Regulation of Energy Metabolism during Early B Lymphocyte Development

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. B Lymphocyte Development
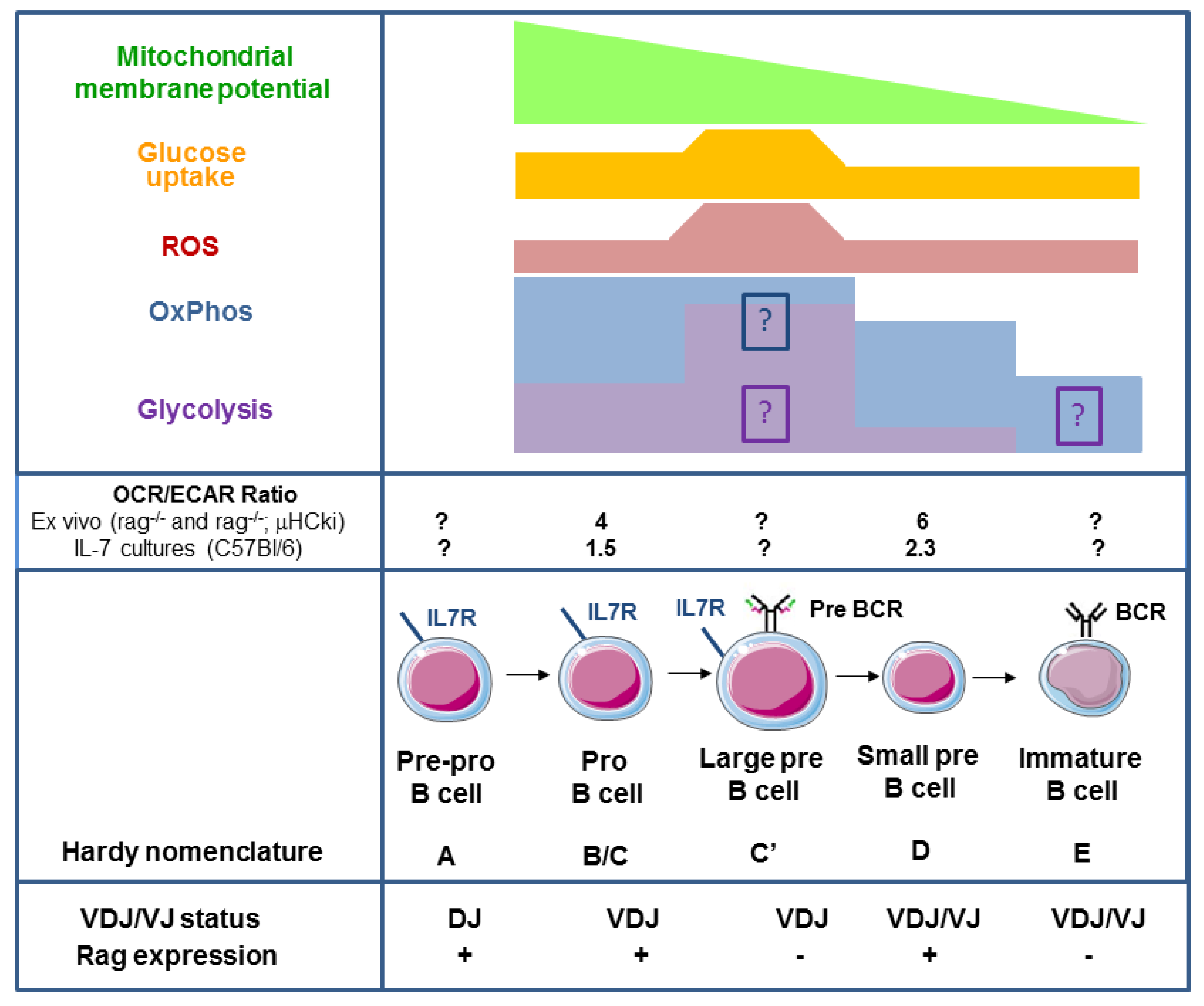
2. Oxidative Phosphorylation and Glycolysis in Pro- and Pre-B Cells
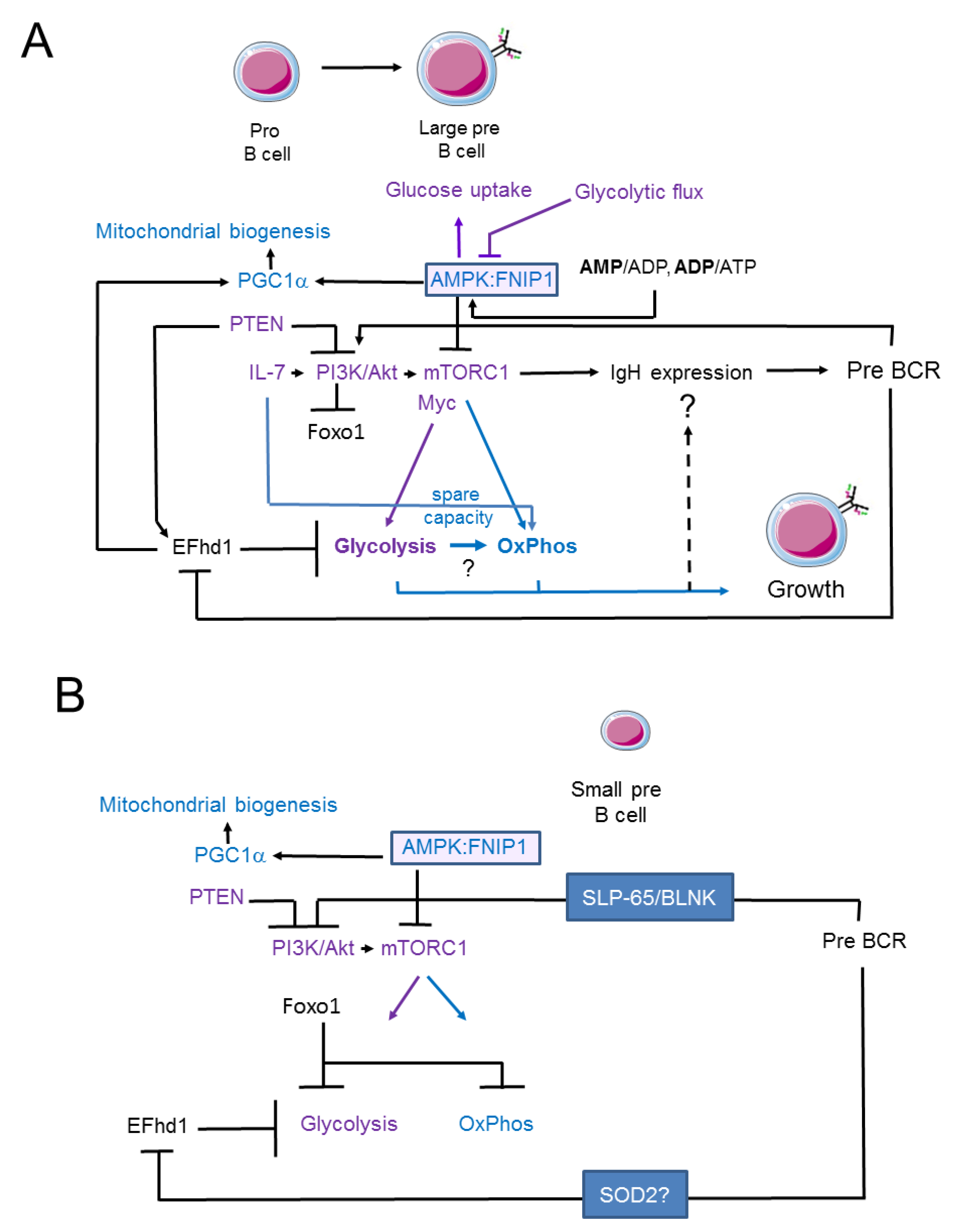
3. Signalling Pathways Linking Membrane Receptor Signals to Glycolysis and Oxidative Phosphorylation in Pro- and Early Pre-B Cells
4. Swiprosin-2/EFhd1 as a Regulator of Glycolysis in Pro-B Cells
5. Signalling Pathways Linking Membrane Receptor Signals to Glycolysis and Oxidative Phosphorylation in Late Pre-B Cells
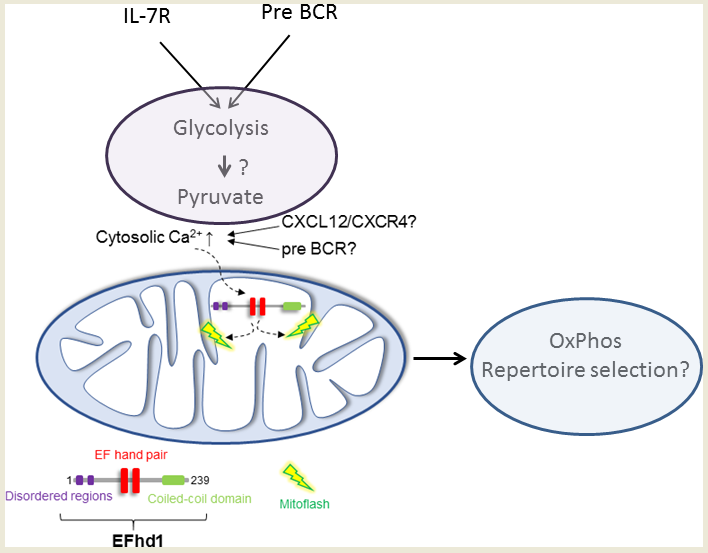
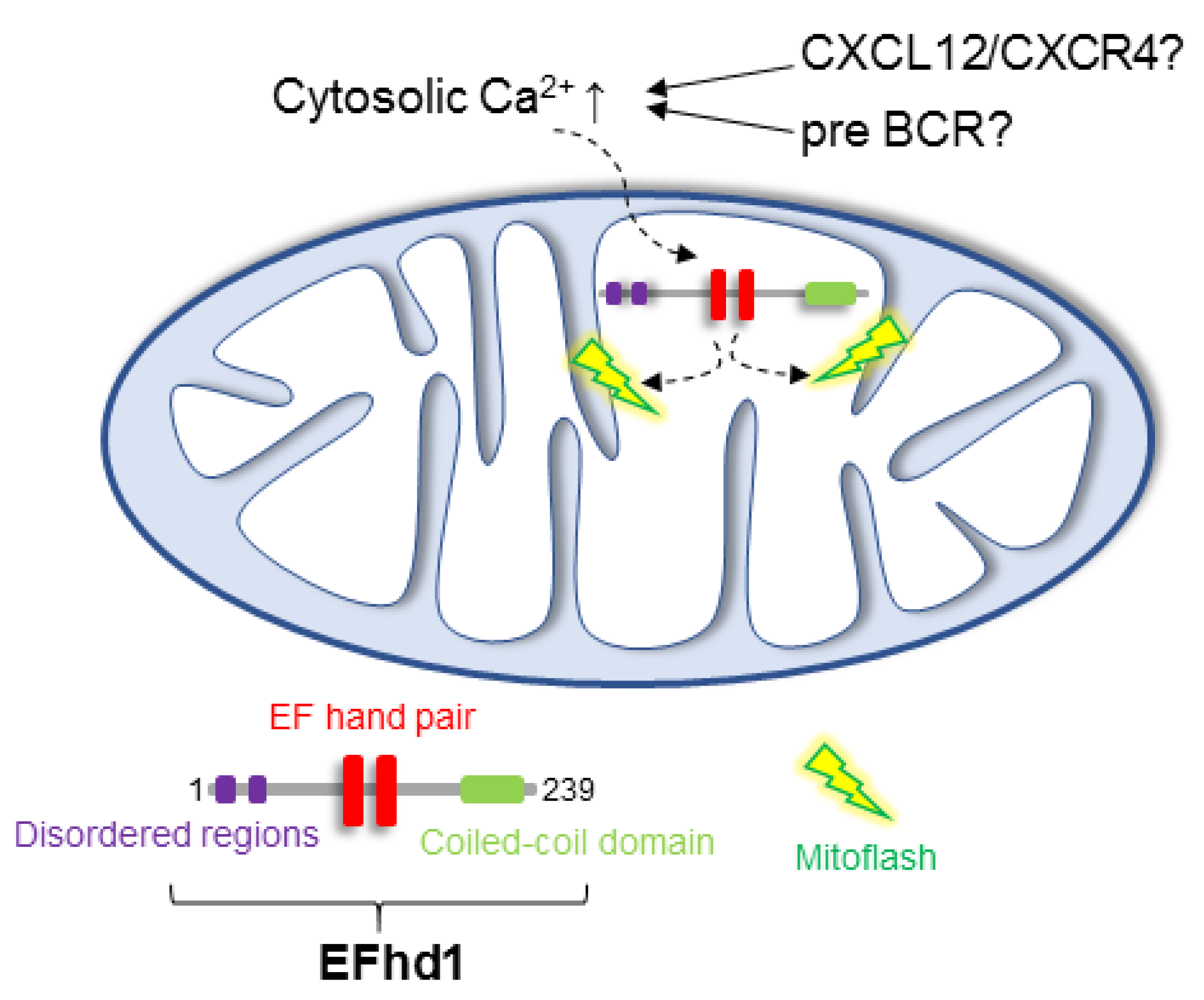
6. Putative Existence of Mitoflashes in Early B Cells—Regulated by EFhd1?
7. Conclusions and Perspectives
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ALL | Acute lymphoblastic leukaemia |
| AMP | Adenosine monophosphate |
| ADP | Adenosine diphosphate |
| ATP | Adenosine triphosphate |
| 6-NBDG | 6-(N-(7-nitrobenz-2-oxa-1, 3-diazol-4-yl)amino)-2-deoxyglucose |
| BLP | B cell-biased lymphoid progenitor |
| BCR | B cell receptor |
| BM | Bone marrow |
| CAR cell | CXCL12-abundant reticular cell |
| CLP | Common lymphoid progenitor |
| ECAR | Extracellular acidification rate |
| EFhd1 | Swiprosin-2/EFhd1 |
| EFhd1tg | EFhd1 transgenic |
| ETC | Electron transport chain |
| Erk | Extracellular signal regulated kinase |
| FA | Fatty acid |
| Flt | Fms-like tyrosine kinase |
| Fnip1 | Folliculin-interacting protein 1 |
| HC | Heavy chain |
| HSC | Hematopoietic stem cell |
| Ig | Immunoglobulin |
| IMM | Inner mitochondrial membrane |
| JAK | Janus kinase |
| Ki | Knock-in |
| LC | Light chain |
| LPS | Lipopolysaccharide |
| ∆ψm | Mitochondrial membrane potential |
| OCR | Oxygen consumption rate |
| OxPhos | Oxidative phosphorylation |
| Pi | Inorganic phosphate |
| PI3K | Phosphatidyl-inositol-3-kinase |
| ∆pH | Proton gradient |
| PTEN | Phosphatase and Tensin homologue |
| ROS | Reactive oxygen species |
| SCF | Stem cell factor |
| SDF-1 | Stromal cell derived factor |
| STAT | Signal transducer of activation and transcription |
| TCA | Tricarbon cycle |
| TF | Transcription factor |
| WT | Wildtype |
References
- Green, A.C.; Rudolph-Stringer, V.; Chantry, A.D.; Wu, J.Y.; Purton, L.E. Mesenchymal lineage cells and their importance in B lymphocyte niches. Bone 2017. [Google Scholar] [CrossRef] [PubMed]
- Boulais, P.E.; Frenette, P.S. Making sense of hematopoietic stem cell niches. Blood 2015, 125, 2621–2630. [Google Scholar] [CrossRef] [PubMed]
- Tokoyoda, K.; Egawa, T.; Sugiyama, T.; Choi, B.; Nagasawa, T. Cellular niches controlling B lymphocyte behavior within bone marrow during development. Immunity 2004, 20, 707–718. [Google Scholar] [CrossRef] [PubMed]
- Morrison, S.J.; Scadden, D.T. The bone marrow niche for haematopoietic stem cells. Nature 2014, 505, 327–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breton, C.; Tellier, J.; Narang, P.; Chasson, L.; Jorquera, A.; Coles, M.; Schiff, C. Galectin-1—Expressing stromal cells constitute a specific niche for pre-BII cell development in mouse bone marrow. Blood 2011, 117, 6552–6561. [Google Scholar] [CrossRef]
