PGC-1α Protects RPE Cells of the Aging Retina against Oxidative Stress-Induced Degeneration through the Regulation of Senescence and Mitochondrial Quality Control. The Significance for AMD Pathogenesis




Abstract
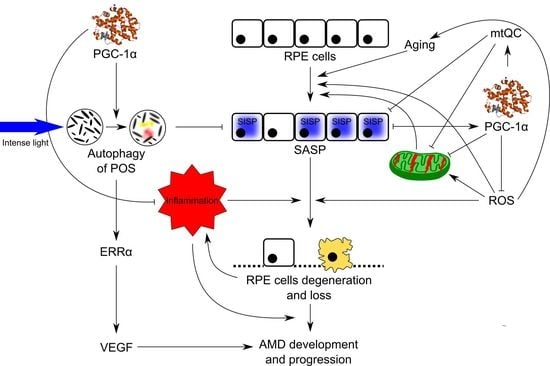
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
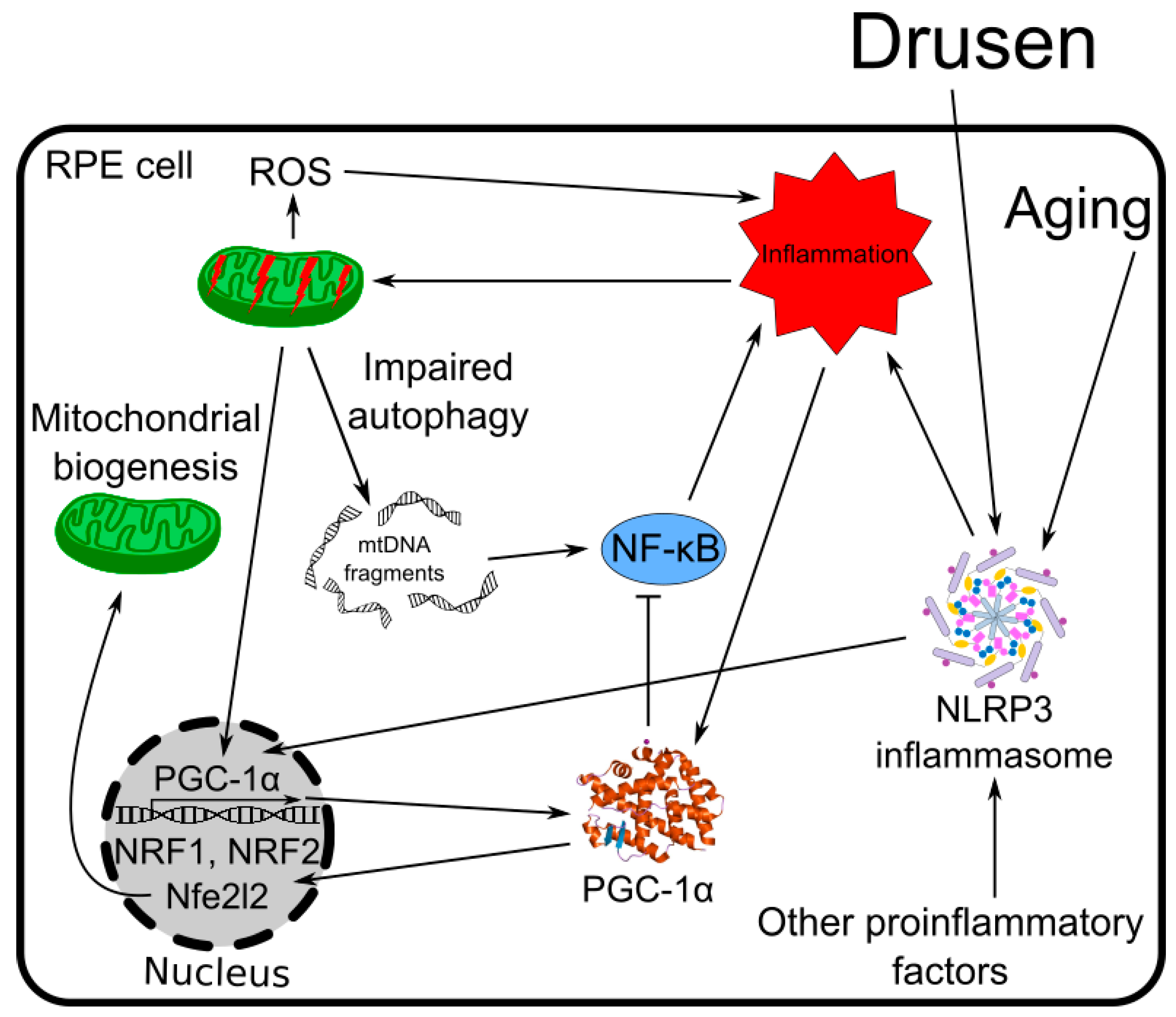{kind=link}
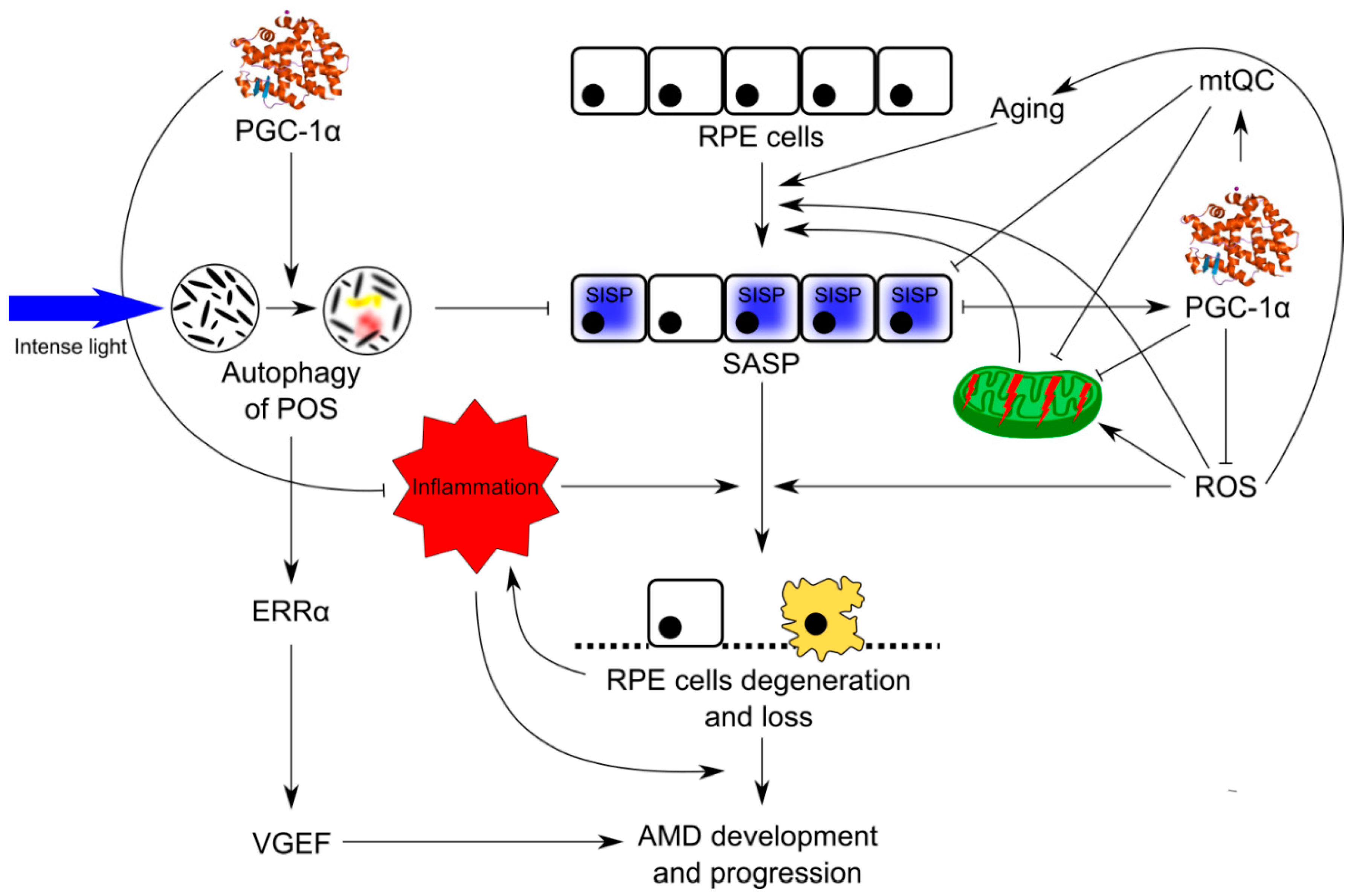{kind=link}
1. Introduction
2. PGC-1α—The Gene and the Protein
3. PGC-1α—A Key Regulator of Mitochondrial Biogenesis and Redox Control
4. Stress-Induced Senescence and Mitochondrial Dysfunction—Major Factors of AMD Pathogenesis
5. Involvement of PGC-1α in Senescence Regulation in RPE Cells
6. Potential of PGC-1α in AMD
7. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Bowes Rickman, C.; Farsiu, S.; Toth, C.A.; Klingeborn, M. Dry age-related macular degeneration: Mechanisms, therapeutic targets, and imaging. Investig. Ophthalmol. Vis. Sci. 2013, 54, ORSF68–ORSF80. [Google Scholar] [CrossRef] [PubMed]
- De Jong, P. Elusive drusen and changing terminology of AMD. Eye, 9 February 2018. [Google Scholar]
- Barot, M.; Gokulgandhi, M.R.; Mitra, A.K. Mitochondrial Dysfunction in Retinal Diseases. Curr. Eye Res. 2011, 36, 1069–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feher, J.; Kovacs, I.; Artico, M.; Cavallotti, C.; Papale, A.; Balacco Gabrieli, C. Mitochondrial alterations of retinal pigment epithelium in age-related macular degeneration. Neurobiol. Aging 2006, 27, 983–993. [Google Scholar] [CrossRef] [PubMed]
- Nordgaard, C.L.; Berg, K.M.; Kapphahn, R.J.; Reilly, C.; Feng, X.; Olsen, T.W.; Ferrington, D.A. Proteomics of the retinal pigment epithelium reveals altered protein expression at progressive stages of age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2006, 47, 815–822. [Google Scholar] [CrossRef] [PubMed]
- Nordgaard, C.L.; Karunadharma, P.P.; Feng, X.; Olsen, T.W.; Ferrington, D.A. Mitochondrial proteomics of the retinal pigment epithelium at progressive stages of age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2008, 49, 2848–2855. [Google Scholar] [CrossRef] [PubMed]
- Puigserver, P.; Adelmant, G.; Wu, Z.; Fan, M.; Xu, J.; O’Malley, B.; Spiegelman, B.M. Activation of PPARgamma coactivator-1 through transcription factor docking. Science 1999, 286, 1368–1371. [Google Scholar] [CrossRef] [PubMed]
