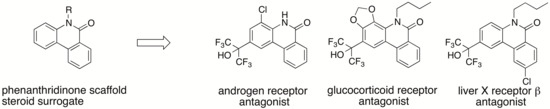
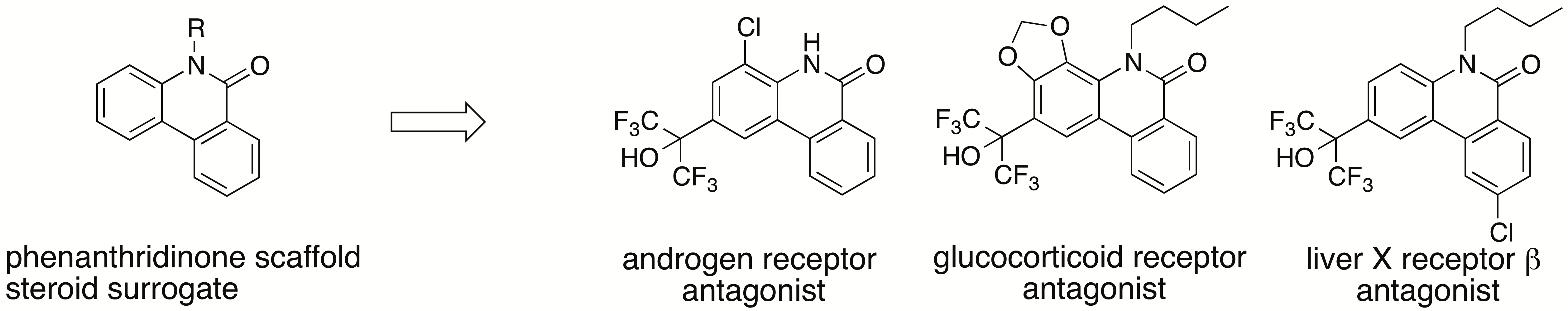
1. Introduction
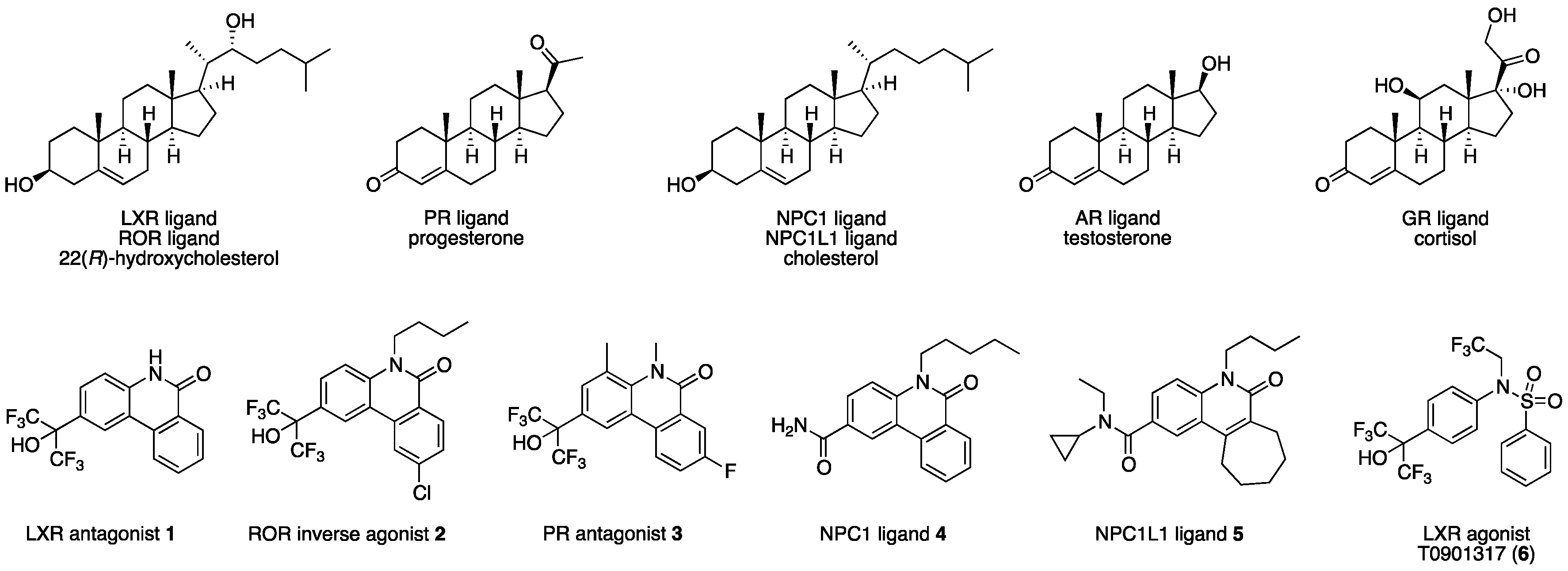
Nuclear receptors (NRs) are ligand-dependent transcription factors that regulate DNA transcription by binding small molecular agonists such as hormones. Forty-eight structurally conserved kinds of NRs have been identified, and many of them recognize endogenous steroidal ligands (
Figure 1). Because NRs control diverse biological functions including reproduction, differentiation, homeostasis, and the immune system, they were the targets of approximately 5% of FDA-approved drugs in 2011 [
1]. However, not only a steroidal ligand, but also its metabolites, brings with it the potential to cross-react with other nuclear receptors, which can result in unwanted side effects, potentially limiting the clinical utility of these agents. Therefore, surrogates for the steroid skeleton are required to develop selective non-steroidal NR ligands for clinical use [
2].
The number of fold structures of human proteins is at least 50 times smaller than the number of human proteins [
2]. This fact indicates that a scaffold that is spatially complementary to one-fold structure might serve as a common scaffold for ligands that would interact specifically with more than 50 different human proteins, neglecting interactions of the peptide sequences of the proteins. In other words, the structures of ligands that bind to one member of fold structures may be useful for the development of novel lead compounds for other proteins with the similar fold structure. However, lead compounds obtained based on this approach are likely to possess polypharmacological character. Therefore, chemical modification of polypharmacological lead compounds, aimed at optimizing selectivity for a particular target, is required. Since the fold structures of NR ligand-binding domains (LBDs) are similar, we have used this strategy to create ligands bearing a diphenylmethane skeleton for several NRs, including farnesoid X receptor [