- Spencer, J.A.; Ferraro, F.; Roussakis, E.; Klein, A.; Wu, J.; Runnels, J.M.; Zaher, W.; Mortensen, L.J.; Alt, C.; Turcotte, R.; et al. Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature 2014, 508, 269–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Apuzzo, M.; Rolink, A.; Loetscher, M.; Hoxie, J.A.; Clark-Lewis, I.; Melchers, F.; Baggiolini, M.; Moser, B. The chemokine SDF-1, stromal cell-derived factor 1, attracts early stage B cell precursors via the chemokine receptor CXCR4. Eur. J. Immunol. 1997, 27, 1788–1793. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-Y.; Wolfram, P.; Canty, K.; Harley, B.; Nombela-Arrieta, C.; Pivarnik, G.; Manis, J.; Beggs, H.E.; Silberstein, L.E. Focal Adhesion Kinase Regulates the Localization and Retention of Pro-B Cells in Bone Marrow Microenvironments. J. Immunol. 2013, 190, 1094–1102. [Google Scholar] [CrossRef] [PubMed]
- Egawa, T.; Kawabata, K.; Kawamoto, H.; Amada, K.; Okamoto, R.; Fujii, N.; Kishimoto, T.; Katsura, Y.; Nagasawa, T. The earliest stages of B cell development require a chemokine stromal cell-derived factor/pre-B cell growth-stimulating factor. Immunity 2001, 15, 323–334. [Google Scholar] [CrossRef]
- Hagman, J.; Lukin, K. Transcription factors drive B cell development. Curr. Opin. Immunol. 2006, 18, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Hardy, R.R.; Carmack, C.E.; Shinton, S.A.; Kemp, J.D.; Hayakawa, K. Resolution and characterization of pro-B and pre-pro-B cell stages in normal mouse bone marrow. J. Exp. Med. 1991, 173, 1213–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vettermann, C.; Herrmann, K.; Jäck, H.M. Powered by pairing: The surrogate light chain amplifies immunoglobulin heavy chain signaling and pre-selects the antibody repertoire. Semin. Immunol. 2006, 18, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Clark, M.R.; Mandal, M.; Ochiai, K.; Singh, H. Orchestrating B cell lymphopoiesis through interplay of IL-7 receptor and pre-B cell receptor signalling. Nat. Rev. Immunol. 2014, 14, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Hamel, K.M.; Mandal, M.; Karki, S.; Clark, M.R. Balancing proliferation with Igκ recombination during B-lymphopoiesis. Front. Immunol. 2014, 5, 139. [Google Scholar] [CrossRef] [PubMed]
- Hedrick, S.M. The cunning little vixen: Foxo and the cycle of life and death. Nat. Immunol. 2009, 10, 1057–1063. [Google Scholar] [CrossRef] [PubMed]
- Prieyl, J.A.; LeBien, T.W. Interleukin 7 independent development of human B cells. Proc. Natl.Acad. Sci. USA 1996, 93, 10348–10353. [Google Scholar] [CrossRef] [PubMed]
- Milne, C.D.; Paige, C.J. IL-7: A key regulator of B lymphopoiesis. Semin. Immunol. 2006, 18, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Milne, C.D.; Fleming, H.E.; Paige, C.J. IL-7 does not prevent pro-B/pre-B cell maturation to the immature/sIgM(+) stage. Eur. J. Immunol. 2004, 34, 2647–2655. [Google Scholar] [CrossRef] [PubMed]