- Egger, A.; Samardzija, M.; Sothilingam, V.; Tanimoto, N.; Lange, C.; Salatino, S.; Fang, L.; Garcia-Garrido, M.; Beck, S.; Okoniewski, M.J.; et al. PGC-1α determines light damage susceptibility of the murine retina. PLoS ONE 2012, 7, e31272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blasiak, J.; Piechota, M.; Pawlowska, E.; Szatkowska, M.; Sikora, E.; Kaarniranta, K. Cellular Senescence in Age-Related Macular Degeneration: Can Autophagy and DNA Damage Response Play a Role? Oxid. Med. Cell. Longev. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Kozlowski, M.R. RPE cell senescence: A key contributor to age-related macular degeneration. Med. Hypotheses 2012, 78, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Wang, K.; Zhang, K.; Zhou, F.; Zhu, L. Induction of oxidative and nitrosative stresses in human retinal pigment epithelial cells by all-trans-retinal. Exp. Cell Res. 2016, 348, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Maeda, A.; Maeda, T.; Golczak, M.; Chou, S.; Desai, A.; Hoppel, C.L.; Matsuyama, S.; Palczewski, K. Involvement of all-trans-retinal in acute light-induced retinopathy of mice. J. Biol. Chem. 2009, 284, 15173–15183. [Google Scholar] [CrossRef] [PubMed]
- Sawada, O.; Perusek, L.; Kohno, H.; Howell, S.J.; Maeda, A.; Matsuyama, S.; Maeda, T. All-trans-retinal induces Bax activation via DNA damage to mediate retinal cell apoptosis. Exp. Eye Res. 2014, 123, 27–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Cai, X.; Xia, Q.; Yao, K.; Chen, J.; Zhang, Y.; Naranmandura, H.; Liu, X.; Wu, Y. Involvement of endoplasmic reticulum stress in all-trans-retinal-induced retinal pigment epithelium degeneration. Toxicol. Sci. 2015, 143, 196–208. [Google Scholar] [CrossRef] [PubMed]
- Popov, D.V.; Lysenko, E.A.; Vepkhvadze, T.F.; Kurochkina, N.S.; Maknovskii, P.A.; Vinogradova, O.L. Promoter-specific regulation of PPARGC1A gene expression in human skeletal muscle. J. Mol. Endocrinol. 2015, 55, 159–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miura, S.; Kai, Y.; Kamei, Y.; Ezaki, O. Isoform-specific increases in murine skeletal muscle peroxisome proliferator-activated receptor-gamma coactivator-1α (PGC-1α) mRNA in response to β2-adrenergic receptor activation and exercise. Endocrinology 2008, 149, 4527–4533. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, T.; Inagaki, K.; Noguchi, T.; Sakai, M.; Ogawa, W.; Hosooka, T.; Iguchi, H.; Watanabe, E.; Matsuki, Y.; Hiramatsu, R.; et al. Identification and characterization of an alternative promoter of the human PGC-1α gene. Biochem. Biophys. Res. Commun. 2009, 381, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Esterbauer, H.; Oberkofler, H.; Krempler, F.; Patsch, W. Human peroxisome proliferator activated receptor γ coactivator 1 (PPARGC1) gene: cDNA sequence, genomic organization, chromosomal localization, and tissue expression. Genomics 1999, 62, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.Z.; Liu, Z.; Liu, J.; Ren, J.X.; Sun, T.S. Mitochondrial DNA induces inflammation and increases TLR9/NF-kappaB expression in lung tissue. Int. J. Mol. Med. 2014, 33, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Puigserver, P.; Wu, Z.; Park, C.W.; Graves, R.; Wright, M.; Spiegelman, B.M. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 1998, 92, 829–839. [Google Scholar] [CrossRef]
