The Winding Roadmap of Biomarkers Toward Clinic: Lessons from Predictors of Resistance to Anti-EGFRs in Metastatic Colorectal Cancer



Abstract
:1. Introduction
2. BRAF V600E Mutation: Going beyond Formal Statistical Demonstrations
3. Atypical RAS and BRAF Mutations: What Do They Mean?
4. HER2: Preliminary Retrospective Evidence
5. How to Deal with Very Rare Alterations? The Example of Gene Fusions
6. Looking for Prospective Evidence: The Case-Control PRESSING Study
7. New Biomarkers on the Horizon? Focus on Microsatellite Instability and Consensus Molecular Subtypes
8. Personalizing the Use of Anti-EGFRs: A Potential Application of Liquid Biopsy
9. Levels of Evidence and Pragmatic Approaches: How to Make the Roadmap Less Winding?
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AKT1 | Protein Kinase B |
| ALK | Anaplastic Lymphoma Kinase |
| AREG | Amphiregulin |
| BRAF | V-raf Murine Sarcoma Viral Oncogene Homolog b1 |
| BSC | Best Supportive Care |
| CMS | Consensus Molecular Subtypes |
| EGFR | Epidermal Growth Factor |
| EREG | Epiregulin |
| HER2 | Human Epidermal Growth Factor Receptor 2 |
| HNF4A | Hepatocyte Nuclear Factor 4α |
| IGF2 | Insulin-Like Growth Factor 2 |
| IRS2 | Insulin Receptor Substrate 2 |
| KRAS | Kirsten Rat Sarcoma Viral Oncogene Homolog |
| mCRC | Metastatic Colorectal Cancer |
| moAbs | Monoclonal Antibodies |
| MET | MET Proto-Oncogene |
| MPE | Molecular Pathological Epidemiology |
| NRAS | Neuroblastoma RAS Viral Oncogene Homolog |
| NTRK 1–3 | Neurotrophic Tyrosine Kinase Receptor 1–3 |
| OS | Overall Survival |
| PI3K | Phosphoinositide 3-Kinase |
| PFS | Progression-Free Survival |
| PTEN | Phosphatase and TENsin Homolog |
| RET | Rearranged during Transfection |
| ROS1 | ROS Proto-Oncogene 1 |
| RR | Response Rate |
| Trk | Tropomyosin Receptor Kinase |
References
- Bokemeyer, C.; Bondarenko, I.; Makhson, A.; Hartmann, J.T.; Aparicio, J.; de Braud, F.; Donea, S.; Ludwig, H.; Schuch, G.; Stroh, C.; et al. Fluorouracil, leucovorin, and oxaliplatin with and without cetuximab in the first-line treatment of metastatic colorectal cancer. J. Clin. Oncol. 2009, 27, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Douillard, J.Y.; Siena, S.; Cassidy, J.; Tabernero, J.; Burkes, R.; Barugel, M.; Humblet, Y.; Bodoky, G.; Cunningham, D.; Jassem, J.; et al. Randomized, phase III trial of panitumumab with infusional fluorouracil, leucovorin, and oxaliplatin (FOLFOX4) versus FOLFOX4 alone as first-line treatment in patients with previously untreated metastatic colorectal cancer: The PRIME study. J. Clin. Oncol. 2010, 28, 4697–4705. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Köhne, C.H.; Hitre, E.; Zaluski, J.; Chang Chien, C.R.; Makhson, A.; D’Haens, G.; Pintér, T.; Lim, R.; Bodoky, G.; et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N. Engl. J. Med. 2009, 360, 1408–1417. [Google Scholar] [CrossRef] [PubMed]
- Maughan, T.S.; Adams, R.A.; Smith, C.G.; Meade, A.M.; Seymour, M.T.; Wilson, R.H.; Idziaszczyk, S.; Harris, R.; Fisher, D.; Kenny, S.L.; et al. Addition of cetuximab to oxaliplatin-based first-line combination chemotherapy for treatment of advanced colorectal cancer: Results of the randomised phase 3 MRC COIN trial. Lancet 2011, 377, 2103–2114. [Google Scholar] [CrossRef]
- Douillard, J.Y.; Oliner, K.S.; Siena, S.; Tabernero, J.; Burkes, R.; Barugel, M.; Humblet, Y.; Bodoky, G.; Cunningham, D.; Jassem, J.; et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N. Engl. J. Med. 2013, 369, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- De Roock, W.; Claes, B.; Bernasconi, D.; De Schutter, J.; Biesmans, B.; Fountzilas, G.; Kalogeras, K.T.; Kotoula, V.; Papamichael, D.; Laurent-Puig, P.; et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: A retrospective consortium analysis. Lancet Oncol. 2010, 11, 753–762. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Cervantes, A.; Adam, R.; Sobrero, A.; Van Krieken, J.H.; Aderka, D.; Aranda Aguilar, E.; Bardelli, A.; Benson, A.; Bodoky, G.; et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann. Oncol. 