- Mombaerts, P.; Johnson, R.S.; Herrup, K.; Tonegawa, S.; Papaioannouo, V.E. RAG-1-Deficient Mice Have No Mature B and T Lymphocytes. Cell 1992, 68, 869–877. [Google Scholar] [CrossRef]
- Young, F.; Ardman, B.; Shinkai, Y.; Lansford, R.; Blackwell, T.K.; Mendelsohn, M.; Rolink, A.; Melchers, F.; Alt, F.W. Influence of immunoglobulin heavy- and light-chain expression on B-cell differentiation. Genes Dev. 1994, 8, 1043–1057. [Google Scholar] [CrossRef] [PubMed]
- Stein, M.; Dütting, S.; Mougiakakos, D.; Bösl, M.; Fritsch, K.; Reimer, D.; Urbanczyk, S.; Steinmetz, T.; Schuh, W.; Bozec, A.; et al. A defined metabolic state in pre B cells governs B-cell development and is counterbalanced by Swiprosin-2/EFhd1. Cell Death Differ. 2017, 24, 1239–1252. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Yu, M.; Tan, H.; Li, Y.; Su, W.; Shi, H.; Dhungana, Y.; Guy, C.; Neale, G.; Cloer, C.; et al. Discrete roles and bifurcation of PTEN signaling and mTORC1-mediated anabolic metabolism underlie IL-7-driven B lymphopoiesis. Sci. Adv. 2018, 4. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.; Kato, R.M.; Garcia-Lloret, M.; Wahl, M.I.; Rawlings, D.J. Engagement of the Human Pre-B Cell Receptor Generates a Lipid Raft-Dependent Calcium Signaling Complex. Immunity 2000, 13, 243–253. [Google Scholar] [CrossRef]
- Su, Y.W.; Jumaa, H. LAT links the pre-BCR to calcium signaling. Immunity 2003, 19, 295–305. [Google Scholar] [CrossRef]
- Taguchi, T.; Kiyokawa, N.; Takenouch, H.; Matsui, J.; Tang, W.R.; Nakajima, H.; Suzuki, K.; Shiozawa, Y.; Saito, M.; Katagiri, Y.U.; et al. Deficiency of BLNK hampers PLC-γ2 phosphorylation and Ca2+ influx induced by the pre-B-cell receptor in human pre-B cells. Immunology 2004, 112, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Feldhahn, N.; Klein, F.; Mooster, J.L.; Hadweh, P.; Sprangers, M.; Wartenberg, M.; Bekhite, M.M.; Hofmann, W.-K.; Herzog, S.; Jumaa, H.; et al. Mimicry of a constitutively active pre–B cell receptor in acute lymphoblastic leukemia cells. J. Exp. Med. 2005, 201, 1837–1852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hess, J.; Werner, A.; Wirth, T.; Melchers, F.; Jack, H.M.; Winkler, T.H. Induction of pre-B cell proliferation after de novo synthesis of the pre-B cell receptor. Proc. Natl. Acad. Sci. USA 2001, 98, 1745–1750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melchers, F. The pre-B-cell receptor: Selector of fitting immunoglobulin heavy chains for the B-cell repertoire. Nat. Rev. Immunol. 2005, 5, 578–584. [Google Scholar] [CrossRef] [PubMed]