- Knutti, D.; Kressler, D.; Kralli, A. Regulation of the transcriptional coactivator PGC-1 via MAPK-sensitive interaction with a repressor. Proc. Natl. Acad. Sci. USA 2001, 98, 9713–9718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallberg, A.E.; Yamamura, S.; Malik, S.; Spiegelman, B.M.; Roeder, R.G. Coordination of p300-mediated chromatin remodeling and TRAP/mediator function through coactivator PGC-1α. Mol. Cell 2003, 12, 1137–1149. [Google Scholar] [CrossRef]
- Jager, S.; Handschin, C.; St-Pierre, J.; Spiegelman, B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1α. Proc. Natl. Acad. Sci. USA 2007, 104, 12017–12022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Monks, B.; Ge, Q.; Birnbaum, M.J. Akt/PKB regulates hepatic metabolism by directly inhibiting PGC-1α transcription coactivator. Nature 2007, 447, 1012–1016. [Google Scholar] [CrossRef] [PubMed]
- Olson, B.L.; Hock, M.B.; Ekholm-Reed, S.; Wohlschlegel, J.A.; Dev, K.K.; Kralli, A.; Reed, S.I. SCFCdc4 acts antagonistically to the PGC-1α transcriptional coactivator by targeting it for ubiquitin-mediated proteolysis. Genes Dev. 2008, 22, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Puigserver, P.; Rhee, J.; Lin, J.; Wu, Z.; Yoon, J.C.; Zhang, C.Y.; Krauss, S.; Mootha, V.K.; Lowell, B.B.; Spiegelman, B.M. Cytokine stimulation of energy expenditure through p38 MAP kinase activation of PPARγ coactivator-1. Mol. Cell 2001, 8, 971–982. [Google Scholar] [CrossRef]
- Canto, C.; Auwerx, J. PGC-1α, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr. Opin. Lipidol. 2009, 20, 98–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Housley, M.P.; Udeshi, N.D.; Rodgers, J.T.; Shabanowitz, J.; Puigserver, P.; Hunt, D.F.; Hart, G.W. A PGC-1α-O-GlcNAc transferase complex regulates FoxO transcription factor activity in response to glucose. J. Biol. Chem. 2009, 284, 5148–5157. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Marcos, P.J.; Auwerx, J. Regulation of PGC-1α, a nodal regulator of mitochondrial biogenesis. Am. J. Clin. Nutr. 2011, 93, 884S–890S. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Huypens, P.; Adamson, A.W.; Chang, J.S.; Henagan, T.M.; Boudreau, A.; Lenard, N.R.; Burk, D.; Klein, J.; Perwitz, N.; et al. Alternative mRNA splicing produces a novel biologically active short isoform of PGC-1α. J. Biol. Chem. 2009, 284, 32813–32826. [Google Scholar] [CrossRef] [PubMed]