2016, 27, 1386–1422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sepulveda, A.R.; Hamilton, S.R.; Allegra, C.J.; Grody, W.; Cushman-Vokoun, A.M.; Funkhouser, W.K.; Kopetz, S.E.; Lieu, C.; Lindor, N.M.; Minsky, B.D.; et al. Molecular Biomarkers for the Evaluation of Colorectal Cancer: Guideline From the American Society for Clinical Pathology, College of American Pathologists, Association for Molecular Pathology, and the American Society of Clinical Oncology. J. Clin. Oncol. 2017, 35, 1453–1486. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Souglakos, J.; Philips, J.; Wang, R.; Marwah, S.; Silver, M.; Tzardi, M.; Silver, J.; Ogino, S.; Hooshmand, S.; Kwak, E.; et al. Prognostic and predictive value of common mutations for treatment response and survival in patients with metastatic colorectal cancer. Br. J. Cancer 2009, 101, 465–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tol, J.; Nagtegaal, I.D.; Punt, C.J.A. BRAF mutation in metastatic colorectal cancer. N. Engl. J. Med. 2009, 361, 98–99. [Google Scholar] [CrossRef] [PubMed]
- Roth, A.D.; Tejpar, S.; Delorenzi, M.; Yan, P.; Fiocca, R.; Klingbiel, D.; Dietrich, D.; Biesmans, B.; Bodoky, G.; Barone, C.; et al. Prognostic role of KRAS and BRAF in stage II and III resected colon cancer: Results of the translational study on the PETACC-3, EORTC 40993, SAKK 60-00 trial. J. Clin. Oncol. 2010, 28, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Richman, S.D.; Seymour, M.T.; Chambers, P.; Elliott, F.; Daly, C.L.; Meade, A.M.; Taylor, G.; Barrett, J.H.; Quirke, P. KRAS and BRAF mutations in advanced colorectal cancer are associated with poor prognosis but do not preclude benefit from oxaliplatin or irinotecan: Results from the MRC FOCUS trial. J. Clin. Oncol. 2009, 27, 5931–5937. [Google Scholar] [CrossRef] [PubMed]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lochhead, P.; Kuchiba, A.; Imamura, Y.; Liao, X.; Yamauchi, M.; Nishihara, R.; Qian, Z.R.; Morikawa, T.; Shen, J.; Meyerhardt, J.A.; et al. Microsatellite instability and BRAF mutation testing in colorectal cancer prognostication. J. Natl. Cancer Inst. 2013, 105, 1151–1156. [Google Scholar] [CrossRef] [PubMed]
- Di Nicolantonio, F.; Martini, M.; Molinari, F.; Sartore-Bianchi, A.; Arena, S.; Saletti, P.; De Dosso, S.; Mazzucchelli, L.; Frattini, M.; Siena, S.; et al. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J. Clin. Oncol. 2008, 26, 5705–5712. [Google Scholar] [CrossRef] [PubMed]
- Loupakis, F.; Ruzzo, A.; Cremolini, C.; Vincenzi, B.; Salvatore, L.; Santini, D.; Masi, G.; Stasi, I.; Canestrari, E.; Rulli, E.; et al. KRAS codon 61, 146 and BRAF mutations predict resistance to cetuximab plus irinotecan in KRAS codon 12 and 13 wild-type metastatic colorectal cancer. Br. J. Cancer 2009, 101, 715–721. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.X.; Wang, X.Y.; Qin, Q.Y.; Chen, D.F.; Zhong, Q.H.; Wang, L.; Wang, J.P. The prognostic role of BRAF mutation in metastatic colorectal cancer receiving anti-EGFR monoclonal antibodies: A meta-analysis. PLoS ONE 2013, 8, e65995. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Xu, A.T.; Zhu, M.M.; Tong, J.L.; Xu, X.T.; Ran, Z.H. Predictive and prognostic roles of BRAF mutation in patients with metastatic colorectal cancer treated with anti-epidermal growth factor receptor monoclonal antibodies: A meta-analysis. J. Dig. Dis. 2013, 14, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Cui, D.; Cao, D.; Yang, Y.; Qiu, M.; Huang, Y.; Yi, C. Effect of BRAF V600E mutation on tumor response of anti-EGFR monoclonal antibodies for first-line metastatic colorectal cancer treatment: A meta-analysis of randomized studies. Mol. Biol. Rep. 2014, 41, 1291–1298. [Google Scholar] [CrossRef] [PubMed]
- Pietrantonio, F.; Petrelli, F.; Coinu, A.; Di Bartolomeo, M.; Borgonovo, K.; Maggi, C.; Cabiddu, M.; Iacovelli, R.; Bossi, I.; Lonati, V.; et al. Predictive role of BRAF mutations in patients with advanced colorectal cancer receiving cetuximab and panitumumab: A meta-analysis. Eur. J. Cancer 2015, 51, 587–594. [Google Scholar] [CrossRef] [PubMed]