- Müschen, M. Autoimmunity checkpoints as therapeutic targets in B cell malignancies. Nat. Rev. Cancer 2018, 18, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Zouali, M. Transcriptional and metabolic pre-B cell receptor-mediated checkpoints: Implications for autoimmune diseases. Mol. Immunol. 2014, 62, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Vettermann, C.; Jäck, H.M. The pre-B cell receptor: Turning autoreactivity into self-defense. Trends Immunol. 2010, 31, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Herzog, S.; Reth, M.; Jumaa, H. Regulation of B-cell proliferation and differentiation by pre-B-cell receptor signalling. Nat. Rev. Immunol. 2009, 9, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Carsetti, R. Characterization of B-Cell Maturation in the Peripheral Immune System. In B Cell Protocols; Humana Press: Totowa, NJ, USA, 2004; pp. 25–36, E-ISBN: 1-59259-796-3. [Google Scholar]
- Reth, M.; Nielsen, P. Signaling Circuits in Early B-Cell Development. Adv. Immunol. 2014, 122, 129–175. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, R.P.; Finlay, D.K. Glucose, glycolysis and lymphocyte responses. Mol. Immunol. 2015, 68, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Santo-Domingo, J.; Demaurex, N. Perspectives on: SGP symposium on mitochondrial physiology and medicine: The renaissance of mitochondrial pH. J. Gen. Physiol. 2012, 139, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G. AMP-activated protein kinase: A key regulator of energy balance with many roles in human disease. J. Intern. Med. 2014, 276, 543–559. [Google Scholar] [CrossRef] [PubMed]
- Andris, F.; Leo, O. AMPK in Lymphocyte Metabolism and Function. Int. Rev. Immunol. 2015, 34, 67–81. [Google Scholar] [CrossRef] [PubMed]
- Iwata, T.N.; Ramírez-Komo, J.A.; Park, H.; Iritani, B.M. Control of B lymphocyte development and functions by the mTOR signaling pathways. Cytokine Growth Factor Rev. 2017, 35, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Kojima, H.; Kobayashi, A.; Sakurai, D.; Kanno, Y.; Hase, H.; Takahashi, R.; Totsuka, Y.; Semenza, G.L.; Sitkovsky, M.V.; Kobata, T. Differentiation stage-specific requirement in hypoxia-inducible factor-1alpha-regulated glycolytic pathway during murine B cell development in bone marrow. J. Immunol. 2010, 184, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Staehling, K.; Tsang, M.; Appleby, M.W.; Brunkow, M.E.; Margineantu, D.; Hockenbery, D.M.; Habib, T.; Liggitt, H.D.; Iritani, B.M. Disruption of Fnip1 reveals a metabolic checkpoint controlling B lymphocyte development. Immunity 2012, 36, 769–781. [Google Scholar] [CrossRef] [PubMed]
- Xi, H.; Kurtoglu, M.; Lampidis, T.J. The wonders of 2-deoxy-D-glucose. IUBMB Life 2014, 66, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.-H.; Liu, G.-Y.; Wang, R.; Moon, S.H.; Gross, R.W.; Patti, G.J. Identifying off-target effects of etomoxir reveals that carnitine palmitoyltransferase I is essential for cancer cell proliferation independent of β-oxidation. PLOS Biol. 2018, 16, e2003782. [Google Scholar] [CrossRef] [PubMed]