- Suliman, H.B.; Piantadosi, C.A. Mitochondrial Quality Control as a Therapeutic Target. Pharmacol. Rev. 2016, 68, 20–48. [Google Scholar] [CrossRef] [PubMed]
- Cherry, A.D.; Piantadosi, C.A. Regulation of mitochondrial biogenesis and its intersection with inflammatory responses. Antioxid. Redox Signal. 2015, 22, 965–976. [Google Scholar] [CrossRef] [PubMed]
- Cherry, A.D.; Suliman, H.B.; Bartz, R.R.; Piantadosi, C.A. Peroxisome proliferator-activated receptor γ co-activator 1-α as a critical co-activator of the murine hepatic oxidative stress response and mitochondrial biogenesis in Staphylococcus aureus sepsis. J. Biol. Chem. 2014, 289, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.Q.; Cammarata, P.R.; Baines, C.P.; Yager, J.D. Regulation of mitochondrial respiratory chain biogenesis by estrogens/estrogen receptors and physiological, pathological and pharmacological implications. Biochim. Biophys. Acta 2009, 1793, 1540–1570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gleyzer, N.; Vercauteren, K.; Scarpulla, R.C. Control of mitochondrial transcription specificity factors (TFB1M and TFB2M) by nuclear respiratory factors (NRF-1 and NRF-2) and PGC-1 family coactivators. Mol. Cell. Biol. 2005, 25, 1354–1366. [Google Scholar] [CrossRef] [PubMed]
- Vercauteren, K.; Gleyzer, N.; Scarpulla, R.C. PGC-1-related coactivator complexes with HCF-1 and NRF-2β in mediating NRF-2(GABP)-dependent respiratory gene expression. J. Biol. Chem. 2008, 283, 12102–12111. [Google Scholar] [CrossRef] [PubMed]
- Vercauteren, K.; Pasko, R.A.; Gleyzer, N.; Marino, V.M.; Scarpulla, R.C. PGC-1-related coactivator: Immediate early expression and characterization of a CREB/NRF-1 binding domain associated with cytochrome c promoter occupancy and respiratory growth. Mol. Cell. Biol. 2006, 26, 7409–7419. [Google Scholar] [CrossRef] [PubMed]
- Eiyama, A.; Okamoto, K. PINK1/Parkin-mediated mitophagy in mammalian cells. Curr. Opin. Cell Biol. 2015, 33, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Kowald, A.; Kirkwood, T.B. The evolution and role of mitochondrial fusion and fission in aging and disease. Commun. Integr. Biol. 2011, 4, 627–629. [Google Scholar] [CrossRef] [PubMed]
- Cartoni, R.; Leger, B.; Hock, M.B.; Praz, M.; Crettenand, A.; Pich, S.; Ziltener, J.L.; Luthi, F.; Deriaz, O.; Zorzano, A.; et al. Mitofusins 1/2 and ERRalpha expression are increased in human skeletal muscle after physical exercise. J. Physiol. 2005, 567 Pt 1, 349–358. [Google Scholar] [CrossRef]