- Rowland, A.; Dias, M.M.; Wiese, M.D.; Kichenadasse, G.; McKinnon, R.A.; Karapetis, C.S.; Sorich, M.J. Meta-analysis of BRAF mutation as a predictive biomarker of benefit from anti-EGFR monoclonal antibody therapy for RAS wild-type metastatic colorectal cancer. Br. J. Cancer 2015, 112, 1888–1894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masi, G.; Loupakis, F.; Salvatore, L.; Fornaro, L.; Cremolini, C.; Cupini, S.; Ciarlo, A.; Del Monte, F.; Cortesi, E.; Amoroso, D.; et al. Bevacizumab with FOLFOXIRI (irinotecan, oxaliplatin, fluorouracil, and folinate) as first-line treatment for metastatic colorectal cancer: A phase 2 trial. Lancet Oncol. 2010, 11, 845–852. [Google Scholar] [CrossRef]
- Cremolini, C.; Loupakis, F.; Antoniotti, C.; Lupi, C.; Sensi, E.; Lonardi, S.; Mezi, S.; Tomasello, G.; Ronzoni, M.; Zaniboni, A.; et al. FOLFOXIRI plus bevacizumab versus FOLFIRI plus bevacizumab as first-line treatment of patients with metastatic colorectal cancer: Updated overall survival and molecular subgroup analyses of the open-label, phase 3 TRIBE study. Lancet Oncol. 2015, 16, 1306–1315. [Google Scholar] [CrossRef]
- Loupakis, F.; Cremolini, C.; Salvatore, L.; Masi, G.; Sensi, E.; Schirripa, M.; Michelucci, A.; Pfanner, E.; Brunetti, I.; Lupi, C.; et al. FOLFOXIRI plus bevacizumab as first-line treatment in BRAF mutant metastatic colorectal cancer. Eur. J. Cancer 2014, 50, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Kopetz, S.; Desai, J.; Chan, E.; Hecht, J.R.; O’Dwyer, P.J.; Maru, D.; Morris, V.; Janku, F.; Dasari, A.; Chung, W.; et al. Phase II Pilot Study of Vemurafenib in Patients With Metastatic BRAF-Mutated Colorectal Cancer. J. Clin. Oncol. 2015, 33, 4032–4038. [Google Scholar] [CrossRef] [PubMed]
- Tabernero, J.; Geel, R.V.; Guren, T.K.; Yaeger, R.D.; Spreafico, A.; Faris, J.E.; Yoshino, T.; Yamada, Y.; Kim, T.W.; Bendell, J.C.; et al. Phase 2 results: Encorafenib (ENCO) and cetuximab (CETUX) with or without alpelisib (ALP) in patients with advanced BRAF-mutant colorectal cancer (BRAFm CRC). J. Clin. Oncol. 2016, 34, 3544. [Google Scholar] [CrossRef]
- Atreya, C.E.; Van Cutsem, E.; Bendell, J.C.; Andre, T.; Schellens, J.H.; Gordon, M.S.; McRee, A.J.; O’Dwyer, P.J.; Muro, K.; Tabernero, J.; et al. Updated efficacy of the MEK inhibitor trametinib (T), BRAF inhibitor dabrafenib (D), and anti-EGFR antibody panitumumab (P) in patients (pts) with BRAF V600E mutated (BRAFm) metastatic colorectal cancer (mCRC). J. Clin. Oncol. 2015, 33. Available online: http://ascopubs.org/doi/abs/10.1200/jco.2015.33.15_suppl.103 (accessed on 2 Jun 2018).
- Van Geel, R.M.J.M.; Tabernero, J.; Elez, E.; Bendell, J.C.; Spreafico, A.; Schuler, M.; Yoshino, T.; Delord, J.P.; Yamada, Y.; Lolkema, M.P.; Faris, J.E.; et al. A Phase Ib Dose-Escalation Study of Encorafenib and Cetuximab with or without Alpelisib in Metastatic BRAF-Mutant Colorectal Cancer. Cancer Discov. 2017, 7, 610–619. [Google Scholar] [CrossRef] [PubMed]
- Study of Encorafenib + Cetuximab Plus or Minus Binimetinib vs. Irinotecan/Cetuximab or Infusional 5-Fluorouracil (5-FU)/Folinic Acid (FA)/Irinotecan (FOLFIRI)/Cetuximab with a Safety Lead-in of Encorafenib + Binimetinib + Cetuximab in Patients with BRAF V600E-mutant Metastatic Colorectal Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT02928224 (accessed on 16 May 2018).
- Han, S.W.; Kim, H.P.; Shin, J.Y.; Jeong, E.G.; Lee, W.C.; Lee, K.H.; Won, J.K.; Kim, T.Y.; Oh, D.Y.; Im, S.A.; et al. Targeted sequencing of cancer-related genes in colorectal cancer using next-generation sequencing. PLoS ONE 2013, 8, e64271. [Google Scholar] [CrossRef] [PubMed]
- Malapelle, U.; Vigliar, E.; Sgariglia, R.; Bellevicine, C.; Colarossi, L.; Vitale, D.; Pallante, P.; Troncone, G. Ion Torrent next-generation sequencing for routine identification of clinically relevant mutations in colorectal cancer patients. J. Clin. Pathol. 2015, 68, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Loree, J.M.; Miron, B.; Holla, V.; Overman, M.J.; Pereira, A.A.L.; Lam, M.; Morris, V.K.; Raghav, K.P.S.; Routbort, M.; Shaw, K.R.; et al. Not all RAS mutations created equal: Functional and clinical characterization of 80 different KRAS and NRAS mutations. J. Clin. Oncol. 2017, 35, 3589. Available online: http://ascopubs.org/doi/abs/10.1200/JCO.2017.35.15_suppl.3589 (accessed on 18 May 2018).