- Shojaee, S.; Chan, L.N.; Buchner, M.; Cazzaniga, V.; Cosgun, K.N.; Geng, H.; Qiu, Y.H.; von Minden, M.D.; Erns, T.; Hochhaus, A.; et al. PTEN opposes negative selection and enables oncogenic transformation of pre-B cells. Nat. Med. 2016, 4, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Wofford, J.A.; Wieman, H.L.; Jacobs, S.R.; Zhao, Y.; Rathmell, J.C.; Jeffrey, C. IL-7 promotes Glut1 trafficking and glucose uptake via STAT5-mediated activation of Akt to support T cell survival IL-7 promotes Glut1 trafficking and glucose uptake via STAT5-mediated activation of Akt to support T cell survival. Blood 2007, 111, 2101–2112. [Google Scholar] [CrossRef] [PubMed]
- Baracho, G.V.; Miletic, A.V.; Omori, S.A.; Cato, M.H.; Rickert, R.C. Emergence of the PI3-kinase pathway as a central modulator of normal and aberrant B cell differentiation. Curr. Opin. Immunol. 2011, 23, 178–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ochiai, K.; Maienschein-Cline, M.; Mandal, M.; Triggs, J.R.; Bertolino, E.; Sciammas, R.; Dinner, A.R.; Clark, M.R.; Singh, H. A self-reinforcing regulatory network triggered by limiting IL-7 activates pre-BCR signaling and differentiation. Nat. Immunol. 2012, 13, 300–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caro-Maldonado, A.; Wang, R.; Nichols, A.G.; Kuraoka, M.; Milasta, S.; Sun, L.D.; Gavin, A.L.; Abel, E.D.; Kelsoe, G.; Green, D.R.; et al. Metabolic Reprogramming Is Required for Antibody Production That Is Suppressed in Anergic but Exaggerated in Chronically BAFF-Exposed B Cells. J. Immunol. 2014, 192, 3626–3636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doughty, C.A.; Bleiman, B.F.; Wagner, D.J.; Dufort, F.J.; Mataraza, J.M.; Roberts, M.F.; Chiles, T.C. Antigen receptor-mediated changes in glucose metabolism in B lymphocytes: Role of phosphatidylinositol 3-kinase signaling in the glycolytic control of growth. Blood 2006, 107, 4458–4465. [Google Scholar] [CrossRef] [PubMed]
- Woodland, R.T.; Fox, C.J.; Schmidt, M.R.; Hammerman, P.S.; Opferman, J.T.; Korsmeyer, S.J.; Hilbert, D.M.; Thompson, C.B. Multiple signaling pathways promote B lymphocyte stimulator dependent B-cell growth and survival. Blood 2008, 111, 750–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, A.J.; Riffelmacher, T.; Braas, D.; Cornall, R.J.; Simon, A.K. B1a B cells require autophagy for metabolic homeostasis and self-renewal. J. Exp. Med. 2018, 215, 399–413. [Google Scholar] [CrossRef] [PubMed]
- Lam, W.Y.; Becker, A.M.; Kennerly, K.M.; Wong, R.; Curtis, J.D.; Llufrio, E.M.; McCommis, K.S.; Fahrmann, J.; Pizzato, H.A.; Nunley, R.M.; et al. Mitochondrial Pyruvate Import Promotes Long-Term Survival of Antibody-Secreting Plasma Cells. Immunity 2016, 45, 60–73. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Medina, K.L.; Lancki, D.W.; Singh, H. IRF-4, 8 orchestrate the pre-B-to-B transition in lymphocyte development. Genes Dev. 2003, 1703–1708. [Google Scholar] [CrossRef] [PubMed]
- Jumaa, H.; Wollscheid, B.; Mitterer, M.; Wienands, J.; Reth, M.; Nielsen, P.J. Abnormal development and function of B lymphocytes in mice deficient for the signaling adaptor protein SLP-65. Immunity 1999, 11, 547–554. [Google Scholar] [CrossRef]
- Zhang, S.; Yang, C.; Yang, Z.; Zhang, D.; Ma, X.; Mills, G.; Liu, Z. Homeostasis of redox status derived from glucose metabolic pathway could be the key to understanding the Warburg effect. Am. J. Cancer Res. 2015, 5, 1265–1280. [Google Scholar] [PubMed]
