, 44, 6900–6909. [Google Scholar] [CrossRef] [PubMed]
- Nilsen, J.; Brinton, R.D. Mitochondria as therapeutic targets of estrogen action in the central nervous system. Curr. Drug Targets CNS Neurol. Disord. 2004, 3, 297–313. [Google Scholar] [CrossRef] [PubMed]
- Tajbakhsh, A.; Pasdar, A.; Rezaee, M.; Fazeli, M.; Soleimanpour, S.; Hassanian, S.M.; FarshchiyanYazdi, Z.; Younesi, R.T.; Ferns, G.A.; Avan, A. The current status and perspectives regarding the clinical implication of intracellular calcium in breast cancer. J. Cell. Physiol. 2018, 233, 5623–5641. [Google Scholar] [CrossRef] [PubMed]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef] [PubMed]
- Byun, J.M.; Kim, S.S.; Kim, K.T.; Kang, M.S.; Jeong, D.H.; Lee, D.S.; Jung, E.J.; Kim, Y.N.; Han, J.; Song, I.S.; et al. Overexpression of peroxiredoxin-3 and -5 is a potential biomarker for prognosis in endometrial cancer. Oncol. Lett. 2018, 15, 5111–5118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, I.S.; Jeong, Y.J.; Seo, Y.J.; Byun, J.M.; Kim, Y.N.; Jeong, D.H.; Han, J.; Kim, K.T.; Jang, S.W. Peroxiredoxin 3 maintains the survival of endometrial cancer stem cells by regulating oxidative stress. Oncotarget 2017, 8, 92788–92800. [Google Scholar] [CrossRef] [PubMed]
- Polimeno, L.; Capuano, F.; Marangi, L.C.; Margiotta, M.; Lisowsky, T.; Ierardi, E.; Francavilla, R.; Francavilla, A. The augmenter of liver regeneration induces mitochondrial gene expression in rat liver and enhances oxidative phosphorylation capacity of liver mitochondria. Dig. Liver Dis. 2000, 32, 510–517. [Google Scholar] [CrossRef]
- Polimeno, L.; Pesetti, B.; Lisowsky, T.; Iannone, F.; Resta, L.; Giorgio, F.; Mallamaci, R.; Buttiglione, M.; Santovito, D.; Vitiello, F.; et al. Protective effect of augmenter of liver regeneration on hydrogen peroxide-induced apoptosis in SH-SY5Y human neuroblastoma cells. Free Radic. Res. 2009, 43, 865–875. [Google Scholar] [PubMed]
- Polimeno, L.; Rossi, M.; Mastrodonato, M.; Montagnani, M.; Piscitelli, D.; Pesetti, B.; de Benedictis, L.; Girardi, B.; Resta, L.; Napoli, A.; et al. Augmenter of liver regeneration, a protective factor against ROS-induced oxidative damage in muscle tissue of mitochondrial myopathy affected patients. Int. J. Biochem. Cell. Biol. 2013, 45, 2410–2419. [Google Scholar] [CrossRef] [PubMed]
- Polimeno, L.; Pesetti, B.; de Santis, F.; Resta, L.; Rossi, R.; de Palma, A.; Girardi, B.; Amoruso, A.; Francavilla, A. Decreased expression of the augmenter of liver regeneration results in increased apoptosis and oxidative damage in human-derived glioma cells. Cell. Death Dis. 2012, 3, e289. [Google Scholar] [CrossRef] [PubMed]
- Fuzio, P.; Valletti, A.; Napoli, A.; Napoli, G.; Cormio, G.; Selvaggi, L.; Liuni, S.; Pesole, G.; Maiorano, E.; Perlino, E. Regulation of the expression of CLU isoforms in endometrial proliferative diseases. Int. J. Oncol. 2013, 42, 1929–1944. [Google Scholar] [CrossRef] [PubMed]
- Drake, L.E.; Springer, M.Z.; Poole, L.P.; Kim, C.J.; Macleod, K.F. Expanding perspectives on the significance of mitophagy in cancer. Semin. Cancer Biol. 2017, 47, 110–124. [Google Scholar] [CrossRef] [PubMed]
- Hamon, M.P.; Bulteau, A.L.; Friguet, B. Mitochondrial proteases and protein quality control in ageing and longevity. Ageing Res. Rev. 2015, 23, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Trotta, A.P.; Chipuk, J.E. Mitochondrial dynamics as regulators of cancer biology. Cell. Mol. Life Sci. 2017, 74, 1999–2017. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Mutation | Gene | Mutation type | Population | Effect | References |
|---|---|---|---|---|---|
| m.16189T>C | D-loop | Point mutation | Chinese | Predispose | [30] |
| m.16223C>A | D-loop | Point mutation | Polish | Predispose | [32] |
| m.207G>A | D-loop | Point mutation | Polish | Predispose | [32] |
| m.16126T>C | D-loop | Point mutation | Polish | Predispose | [32] |
| m.5178A>C | ND2 | Point mutation | Chinese | Predispose | [31] |
| m.7028C>T | COI | Point mutation | Polish | Protect | [32] |
| Mutation | Gene | Mutation type | AA | References |
|---|---|---|---|---|
| m.152T>C | D-loop | Point mutation | - | [33] |