- Cremolini, C.; Di Bartolomeo, M.; Amatu, A.; Antoniotti, C.; Moretto, R.; Berenato, R.; Perrone, F.; Tamborini, E.; Aprile, G.; Lonardi, S.; et al. BRAF codons 594 and 596 mutations identify a new molecular subtype of metastatic colorectal cancer at favorable prognosis. Ann. Oncol. 2015, 26, 2092–2097. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.C.; Renfro, L.A.; Al-Shamsi, H.O.; Schrock, A.B.; Rankin, A.; Zhang, B.Y.; Kasi, P.M.; Voss, J.S.; Leal, A.D.; Sun, J.; et al. Non-V600 BRAF Mutations Define a Clinically Distinct Molecular Subtype of Metastatic Colorectal Cancer. J. Clin. Oncol. 2017, 35, 2624–2630. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Dekervel, J. Not All BRAF-Mutant Metastatic Colorectal Cancers Are Identical: Distinct Clinical Consequences of non-V600 BRAF Mutations. J. Clin. Oncol. 2017, 35, 2598–2599. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Yaeger, R.; Rodrik-Outmezguine, V.S.; Tao, A.; Torres, N.M.; Chang, M.T.; Drosten, M.; Zhao, H.; Cecchi, F.; Hembrough, T.; et al. Tumors with class 3 BRAF mutants are sensitive to the inhibition of activated RAS. Nature 2017, 548, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Bertotti, A.; Migliardi, G.; Galimi, F.; Sassi, F.; Torti, D.; Isella, C.; Corà, D.; Di Nicolantonio, F.; Buscarino, M.; Petti, C.; et al. A molecularly annotated platform of patient-derived xenografts (“xenopatients”) identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer Discov. 2011, 1, 508–523. [Google Scholar] [CrossRef] [PubMed]
- Yonesaka, K.; Zejnullahu, K.; Okamoto, I.; Satoh, T.; Cappuzzo, F.; Souglakos, J.; Ercan, D.; Rogers, A.; Roncalli, M.; Takeda, M.; et al. Activation of ERBB2 signaling causes resistance to the EGFR-directed therapeutic antibody cetuximab. Sci. Transl. Med. 2011, 3, 99ra86. [Google Scholar] [CrossRef] [PubMed]
- Martin, V.; Landi, L.; Molinari, F.; Fountzilas, G.; Geva, R.; Riva, A.; Saletti, P.; De Dosso, S.; Spitale, A.; Tejpar, S.; et al. HER2 gene copy number status may influence clinical efficacy to anti-EGFR monoclonal antibodies in metastatic colorectal cancer patients. Br. J. Cancer 2013, 108, 668–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raghav, K.P.S.; Overman, M.J.; Yu, R.; Meric-Bernstam, F.; Menter, D.; Kee, B.K.; Muranyi, A.; Singh, S.; Routbort, M.; Chen, K.; et al. HER2 amplification as a negative predictive biomarker for anti-epidermal growth factor receptor antibody therapy in metastatic colorectal cancer. J. Clin. Oncol. 2016, 34, 3517. [Google Scholar] [CrossRef]
- Sartore-Bianchi, A.; Amatu, A.; Porcu, L.; Ghezzi, S.; Lonardi, S.; Bergamo, F.; Bonazzina, E.F.; Mauri, G.; Cremolini, C.; Ciardiello, F.; et al. Clinicopathological characteristics and HER2 status in metastatic colorectal cancer patients: Results of a diagnostic model development study. J. Clin. Oncol. 2018, 36, 581. [Google Scholar] [CrossRef]
- Sawada, K.; Nakamura, Y.; Yamanaka, T.; Kuboki, Y.; Yamaguchi, D.; Yuki, S.; Yoshino, T.; Komatsu, Y.; Sakamoto, N.; Okamoto, W.; et al. Prognostic and Predictive Value of HER2 Amplification in Patients with Metastatic Colorectal Cancer. Clin. Colorectal Cancer 2018. [Google Scholar] [CrossRef] [PubMed]
- Cremolini, C.; Morano, F.; Moretto, R.; Berenato, R.; Tamborini, E.; Perrone, F.; Rossini, D.; Gloghini, A.; Busico, A.; Zucchelli, G.; et al. Negative hyper-selection of metastatic colorectal cancer patients for anti-EGFR monoclonal antibodies: The PRESSING case-control study. Ann. Oncol. 2017, 28, 3009–3014. [Google Scholar] [CrossRef] [PubMed]
- Sartore-Bianchi, A.; Trusolino, L.; Martino, C.; Bencardino, K.; Lonardi, S.; Bergamo, F.; Zagonel, V.; Leone, F.; Depetris, I.; Martinelli, E.; et al. Dual-targeted therapy with trastuzumab and lapatinib in treatment-refractory, KRAS codon 12/13 wild-type, HER2-positive metastatic colorectal cancer (HERACLES): A proof-of-concept, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016, 17, 738–746. [Google Scholar] [CrossRef]
- Hainsworth, J.D.; Meric-Bernstam, F.; Swanton, C.; Hurwitz, H.; Spigel, D.R.; Sweeney, C.; Burris, H.; Bose, R.; Yoo, B.; Stein, A.; et al. Targeted Therapy for Advanced Solid Tumors on the Basis of Molecular Profiles: Results From MyPathway, an Open-Label, Phase IIa Multiple Basket Study. J. Clin. Oncol. 2018, 36, 536–542. [Google Scholar] [CrossRef] [PubMed]
- Medico, E.; Russo, M.; Picco, G.; Cancelliere, C.; Valtorta, E.; Corti, G.; Buscarino, M.; Isella, C.; Lamba, S.; Martinoglio, B.; et al. The molecular landscape of colorectal cancer cell lines unveils clinically actionable kinase targets. Nat. Commun. 2015, 6, 7002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amatu, A.; Sartore-Bianchi, A.; Siena, S. NTRK gene fusions as novel targets of cancer therapy across multiple tumor types. ESMO Open 2016, 1, e000023. [Google Scholar] [CrossRef] [PubMed]
- Lipson, D.; Capelletti, M.; Yelensky, R.; Otto, G.; Parker, A.; Jarosz, M.; Curran, J.A.; Balasubramanian, S.; Bloom, T.; Brennan, K.W.; et al. Identification of new ALK and RET gene fusions from colorectal and lung cancer biopsies. Nat. Med. 2012, 18, 382–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houang, M.; Toon, C.W.; Clarkson, A.; Sioson, L.; de Silva, K.; Watson, N.; Singh, N.R.; Chou, A.; Gill, A.J. ALK and ROS1 overexpression is very rare in colorectal adenocarcinoma. Appl. Immunohistochem. Mol. Morphol. 2015, 23, 134–138. [Google Scholar] [CrossRef] [PubMed]