- Sitkovsky, M.; Lukashev, D. Regulation of immune cells by local-tissue oxygen tension: HIF1 alpha and adenosine receptors. Nat. Rev. Immunol. 2005, 5, 712–721. [Google Scholar] [CrossRef] [PubMed]
- Kojima, H.; Gu, H.; Nomura, S.; Caldwell, C.C.; Kobata, T.; Carmeliet, P.; Semenza, G.L.; Sitkovsky, M.V. Abnormal B lymphocyte development and autoimmunity in hypoxia-inducible factor 1α-deficient chimeric mice. Proc. Natl. Acad. Sci. USA 2002, 99, 2170–2174. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Grötsch, B.; Luo, Y.; Knaup, K.X.; Wiesener, M.S.; Chen, X.X.; Jantsch, J.; Fillatreau, S.; Schett, G.; Bozec, A. Hypoxia-inducible factor-1α is a critical transcription factor for IL-10-producing B cells in autoimmune disease. Nat. Commun. 2018, 9, 251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraus, H.; Kaiser, S.; Aumann, K.; Bonelt, P.; Salzer, U.; Vestweber, D.; Erlacher, M.; Kunze, M.; Burger, M.; Pieper, K.; et al. A Feeder-Free Differentiation System Identifies Autonomously Proliferating B Cell Precursors in Human Bone Marrow. J. Immunol. 2014, 192, 1044–1054. [Google Scholar] [CrossRef] [PubMed]
- Schuh, W.; Meister, S.; Herrmann, K.; Bradl, H.; Jäck, H.M. Transcriptome analysis in primary B lymphoid precursors following induction of the pre-B cell receptor. Mol. Immunol. 2008, 45, 362–375. [Google Scholar] [CrossRef] [PubMed]
- Chan, L.N.; Müschen, M. B-cell identity as a metabolic barrier against malignant transformation. Exp. Hematol. 2017, 53, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, T.; Sanjo, H.; Pagès, G.; Kawano, Y.; Karasuyama, H.; Pouysségur, J.; Ogata, M.; Kurosaki, T. Erk Kinases Link Pre-B Cell Receptor Signaling to Transcriptional Events Required for Early B Cell Expansion. Immunity 2008, 28, 499–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowh, M.A.W.; Bassing, C.H. Foxos around make B cells tolerable. Nat. Immunol. 2008, 9, 586–588. [Google Scholar] [CrossRef] [PubMed]
- Klotz, L.O.; Sánchez-Ramos, C.; Prieto-Arroyo, I.; Urbánek, P.; Steinbrenner, H.; Monsalve, M. Redox regulation of FoxO transcription factors. Redox Biol. 2015, 6, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Siggs, O.M.; Stockenhuber, A.; Deobagkar-Lele, M.; Bull, K.R.; Crockford, T.L.; Kingston, B.L.; Crawford, G.; Anzilotti, C.; Steeples, V.; Ghaffari, S.; et al. Mutation of Fnip1 is associated with B-cell deficiency, cardiomyopathy, and elevated AMPK activity. Proc. Natl. Acad. Sci. USA 2016, 113, E3706–E3715. [Google Scholar] [CrossRef] [PubMed]