- Martin, O.J.; Lai, L.; Soundarapandian, M.M.; Leone, T.C.; Zorzano, A.; Keller, M.P.; Attie, A.D.; Muoio, D.M.; Kelly, D.P. A role for peroxisome proliferator-activated receptor γ coactivator-1 in the control of mitochondrial dynamics during postnatal cardiac growth. Circ. Res. 2014, 114, 626–636. [Google Scholar] [CrossRef] [PubMed]
- Mihaylova, M.M.; Shaw, R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011, 13, 1016–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mailloux, R.J.; Harper, M.E. Uncoupling proteins and the control of mitochondrial reactive oxygen species production. Free Radic. Biol. Med. 2011, 51, 1106–1115. [Google Scholar] [CrossRef] [PubMed]
- Mattiasson, G.; Shamloo, M.; Gido, G.; Mathi, K.; Tomasevic, G.; Yi, S.; Warden, C.H.; Castilho, R.F.; Melcher, T.; Gonzalez-Zulueta, M.; et al. Uncoupling protein-2 prevents neuronal death and diminishes brain dysfunction after stroke and brain trauma. Nat. Med. 2003, 9, 1062–1068. [Google Scholar] [CrossRef] [PubMed]
- Rigoulet, M.; Yoboue, E.D.; Devin, A. Mitochondrial ROS generation and its regulation: Mechanisms involved in H2O2 signaling. Antioxid. Redox Signal. 2011, 14, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Barnstable, C.J.; Tombran-Tink, J. Molecular mechanisms of neuroprotection in the eye. Adv. Exp. Med. Biol. 2006, 572, 291–295. [Google Scholar] [PubMed]
- Borniquel, S.; Valle, I.; Cadenas, S.; Lamas, S.; Monsalve, M. Nitric oxide regulates mitochondrial oxidative stress protection via the transcriptional coactivator PGC-1α. FASEB J. 2006, 20, 1889–1891. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Diaz, F.; Iommarini, L.; Aure, K.; Lombes, A.; Moraes, C.T. PGC-1α/β induced expression partially compensates for respiratory chain defects in cells from patients with mitochondrial disorders. Hum. Mol. Genet. 2009, 18, 1805–1812. [Google Scholar] [CrossRef] [PubMed]
- Jeyapalan, J.C.; Sedivy, J.M. Cellular senescence and organismal aging. Mech. Ageing Dev. 2008, 129, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Korolchuk, V.I.; Miwa, S.; Carroll, B.; von Zglinicki, T. Mitochondria in Cell Senescence: Is Mitophagy the Weakest Link? EBioMedicine 2017, 21, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Bonafe, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Campisi, J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69 (Suppl. 1), S4–S9. [Google Scholar] [CrossRef] [PubMed]
- Sikora, E.; Arendt, T.; Bennett, M.; Narita, M. Impact of cellular senescence signature on ageing research. Ageing Res. Rev. 2011, 10, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Al-Hussaini, H.; Kam, J.H.; Vugler, A.; Semo, M.; Jeffery, G. Mature retinal pigment epithelium cells are retained in the cell cycle and proliferate in vivo. Mol. Vis. 2008, 14, 1784–1791. [Google Scholar] [PubMed]