- Créancier, L.; Vandenberghe, I.; Gomes, B.; Dejean, C.; Blanchet, J.-C.; Meilleroux, J.; Guimbaud, R.; Selves, J.; Kruczynski, A. Chromosomal rearrangements involving the NTRK1 gene in colorectal carcinoma. Cancer Lett. 2015, 365, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Lin, E.; Li, L.; Guan, Y.; Soriano, R.; Rivers, C.S.; Mohan, S.; Pandita, A.; Tang, J.; Modrusan, Z. Exon array profiling detects EML4-ALK fusion in breast, colorectal, and non-small cell lung cancers. Mol. Cancer Res. MCR 2009, 7, 1466–1476. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Hsu, P.P.; Awad, M.M.; Engelman, J.A. Tyrosine kinase gene rearrangements in epithelial malignancies. Nat. Rev. Cancer 2013, 13, 772–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietrantonio, F.; Di Nicolantonio, F.; Schrock, A.B.; Lee, J.; Tejpar, S.; Sartore-Bianchi, A.; Hechtman, J.F.; Christiansen, J.; Novara, L.; Tebbutt, N.; et al. ALK, ROS1, and NTRK Rearrangements in Metastatic Colorectal Cancer. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef] [PubMed]
- Pietrantonio, F.; Di Nicolantonio, F.; Schrock, A.B.; Lee, J.; Morano, F.; Fucà, G.; Nikolinakos, P.; Drilon, A.; Hechtman, J.F.; Christiansen, J.; et al. RET fusions in a small subset of advanced colorectal cancers at risk of being neglected. Ann. Oncol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Rolfo, C.; Ruiz, R.; Giovannetti, E.; Gil-Bazo, I.; Russo, A.; Passiglia, F.; Giallombardo, M.; Peeters, M.; Raez, L. Entrectinib: A potent new TRK, ROS1, and ALK inhibitor. Expert Opin. Investig. Drugs 2015, 24, 1493–1500. [Google Scholar] [CrossRef] [PubMed]
- Sartore-Bianchi, A.; Ardini, E.; Bosotti, R.; Amatu, A.; Valtorta, E.; Somaschini, A.; Raddrizzani, L.; Palmeri, L.; Banfi, P.; Bonazzina, E.; et al. Sensitivity to Entrectinib Associated with a Novel LMNA-NTRK1 Gene Fusion in Metastatic Colorectal Cancer. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A.; Nagasubramanian, R.; Blake, J.F.; Ku, N.; Tuch, B.B.; Ebata, K.; Smith, S.; Lauriault, V.; Kolakowski, G.R.; Brandhuber, B.J.; et al. A Next-Generation TRK Kinase Inhibitor Overcomes Acquired Resistance to Prior TRK Kinase Inhibition in Patients with TRK Fusion-Positive Solid Tumors. Cancer Discov. 2017, 7, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A.; Laetsch, T.W.; Kummar, S.; DuBois, S.G.; Lassen, U.N.; Demetri, G.D.; Nathenson, M.; Doebele, R.C.; Farago, A.F.; Pappo, A.S.; et al. Efficacy of Larotrectinib in TRK Fusion-Positive Cancers in Adults and Children. N. Engl. J. Med. 2018, 378, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Basket Study of Entrectinib (RXDX-101) for the Treatment of Patients With Solid Tumors Harboring NTRK 1/2/3 (Trk A/B/C), ROS1, or ALK Gene Rearrangements (Fusions). Available online: https://clinicaltrials.gov/ct2/show/NCT02568267 (accessed on 18 May 2018).
- Burki, T.K. Larotrectinib in TRK fusion-positive cancers. Lancet Oncol. 2018, 19, e187. [Google Scholar] [CrossRef]
- Subbiah, V.; Velcheti, V.; Tuch, B.B.; Ebata, K.; Busaidy, N.L.; Cabanillas, M.E.; Wirth, L.J.; Stock, S.; Smith, S.; Lauriault, V.; Corsi-Travali, S.; et al. Selective RET Kinase Inhibition for Patients with RET-Altered Cancers. Ann. Oncol. [CrossRef] [PubMed]
- Holch, J.W.; Ricard, I.; Stintzing, S.; Modest, D.P.; Heinemann, V. The relevance of primary tumor location in patients with metastatic colorectal cancer: A meta-analysis of first-line clinical trials. Eur. J. Cancer 2017, 70, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Benson, A.B.; Venook, A.P.; Al-Hawary, M.M.; Cederquist, L.; Chen, Y.J.; Ciombor, K.K.; Cohen, S.; Cooper, H.S.; Deming, D.; Engstrom, P.F.; et al. NCCN Guidelines Insights: Colon Cancer, Version 2. 2018. J. Natl. Compr. Cancer Netw. 2018, 16, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Overman, M.J.; McDermott, R.; Leach, J.L.; Lonardi, S.; Lenz, H.J.; Morse, M.A.; Desai, J.; Hill, A.; Axelson, M.; Moss, R.A.; et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): An open-label, multicentre, phase 2 study. Lancet Oncol. 2017, 18, 1182–1191. [Google Scholar] [CrossRef]
- Overman, M.J.; Lonardi, S.; Wong, K.Y.M.; Lenz, H.J.; Gelsomino, F.; Aglietta, M.; Morse, M.A.; Van Cutsem, E.; McDermott, R.; Hill, A.; et al. Durable Clinical Benefit With Nivolumab Plus Ipilimumab in DNA Mismatch Repair-Deficient/Microsatellite Instability-High Metastatic Colorectal Cancer. J. Clin. Oncol. 2018, 36, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Innocenti, F.; Ou, F.S.; Zemla, T.; Niedzwiecki, D.; Qu, X.; Tam, R.; Mahajan, S.; Goldberg, R.M.; Mayer, R.J.; Bertagnolli, M.M. Somatic DNA mutations, MSI status, mutational load (ML): Association with overall survival (OS) in patients (pts) with metastatic colorectal cancer (mCRC) of CALGB/SWOG 80405 (Alliance). J. Clin. Oncol. 2017, 35, 3504. Available online: http://ascopubs.org/doi/abs/10.1200/JCO.2017.35.15_suppl.3504 (accessed on 3 June 2018).