- Hobeika, E.; Thiemann, S.; Storch, B.; Jumaa, H.; Nielsen, P.J.; Pelanda, R.; Reth, M. Testing gene function early in the B cell lineage in mb1-cre mice. Proc. Natl. Acad. Sci. USA 2006, 103, 13789–13794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwata, T.N.; Ramírez, J.A.; Tsang, M.; Park, H.; Margineantu, D.H.; Hockenbery, D.M.; Iritani, B.M. Conditional Disruption of Raptor Reveals an Essential Role for mTORC1 in B Cell Development, Survival, and Metabolism. J. Immunol. 2016, 197, 2250–2260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dütting, S.; Brachs, S.; Mielenz, D. Fraternal twins: Swiprosin-1/EFhd2 and Swiprosin-2/EFhd1, two homologous EF-hand containing calcium binding adaptor proteins with distinct functions. Cell Commun. Signal. 2011, 9, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajnóczky, G.; Booth, D.; Csordás, G.; Debattisti, V.; Golenár, T.; Naghdi, S.; Niknejad, N.; Paillard, M.; Seifert, E.L.; Weaver, D. Reliance of ER-mitochondrial calcium signaling on mitochondrial EF-hand Ca2+ binding proteins: Miros, MICUs, LETM1 and solute carriers. Curr. Opin. Cell Biol. 2014, 29, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Mootha, V.K.; Bunkenborg, J.; Olsen, J.V.; Hjerrild, M.; Wisniewski, J.R.; Stahl, E.; Bolouri, M.S.; Ray, H.N.; Sihag, S.; Kamal, M.; et al. Integrated analysis of protein composition, tissue diversity, and gene regulation in mouse mitochondria. Cell 2003, 115, 629–640. [Google Scholar] [CrossRef]
- Tominaga, M.; Kurihara, H.; Honda, S.; Amakawa, G.; Sakai, T.; Tomooka, Y. Molecular characterization of mitocalcin, a novel mitochondrial Ca2+-binding protein with EF-hand and coiled-coil domains. J. Neurochem. 2006, 96, 292–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bossen, C.; Murre, C.S.; Chang, A.N.; Mansson, R.; Rodewald, H.-R.; Murre, C. The chromatin remodeler Brg1 activates enhancer repertoires to establish B cell identity and modulate cell growth. Nat. Immunol. 2015, 16, 775–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, K.T.; Timblin, G.A.; McWhirter, S.M.; Schlissel, M.S. MK5 activates Rag transcription via Foxo1 in developing B cells. J. Exp. Med. 2013, 210, 1621–1634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henzi, T.; Schwaller, B. Antagonistic Regulation of Parvalbumin Expression and Mitochondrial Calcium Handling Capacity in Renal Epithelial Cells. PLoS ONE 2015, 10, e0142005. [Google Scholar] [CrossRef] [PubMed]
- Takane, K.; Midorikawa, Y.; Yagi, K.; Sakai, A.; Aburatani, H.; Takayama, T.; Kaneda, A. Aberrant promoter methylation of PPP1R3C and EFHD1 in plasma of colorectal cancer patients. Cancer Med. 2014, 3, 1235–1245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleming, H.E.; Paige, C.J. Cooperation between IL-7 and the pre-B cell receptor: A key to B cell selection. Semin. Immunol. 2002, 14, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Flemming, A.; Brummer, T.; Reth, M.; Jumaa, H. The adaptor protein SLP-65 acts as a tumor suppressor that limits pre-B cell expansion. Nat. Immunol. 2003, 4, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Amin, R.H.; Schlissel, M.S. Foxo1 directly regulates the trancription of recombination activating genes during B cell development. Nat. Immunol. 