3], liver X receptor (LXR) [
4], vitamin D receptor [
5], androgen receptor (AR) [
6], and estrogen receptor [
7].
We have previously developed LXRs antagonist
1 [
8], retinoic acid receptor-related orphan receptors (RORs) inverse agonist
2 [
9], progesterone receptor (PR) antagonist
3 [
10], and pharmacological chaperones
4 and
5 for Niemann-Pick disease type C1 (NPC1) [
11] and Niemann-Pick type C1-like 1 (NPC1L1) (
Figure 1) [
12]. These compounds all contain a phenanthridin-6(5
H)-one skeleton as a cyclized carba-analog of the skeleton of LXRs pan agonist T0901317 (
6). Because endogenous ligands for the above proteins (LXRs, RORs, PR, NPLC1 and NPC1L1) all have a steroid skeleton, we hypothesized that if the phenanthridin-6(5
H)-one scaffold acts as a steroid surrogate, phenanthridinone derivatives would also bind to other NRs that recognize endogenous steroidal ligands. The approach described here is expected to be useful in the development of novel steroid surrogates, such as the phenanthridinone skeleton, enabling efficient lead finding of ligands for target proteins that recognize endogenous steroidal ligands, and the generation of more selective ligands by further molecular modifications of new and convenient scaffolds (easy to synthesis, easy to introduce substitutent(s) at any position, etc.), compared to the steroid skeleton. Here, we show that application of this approach to our small library of phenanthridinone-based RORs inverse agonists and PR antagonists led to several selective LXRβ, AR and glucocorticoid receptor (GR) antagonists.