| m.251G>A | D-loop | Point mutation | - | [33] |
| m.294T>C | D-loop | Point mutation | - | [33] |
| m.289-346del | D-loop | 50bp deletion | - | [33] |
| m.305C>A | D-loop | Point mutation | - | [34] |
| m.306C>G | D-loop | Point mutation | - | [34] |
| m.303-309 | D-loop | mtMSI | - | [33,34,35,36,37] |
| m.309C>A | D-loop | Point mutation | - | [34] |
| m.514-523 | D-loop | mtMSI | - | [33,34,35,37] |
| m.16153G>A | D-loop | Point mutation | - | [32] |
| m.16182A>C | D-loop | Point mutation | - | [34] |
| m.16183A>C | D-loop | Point mutation | - | [34] |
| m.16184-16193 | D-loop | mtMSI | - | [33,34,35,37] |
| m.16188A>C | D-loop | Point mutation | - | [32] |
| m.16189T>C | D-loop | Point mutation | - | [34] |
| m.650T>C | 12S rRNA | Point mutation | - | [33] |
| m.817G>A | 12S rRNA | Point mutation | - | [33] |
| m.879T>C | 12S rRNA | Point mutation | - | [33] |
| m.956-965 | 12S rRNA | mtMSI | - | [33,34,35,37] |
| m.961T>C | 12S rRNA | Point mutation | - | [34] |
| m.1474G>A | 12S rRNA | Point mutation | - | [38] |
| m.3163G>A | 16S rRNA | Point mutation | - | [33] |
| m.3470T>Y | ND1 | Point mutation | L55P | [39] |
| m.3730T>Y | ND1 | Point mutation | Y142H | [39] |
| m.3670G>A | ND1 | Point mutation | A122T | [39] |
| m.3425T>Y | ND1 | Point mutation | V40A | [39] |
| m.4722A>G | ND2 | Point mutation | Y85A | [38] |
| m.5212T>C | ND2 | Point mutation | L248P | [39] |
| m.5567T>C | TW | Point mutation | - | [38] |
| m.6129G>R | COI | Point mutation | G76stop codon | [39] |
| m.6562T>C | COI | Point mutation | F220S | [39] |
| m.6822T>A | COI | Point mutation | S307T | [39] |
| m.6991T>Y | COI | Point mutation | L363P | [39] |
| m.7962T>Y | COII | Point mutation | L126S | [39] |
| m.8545G>A | ATP6 | Point mutation | A7T | [39] |
| m.10290G>A | ND3 | Point mutation | A78T | [39] |
| m.11863insC | ND4 | Point mutation | - | [38] |
| m.11873insC | ND4 | Point mutation | - | [38] |
| m.12425insA | ND5 | Point mutation | - | [38] |
| m.12439T>C | ND5 | Point mutation | Y35H | [39] |
| m.13718G>A | ND5 | Point mutation | S461N | [39] |
| m.13994T>C | ND5 | Point mutation | L553P | [38] |
| m.14279G>A | ND6 | Point mutation | S132L | [39] |
| m.14510delA | ND6 | Point mutation | - | [38] |
| m.15172G>A | CYB | Point mutation | S | [38] |
| m.15247C>T | CYB | Point mutation | S | [38] |
| m.15573T>C | CYB | Point mutation | F276S | [38] |
| m.15831T>C | CYB | Point mutation | I362T | [39] |
| Mutation | Gene | Mutation type | AA | References |
|---|---|---|---|---|
| m.3730T>Y | ND1 | Point mutation | Y142H | [39,48] |
| m.3425T>Y | ND1 | Point mutation | V40A | [39,48] |
| m.5212T>C | ND2 | Point mutation | L248P | [39] |
| m.10844A>C | ND4 | Point mutation | T29P | [39] |
| m.14510delA | ND6 | Point mutation | - | [39] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Musicco, C.; Cormio, G.; Pesce, V.; Loizzi, V.; Cicinelli, E.; Resta, L.; Ranieri, G.; Cormio, A. Mitochondrial Dysfunctions in Type I Endometrial Carcinoma: Exploring Their Role in Oncogenesis and Tumor Progression. Int. J. Mol. Sci. 2018, 19, 2076. https://doi.org/10.3390/ijms19072076
Musicco C, Cormio G, Pesce V, Loizzi V, Cicinelli E, Resta L, Ranieri G, Cormio A. Mitochondrial Dysfunctions in Type I Endometrial Carcinoma: Exploring Their Role in Oncogenesis and Tumor Progression. International Journal of Molecular Sciences. 2018; 19(7):2076. https://doi.org/10.3390/ijms19072076
Chicago/Turabian StyleMusicco, Clara, Gennaro Cormio, Vito Pesce, Vera Loizzi, Ettore Cicinelli, Leonardo Resta, Girolamo Ranieri, and Antonella Cormio. 2018. "Mitochondrial Dysfunctions in Type I Endometrial Carcinoma: Exploring Their Role in Oncogenesis and Tumor Progression" International Journal of Molecular Sciences 19, no. 7: 2076. https://doi.org/10.3390/ijms19072076
APA StyleMusicco, C., Cormio, G., Pesce, V., Loizzi, V., Cicinelli, E., Resta, L., Ranieri, G., & Cormio, A. (2018). Mitochondrial Dysfunctions in Type I Endometrial Carcinoma: Exploring Their Role in Oncogenesis and Tumor Progression. International Journal of Molecular Sciences, 19(7), 2076. https://doi.org/10.3390/ijms19072076