- Dienstmann, R.; Vermeulen, L.; Guinney, J.; Kopetz, S.; Tejpar, S.; Tabernero, J. Consensus molecular subtypes and the evolution of precision medicine in colorectal cancer. Nat. Rev. Cancer 2017, 17, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Sveen, A.; Bruun, J.; Eide, P.W.; Eilertsen, I.A.; Ramirez, L.; Murumägi, A.; Arjama, M.; Danielsen, S.A.; Kryeziu, K.; Elez, E.; et al. Colorectal Cancer Consensus Molecular Subtypes Translated to Preclinical Models Uncover Potentially Targetable Cancer Cell Dependencies. Clin. Cancer Res. 2018, 24, 794–806. [Google Scholar] [CrossRef] [PubMed]
- Lenz, H.J.; Ou, F.S.; Venook, A.P.; Hochster, H.S.; Niedzwiecki, D.; Goldberg, R.M.; Mayer, R.J.; Bertagnolli, M.M.; Blanke, C.D.; Zemla, T.; et al. Impact of consensus molecular subtyping (CMS) on overall survival (OS) and progression free survival (PFS) in patients (pts) with metastatic colorectal cancer (mCRC): Analysis of CALGB/SWOG 80405 (Alliance). J. Clin. Oncol. 2017, 35, 3511. [Google Scholar] [CrossRef]
- Stintzing, S.; Wirapati, P.; Lenz, H.J.; Neureiter, D.; Fischer von Weikersthal, L.; Decker, T.; Kiani, A.; Vehling-Kaiser, U.; Al-Batran, S.E.; Heintges, T.; et al. Consensus molecular subgroups (CMS) of colorectal cancer (CRC) and first-line efficacy of FOLFIRI plus cetuximab or bevacizumab in the FIRE3 (AIO KRK-0306) trial. J. Clin. Oncol. 2017, 35, 3510. [Google Scholar] [CrossRef]
- Vidal, J.; Muinelo, L.; Dalmases, A.; Jones, F.; Edelstein, D.; Iglesias, M.; Orrillo, M.; Abalo, A.; Rodríguez, C.; Brozos, E.; et al. Plasma ctDNA RAS mutation analysis for the diagnosis and treatment monitoring of metastatic colorectal cancer patients. Ann. Oncol. 2017, 28, 1325–1332. [Google Scholar] [CrossRef] [PubMed]
- Bachet, J.B.; Bouché, O.; Taieb, J.; Dubreuil, O.; Garcia, M.L.; Meurisse, A.; Normand, C.; Gornet, J.M.; Artru, P.; Louafi, S.; et al. RAS mutation analysis in circulating tumor DNA from patients with metastatic colorectal cancer: The AGEO RASANC prospective multicenter study. Ann. Oncol. 2018, 29, 1211–1219. [Google Scholar] [CrossRef] [PubMed]
- Siravegna, G.; Mussolin, B.; Buscarino, M.; Corti, G.; Cassingena, A.; Crisafulli, G.; Ponzetti, A.; Cremolini, C.; Amatu, A.; Lauricella, C.; et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat. Med. 2015, 21, 827. [Google Scholar] [CrossRef] [PubMed]
- Schmiegel, W.; Scott, R.J.; Dooley, S.; Lewis, W.; Meldrum, C.J.; Pockney, P.; Draganic, B.; Smith, S.; Hewitt, C.; Philimore, H.; et al. Blood-based detection of RAS mutations to guide anti-EGFR therapy in colorectal cancer patients: Concordance of results from circulating tumor DNA and tissue-based RAS testing. Mol. Oncol. 2017, 11, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Jones, F.S.; Edelstein, D.; Wichner, K.; Ross, C.; Stieler, K.; Boehm, V.; Lenfert, E.; Bien, J.; Lukas, A.; Macioszek, J. Performance of standardized BEAMing platform for detecting RAS mutations in the blood of metastatic colorectal cancer (mCRC) patients. J. Clin. Oncol. 2016, 34, 1153. Available online: http://ascopubs.org/doi/abs/10.1200/JCO.2016.34.15_suppl.11538 (accessed on 30 Jul 2018).