2. Results and Discussion
AR and GR, members of NRs, recognize endogenous steroidal ligands as shown in
Figure 1. In addition, because LXRs antagonistic activities of only limited numbers of phenanthridinone analog have been reported [
8], precise structure-activity relationships (SAR) remain unclear. Therefore, we exhaustively evaluated our small library of RORs inverse agonists and/or PR antagonists bearing a phenanthridinone skeleton for LXRα, LXRβ, AR and GR antagonistic activities, and examined their SAR. LXRs regulate ATP-binding cassette proteins (ABCs), ApoE and glucose transporter 4 (GLUT4), are involved in lipid metabolism, reverse cholesterol transport, and glucose transport, so LXRs agonists are likely to be of therapeutic value in the treatment of atherosclerosis, hyperlipidemia, and metabolic syndrome. They are also involved in the upregulation of sterol regulatory element-binding protein-1c (SREBP-1c) and fatty acid synthase (FAS), so LXRs antagonists might have therapeutic value for hepatic steatosis [
13]. AR is a receptor of androgens essential for the development and maintenance of the male reproductive system and secondary male sex characteristics. AR antagonists including hydroxyflutamide (OHF) are used for treatment of androgen-dependent tumors, especially prostate tumors. GR is a receptor of cortisol, and selective antagonists of GR could be useful in treating hypercortisolemia associated with Cushing’s syndrome and other conditions in which the endogenous GR is hyperactivated either through higher glucocorticoid levels or increased receptor sensitivity [
14]. A steroidal GR antagonist, mifepristone (RU486), shows potent activities toward other steroid receptors.
For this work, compounds
1–
3,
7–
42 and
44 were prepared as described previously [
9,
10,
15]. PR-antagonistic activity was evaluated by assay of PR-regulated alkaline phosphatase activity in human breast cancer cell line T47D [
10,
16] while RORα, RORβ and RORγ inverse agonistic activities, and LXRα, LXRβ, AR and GR antagonistic activities were evaluated by reporter gene assay in HEK293 cell line [
9,
10]. Concentration of the agonists were set as around their EC
50 values. Reproducibility of our assay systems is shown in
Table S1. First, we focused on the SAR at the 2-position of phenanthridinone. The results, including the activities of positive controls T0901317 (
6), RU486, and OHF, are shown in
Table 1. We have reported that introduction of alkyl groups (
8–
10) or a hydroxymethyl group (
11) at the 2-position of
7 resulted in retention or decrease of the PR-antagonistic activity [
10] and RORs inverse agonistic activity [
9]. Introduction of a hexafluoropropanol moiety somewhat increased the RORs activity and PR activity. According to the reported X-ray crystal structures of compound
6 and human LXRs LBD, the hydroxyl group of
6 forms a hydrogen bond with a histidine residue (His421 for LXRα and His435 for LXRβ) in helix 11 [
17,
18]. The SARs for RORs and PR are broadly consistent with those of LXRs, AR, and GR. For AR and PR, introduction of a hydroxymethyl group (
11) resulted in retention of the activity, whereas introduction of hexafluoropropanol (
12) resulted in 6-fold and 9-fold increases of PR and AR activity, respectively. On the other hand, for LXRs and RORs,
11 showed weaker activity than
7 whereas
12 showed stronger activity than
7, suggesting that the two trifluoromethyl groups might be associated with more potent activity and the hydroxyl group might not form a strong hydrogen bond with histidine in these receptors, in contrast to the other receptors. This idea is consistent with the reported co-crystal structure of T0901317 (
6) complexed with RORγ [
19]. Overall, we found that compound
12 showed not only PR-antagonistic activity and RORs inverse agonistic activity, but also LXRα, LXRβ, AR and GR antagonistic activity (
Table 1). These results support our view that the phenanthridin-6(5
H)-one scaffold acts as a steroid surrogate.