- Xia, H.; Krebs, M.P.; Kaushal, S.; Scott, E.W. Enhanced retinal pigment epithelium regeneration after injury in MRL/MpJ mice. Exp. Eye Res. 2011, 93, 862–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, D.Y.; Cringle, S.J. Oxygen distribution and consumption within the retina in vascularised and avascular retinas and in animal models of retinal disease. Prog. Retin. Eye Res. 2001, 20, 175–208. [Google Scholar] [CrossRef]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.E.; Lewin, A.S.; Ash, J.D. Mitochondria: Potential Targets for Protection in Age-Related Macular Degeneration. Adv. Exp. Med. Biol. 2018, 1074, 11–17. [Google Scholar] [PubMed]
- Ferrington, D.A.; Ebeling, M.C.; Kapphahn, R.J.; Terluk, M.R.; Fisher, C.R.; Polanco, J.R.; Roehrich, H.; Leary, M.M.; Geng, Z.; Dutton, J.R.; et al. Altered bioenergetics and enhanced resistance to oxidative stress in human retinal pigment epithelial cells from donors with age-related macular degeneration. Redox Biol. 2017, 13, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Ferrington, D.A.; Kapphahn, R.J.; Leary, M.M.; Atilano, S.R.; Terluk, M.R.; Karunadharma, P.; Chen, G.K.; Ratnapriya, R.; Swaroop, A.; Montezuma, S.R.; et al. Increased retinal mtDNA damage in the CFH variant associated with age-related macular degeneration. Exp. Eye Res. 2016, 145, 269–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyttinen, J.M.T.; Viiri, J.; Kaarniranta, K.; Blasiak, J. Mitochondrial quality control in AMD: Does mitophagy play a pivotal role? Cell. Mol. Life Sci. 2018, 75, 2991–3008. [Google Scholar] [CrossRef] [PubMed]
- Nashine, S.; Cohen, P.; Chwa, M.; Lu, S.; Nesburn, A.B.; Kuppermann, B.D.; Kenney, M.C. Humanin G (HNG) protects age-related macular degeneration (AMD) transmitochondrial ARPE-19 cybrids from mitochondrial and cellular damage. Cell Death Dis. 2017, 8, e2951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riazi-Esfahani, M.; Kuppermann, B.D.; Kenney, M.C. The Role of Mitochondria in AMD: Current Knowledge and Future Applications. J. Ophthalmic Vis. Res. 2017, 12, 424–428. [Google Scholar] [PubMed]
- Finnemann, S.C. Focal adhesion kinase signaling promotes phagocytosis of integrin-bound photoreceptors. EMBO J. 2003, 22, 4143–4154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finnemann, S.C.; Silverstein, R.L. Differential roles of CD36 and αβ5 integrin in photoreceptor phagocytosis by the retinal pigment epithelium. J. Exp. Med. 2001, 194, 1289–1298. [Google Scholar] [CrossRef] [PubMed]