- Normanno, N.; Esposito Abate, R.; Lambiase, M.; Forgione, L.; Cardone, C.; Iannaccone, A.; Sacco, A.; Rachiglio, A.M.; Martinelli, E.; Rizzi, D.; et al. CAPRI-GOIM Investigators RAS testing of liquid biopsy correlates with the outcome of metastatic colorectal cancer patients treated with first-line FOLFIRI plus cetuximab in the CAPRI-GOIM trial. Ann. Oncol. 2018, 29, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Grasselli, J.; Elez, E.; Caratù, G.; Matito, J.; Santos, C.; Macarulla, T.; Vidal, J.; Garcia, M.; Viéitez, J.M.; Paéz, D.; et al. Concordance of blood- and tumor-based detection of RAS mutations to guide anti-EGFR therapy in metastatic colorectal cancer. Ann. Oncol. 2017, 28, 1294–1301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misale, S.; Di Nicolantonio, F.; Sartore-Bianchi, A.; Siena, S.; Bardelli, A. Resistance to anti-EGFR therapy in colorectal cancer: From heterogeneity to convergent evolution. Cancer Discov. 2014, 4, 1269–1280. [Google Scholar] [CrossRef] [PubMed]
- Arena, S.; Bellosillo, B.; Siravegna, G.; Martínez, A.; Cañadas, I.; Lazzari, L.; Ferruz, N.; Russo, M.; Misale, S.; González, I.; et al. Emergence of Multiple EGFR Extracellular Mutations during Cetuximab Treatment in Colorectal Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 2157–2166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bardelli, A.; Corso, S.; Bertotti, A.; Hobor, S.; Valtorta, E.; Siravegna, G.; Sartore-Bianchi, A.; Scala, E.; Cassingena, A.; Zecchin, D.; et al. Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov. 2013, 3, 658–673. [Google Scholar] [CrossRef] [PubMed]
- Misale, S.; Yaeger, R.; Hobor, S.; Scala, E.; Janakiraman, M.; Liska, D.; Valtorta, E.; Schiavo, R.; Buscarino, M.; Siravegna, G.; et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature 2012, 486, 532–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montagut, C.; Dalmases, A.; Bellosillo, B.; Crespo, M.; Pairet, S.; Iglesias, M.; Salido, M.; Gallen, M.; Marsters, S.; Tsai, S.P.; et al. Identification of a mutation in the extracellular domain of the Epidermal Growth Factor Receptor conferring cetuximab resistance in colorectal cancer. Nat. Med. 2012, 18, 221–223. [Google Scholar] [CrossRef] [PubMed]
- Rossini, D.; Cremolini, C.; Conca, E.; Del Re, M.; Busico, A.; Pietrantonio, F.; Bergamo, F.; Danesi, R.; Casagrande, M.; Tamburini, E.; et al. Liquid biopsy to predict benefit from rechallenge with cetuximab (cet) + irinotecan (iri) in RAS/BRAF wild-type metastatic colorectal cancer patients (pts) with acquired resistance to first-line cet+iri: Final results and translational analyses of the CRICKET study by GONO. J. Clin. Oncol. 2018, 36, 12007. [Google Scholar]
- Petrelli, F.; Borgonovo, K.; Barni, S. The predictive role of skin rash with cetuximab and panitumumab in colorectal cancer patients: A systematic review and meta-analysis of published trials. Target. Oncol. 2013, 8, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, M.C.; Wu, C.F.; Chen, C.W.; Shi, C.S.; Huang, W.S.; Kuan, F.C. Hypomagnesemia and clinical benefits of anti-EGFR monoclonal antibodies in wild-type KRAS metastatic colorectal cancer: A systematic review and meta-analysis. Sci. Rep. 2018, 8, 2047. [Google Scholar] [CrossRef] [PubMed]