Next, SAR at the nitrogen atom is shown in
Table 2. Similar to the SAR for RORs, introduction of a longer-chain alkyl group on the nitrogen atom (
12 and
15) resulted in enhancement of the GR antagonistic activity. SAR for AR was similar to that for PR, that is, hydrogen analog
12 showed potent AR antagonistic activity with an IC
50 value of 0.10 μM, and more than 200-fold selectivity for AR over RORs and GR, and about 7.8- and 50-fold selectivity over PR and LXRs, respectively. The longest alkyl analog
17 showed decreased activity toward all NRs, suggesting that the binding pocket hosting the N-alkylated derivatives might not be able to accommodate a group larger than a hexyl group.
Next, SAR at the nitrogen atom is shown in
Table 2. Similar to the SAR for RORs, introduction of a longer-chain alkyl group on the nitrogen atom (
12 and
15) resulted in enhancement of the GR antagonistic activity. SAR for AR was similar to that for PR, that is, hydrogen analog
12 showed potent AR antagonistic activity with an IC
50 value of 0.10 μM, and more than 200-fold selectivity for AR over RORs and GR, and about 7.8- and 50-fold selectivity over PR and LXRs, respectively. The longest alkyl analog
17 showed decreased activity towards all NRs, suggesting that the binding pocket hosting the N-alkylated derivatives might not be able to accommodate a group larger than a hexyl group.
Next, the effect of introduction of a methoxy group at every position (methoxy scanning) was investigated (
18–
24;
Table 3) because many types of NR ligands possess a methoxy group(s) [
20]. We have reported that 3-methoxy and 4-methoxy analogs
19 and
20 showed more than 3-fold and 2-fold enhanced PR activity compared with the unsubstituted analog
12, respectively [
10]. In contrast, 9-methoxy analog
23 showed 5-fold weaker PR activity than
12. In the case of RORs, we have reported that introduction of a methoxy group at the 9-position (
23) enhanced ROR activities [
9]. The activities toward LXRs, AR and GR also depended on the position of the methoxy group. 1-Methoxy analog
18 showed decreased activity for all receptors tested. Enhanced activity was observed when a methoxy group was introduced at the 4-, 9- or 10-position for LXRα, 9- or 10-position for LXRβ, 3- and 8-position for AR, and 3- or 4-position for GR. Especially, 3-methoxy analog
19 showed 5.7-fold improved GR activity compared with
12. Overall, introduction of a methoxy group resulted in increased activity in almost all cases except RORβ, but the most suitable position depended on the NR (3-position: PR and GR, 8-position: AR, 9-position: RORα, RORγ and LXRβ, 10-position: LXRα). The introduction of a substituent at every position is useful to investigate substituent effects. In this context, it is important to note that phenanthridin-6(5
H)-one analogs bearing substituent(s) at any position can be quite easily synthesized, and this represents a considerable advantage compared to the steroid skeleton.
The results of methoxy scanning prompted us to investigate the effect of introduction of two methoxy groups (
25–
31 Table 3). In the cases of RORα, PR, AR and GR, 3,4-dimethoxy analog
25 exhibited similar or weaker activity to the 3-methoxy and 4-methoxy analogs
19 and
20. A possible explanation might be the direction of the two methoxy groups, that is, one or both might occupy a space not suitable for receptor interaction due to steric hindrance. This idea is supported by the fact that 3,4-dioxolane analog
26 showed stronger activity than
25 toward PR, AR and GR. Compound
26 was the most potent GR antagonist with an IC
50 value of 0.76 μM, although this compound also showed PR and AR antagonistic activities with IC
50 values of 0.23 and 0.54 μM, respectively. When two methoxy groups were introduced at distal positions, the activity was improved. Thus, 3,8-dimethoxy analog
27 and 4,8-dimethoxy analog
28 showed more potent PR-antagonistic activity than monomethoxy analogs
19,
20, and
22, and 3,8-dimethoxy analog
27 showed more potent AR antagonistic activity than monomethoxy analogs
19 and
22.