2008, 9, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Hurt, E.M.; Thomas, S.B.; Peng, B.; Farrar, W.L. Molecular consequences of SOD2 expression in epigenetically silenced pancreatic carcinoma cell lines. Br. J. Cancer 2007, 97, 1116–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagur, R.; Hajnóczky, G. Intracellular Ca2+ Sensing: Its Role in Calcium Homeostasis and Signaling. Mol. Cell 2017, 66, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Rosselin, M.; Santo-Domingo, J.; Bermont, F.; Giacomello, M.; Demaurex, N. L-OPA1 regulates mitoflash biogenesis independently from membrane fusion. EMBO Rep. 2017, 18, 451–463. [Google Scholar] [CrossRef] [PubMed]
- Hou, T.; Wang, X.; Ma, Q.; Cheng, H. Mitochondrial flashes: New insights into mitochondrial ROS signaling and beyond. J. Physiol. 2014, 592, 3703–3713. [Google Scholar] [CrossRef] [PubMed]
- Schwarzländer, M.; Wagner, S.; Ermakova, Y.G.; Belousov, V.V.; Radi, R.; Beckman, J.S.; Buettner, G.R.; Demaurex, N.; Duchen, M.R.; Forman, H.J.; et al. The ‘mitoflash’ probe cpYFP does not respond to superoxide. Nature 2014, 514, E12–E14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarzländer, M.; Murphy, M.P.; Duchen, M.R.; Logan, D.C.; Fricker, M.D.; Halestrap, A.P.; Müller, F.L.; Rizzuto, R.; Dick, T.P.; Meyer, A.J.; et al. Mitochondrial “flashes”: A radical concept repHined. Trends Cell Biol. 2012, 22, 503–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, T.; Jian, C.; Xu, J.; Huang, A.Y.; Xi, J.; Hu, K.; Wei, L.; Cheng, H.; Wang, X. Identification of EFHD1 as a novel Ca2+ sensor for mitoflash activation. Cell Calcium 2016, 59, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Hagen, S.; Brachs, S.; Kroczek, C.; Fürnrohr, B.G.; Lang, C.; Mielenz, D. The B cell receptor-induced calcium flux involves a calcium mediated positive feedback loop. Cell Calcium 2012, 51, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Poburko, D.; Santo-Domingo, J.; Demaurex, N. Dynamic regulation of the mitochondrial proton gradient during cytosolic calcium elevations. J. Biol. Chem. 2011, 286, 11672–11684. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Urbanczyk, S.; Stein, M.; Schuh, W.; Jäck, H.-M.; Mougiakakos, D.; Mielenz, D. Regulation of Energy Metabolism during Early B Lymphocyte Development. Int. J. Mol. Sci. 2018, 19, 2192. https://doi.org/10.3390/ijms19082192
Urbanczyk S, Stein M, Schuh W, Jäck H-M, Mougiakakos D, Mielenz D. Regulation of Energy Metabolism during Early B Lymphocyte Development. International Journal of Molecular Sciences. 2018; 19(8):2192. https://doi.org/10.3390/ijms19082192
Chicago/Turabian StyleUrbanczyk, Sophia, Merle Stein, Wolfgang Schuh, Hans-Martin Jäck, Dimitrios Mougiakakos, and Dirk Mielenz. 2018. "Regulation of Energy Metabolism during Early B Lymphocyte Development" International Journal of Molecular Sciences 19, no. 8: 2192. https://doi.org/10.3390/ijms19082192
APA StyleUrbanczyk, S., Stein, M., Schuh, W., Jäck, H. -M., Mougiakakos, D., & Mielenz, D. (2018). Regulation of Energy Metabolism during Early B Lymphocyte Development. International Journal of Molecular Sciences, 19(8), 2192. https://doi.org/10.3390/ijms19082192