- Ogino, S.; Chan, A.T.; Fuchs, C.S.; Giovannucci, E. Molecular pathological epidemiology of colorectal neoplasia: An emerging transdisciplinary and interdisciplinary field. Gut 2011, 60, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Ogino, S.; Nowak, J.A.; Hamada, T.; Phipps, A.I.; Peters, U.; Milner, D.A.; Giovannucci, E.L.; Nishihara, R.; Giannakis, M.; Garrett, W.S.; et al. Integrative analysis of exogenous, endogenous, tumor and immune factors for precision medicine. Gut 2018, 67, 1168–1180. [Google Scholar] [CrossRef] [PubMed]


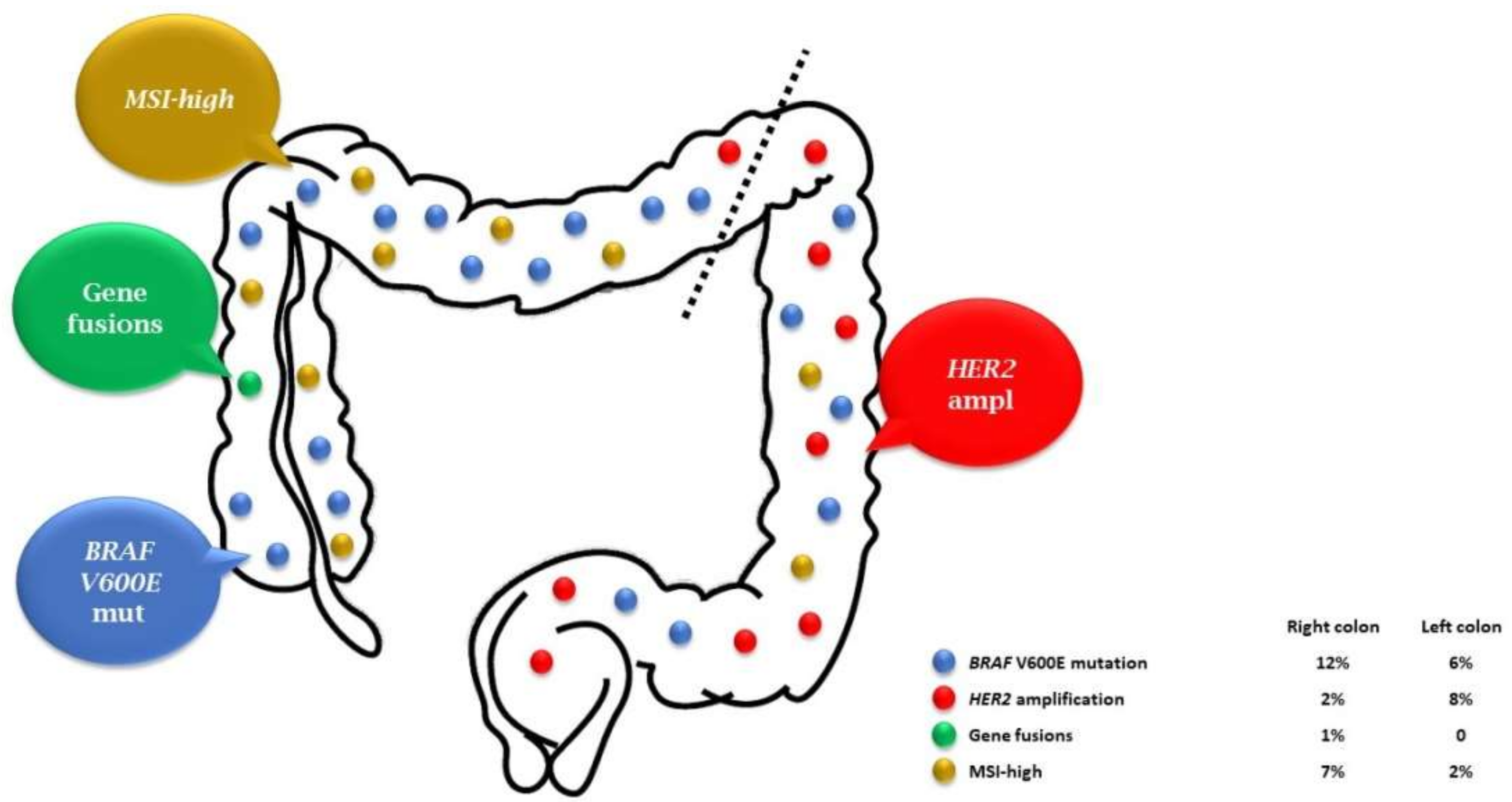{kind=link}
| Reference | Study Design | Population | Main Results |
|---|---|---|---|
| Bertotti et al. [38] | Preclinical | 85 xenopatients a, expanded in two molecularly unselected cohorts; randomized to receive or not cetuximab | HER2 amplification or overexpression in 6 cases out of 44 KRAS wild-type patients resistant to anti-EGFR vs. 0 out of 45 KRAS wild-type patients with objective response to anti-EGFR (p < 0.05) |
| Yonesaka et al. [39] | Retrospective | 182 KRAS wild-type patients treated with cetuximab-based therapy | Worse outcome (PFS and OS) for patients with HER2-amplified vs. HER2-nonamplified tumors |
| Martin et al. [40] | Retrospective | 162 KRAS wild-type patients treated with anti-EGFR | Worse outcome (RR, PFS and OS) for patients with HER2 FISH+ vs. HER2 FISH− tumors |
| Raghav et al. [41] | Retrospective | 196 RAS and BRAF wild-type mCRC patients treated with anti-EGFR therapy | Worse outcome (PFS) for patients with HER2-amplified vs. HER2-nonamplified tumors |
| Sartore-Bianchi et al. [42] | Retrospective | 80 patients with HER2-amplified and KRAS wild-type tumors | Worse outcome (RR and PFS) for patients treated with anti-EGFR vs. patient not treated with anti-EGFR |
| Sawada et al. [43] | Retrospective | 11 patients with HER2-amplified and RAS and BRAF wild-type tumors | Worse outcome (RR, PFS and OS) for patients with HER2-amplified and RAS/BRAF wild-type vs. HER2-nonamplified and RAS/BRAF wild-type tumors |
| Cremolini et al. [44] | Prospectivecase-control | 94 RAS/BRAF wild-type patients: 47 patients resistant and 47 patients sensitive to anti-EGFR-based therapy | HER amplification in 7 cases out of 47 resistant patients vs. 0 out 47 sensitive patients (p = 0.01) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Antoniotti, C.; Ongaro, E.; Falcone, A.; Cremolini, C. The Winding Roadmap of Biomarkers Toward Clinic: Lessons from Predictors of Resistance to Anti-EGFRs in Metastatic Colorectal Cancer. Int. J. Mol. Sci. 2018, 19, 2298. https://doi.org/10.3390/ijms19082298
Antoniotti C, Ongaro E, Falcone A, Cremolini C. The Winding Roadmap of Biomarkers Toward Clinic: Lessons from Predictors of Resistance to Anti-EGFRs in Metastatic Colorectal Cancer. International Journal of Molecular Sciences. 2018; 19(8):2298. https://doi.org/10.3390/ijms19082298
Chicago/Turabian StyleAntoniotti, Carlotta, Elena Ongaro, Alfredo Falcone, and Chiara Cremolini. 2018. "The Winding Roadmap of Biomarkers Toward Clinic: Lessons from Predictors of Resistance to Anti-EGFRs in Metastatic Colorectal Cancer" International Journal of Molecular Sciences 19, no. 8: 2298. https://doi.org/10.3390/ijms19082298
APA StyleAntoniotti, C., Ongaro, E., Falcone, A., & Cremolini, C. (2018). The Winding Roadmap of Biomarkers Toward Clinic: Lessons from Predictors of Resistance to Anti-EGFRs in Metastatic Colorectal Cancer. International Journal of Molecular Sciences, 19(8), 2298. https://doi.org/10.3390/ijms19082298