We next examined the SAR of fluoro derivatives
32–
35 (
Table 4) to investigate the effect of this electron-withdrawing substituent. For PR, the preferences for fluorine substitution position were roughly consistent with the results of the methoxy scanning, that is, 4- and 8-substitued analogs
32 and
34 showed enhanced PR activity, whereas 9-substituted analogs
35 showed weaker PR activity [
10]. For AR, all fluoro analogs
32–
35 showed enhanced activity compared with
12, suggesting that an electron-withdrawing effect at these positions is important for AR antagonistic activity. For LXRα, 4- and 7-fluoro analogs
32 and
33 showed enhanced activity compared with
12.
We then examined various substituents at the 9-position (
Table 5). We have reported that introduction of a chlorine atom at the 9-position (
2) enhanced inverse agonistic activity toward RORγ [
9]. The SARs of RORγ were roughly consistent with the results for LXRβ, and
2 also exhibited potent LXRβ antagonistic activity with the IC
50 value of 0.88 μM. Replacement of fluorine at the 9-position (
35) with chlorine (
2) caused enhanced activity toward RORs and LXRβ, but decreased activity toward PR, AR, and GR, suggesting that molecular modification at the 9-position would be important to improve selectivity for metabolic NRs over steroid receptors.
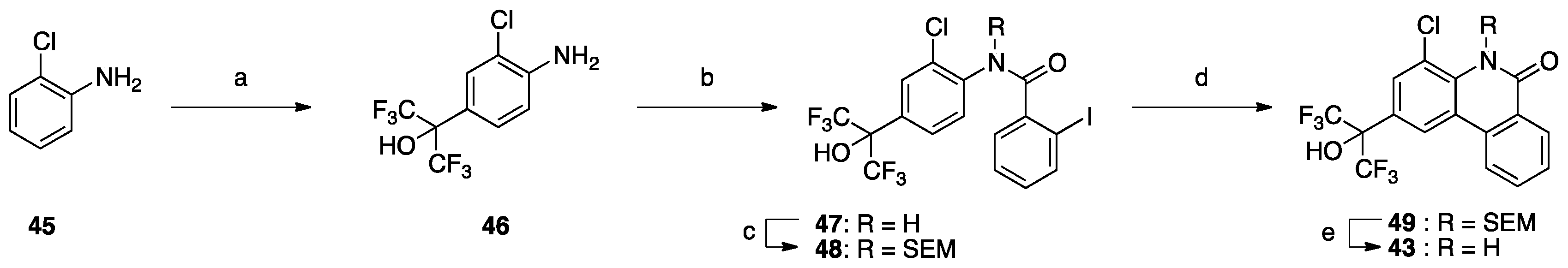
Next, we focused on the 4-position (
Table 6), because 4-methoxy analog
20 showed enhanced RORα, LXRα and PR activity, and 4-fluoro analog
32 showed enhanced LXRα, PR and AR activity. Compounds
39–
44 and
2 showed weak activity toward RORs and GR, indicating that the alkyl group on the nitrogen atom is important for activity toward these receptors. We have reported that 4-alkyl analogs
40–
42 showed greater PR activity than the unsubstituted analog
1, and 4-methyl analog
40 had an IC
50 value of 0.15 µM [
10]. As for LXRs and AR, 4-alkyl analogs
40–
42 showed decreased activity compared with the unsubstituted analog
1, whereas 4-fluoro analog
32 showed greater activity than
1. Based on the reported SARs of non-steroidal AR antagonists, an electron-withdrawing substituent neighboring the amide bond is expected to be important for potent AR antagonism [
21]. Therefore, 4-chloro analog
43 was designed and synthesized as shown in
Scheme 1. Interestingly,
43 showed greater AR activity and weaker RORs, LXRs, PR and GR activity than 4-fluoro analog
32. Compound
43 showed the IC
50 value of 0.059 μM, being 2.9-fold more potent than clinically used OHF under our assay conditions. Compound
43 showed more than 340-fold selectivity over RORs and LXRβ, and 90-fold and 6-fold selectivity over LXRα and PR, respectively. This result indicates that molecular modification at the 4-position can increase selectivity for AR over the other NRs.
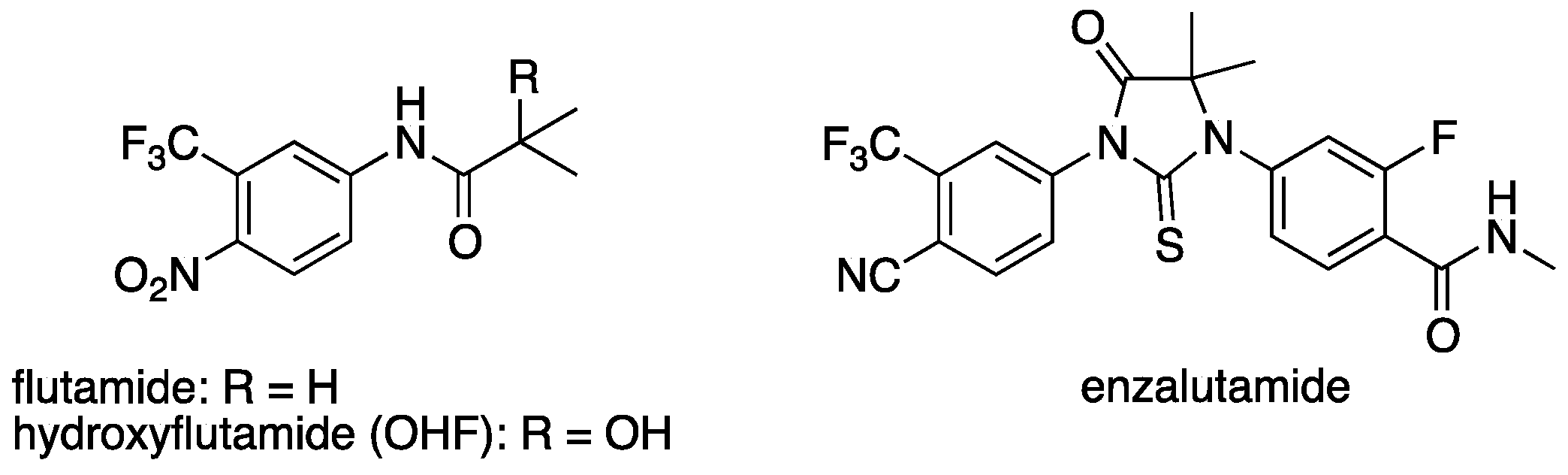
Chronic administration of AR antagonists often leads to the development of resistance. Mutations in the LBD of AR, such as T877A, have been identified in patients treated with flutamide (
Figure 2), and the activated metabolite, hydroxyflutamide, is an agonist of AR bearing T877A [
22]. In contrast, another AR antagonist, enzalutamide, was reported to antagonize AR mutant T877A [
23]. Thus, development of a novel chemical class of AR antagonists, different from flutamide analogs seems an attractive approach for the treatment of AR antagonist-resistant cancer, providing further motivation to evaluate steroid surrogates. Therefore, we investigated the antiandrogenic activity of several potent AR antagonists by means of PSA (prostate-specific antigen, an AR-regulated gene) ELISA (enzyme-linked immunosorbent assay) in human prostate cancer cell line LNCaP, which has T877A mutation in the AR LBD. Potent AR antagonist
43 did not show antiandrogenic activity in LNCaP, whereas several other AR antagonists including
33 decreased the PSA level with an IC
50 of 190 nM (
Table 7). This result suggests that the new non-flutamide type AR antagonist
33 is a promising candidate for antiandrogen therapy of prostate cancer.






{kind=link}
{kind=link}
{kind=link}
{kind=link}