- Kaarniranta, K.; Sinha, D.; Blasiak, J.; Kauppinen, A.; Vereb, Z.; Salminen, A.; Boulton, M.E.; Petrovski, G. Autophagy and heterophagy dysregulation leads to retinal pigment epithelium dysfunction and development of age-related macular degeneration. Autophagy 2013, 9, 973–984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, M.; Finnemann, S.C.; Febbraio, M.; Shan, L.; Annangudi, S.P.; Podrez, E.A.; Hoppe, G.; Darrow, R.; Organisciak, D.T.; Salomon, R.G.; et al. Light-induced oxidation of photoreceptor outer segment phospholipids generates ligands for CD36-mediated phagocytosis by retinal pigment epithelium: A potential mechanism for modulating outer segment phagocytosis under oxidant stress conditions. J. Biol. Chem. 2006, 281, 4222–4230. [Google Scholar] [CrossRef] [PubMed]
- Sparrow, J.R.; Gregory-Roberts, E.; Yamamoto, K.; Blonska, A.; Ghosh, S.K.; Ueda, K.; Zhou, J. The bisretinoids of retinal pigment epithelium. Prog. Retin. Eye Res. 2012, 31, 121–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valapala, M.; Wilson, C.; Hose, S.; Bhutto, I.A.; Grebe, R.; Dong, A.; Greenbaum, S.; Gu, L.; Sengupta, S.; Cano, M.; et al. Lysosomal-mediated waste clearance in retinal pigment epithelial cells is regulated by CRYBA1/βA3/A1-crystallin via V-ATPase-MTORC1 signaling. Autophagy 2014, 10, 480–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, P.K.; Marcheselli, V.L.; de Rivero Vaccari, J.C.; Gordon, W.C.; Jackson, F.E.; Bazan, N.G. Photoreceptor outer segment phagocytosis attenuates oxidative stress-induced apoptosis with concomitant neuroprotectin D1 synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 13158–13163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nandrot, E.F.; Kim, Y.; Brodie, S.E.; Huang, X.; Sheppard, D.; Finnemann, S.C. Loss of synchronized retinal phagocytosis and age-related blindness in mice lacking αβ5 integrin. J. Exp. Med. 2004, 200, 1539–1545. [Google Scholar] [CrossRef] [PubMed]
- Bringold, F.; Serrano, M. Tumor suppressors and oncogenes in cellular senescence. Exp. Gerontol. 2000, 35, 317–329. [Google Scholar] [CrossRef]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [PubMed]
- Cristofalo, V.J.; Lorenzini, A.; Allen, R.G.; Torres, C.; Tresini, M. Replicative senescence: A critical review. Mech. Ageing Dev. 2004, 125, 827–848. [Google Scholar] [CrossRef] [PubMed]
- Sin, T.K.; Yu, A.P.; Yung, B.Y.; Yip, S.P.; Chan, L.W.; Wong, C.S.; Ying, M.; Rudd, J.A.; Siu, P.M. Modulating effect of SIRT1 activation induced by resveratrol on Foxo1-associated apoptotic signalling in senescent heart. J. Physiol. 2014, 592, 2535–2548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, S.; Salazar, G.; Patrushev, N.; Ma, M.; Forouzandeh, F.; Hilenski, L.; Alexander, R.W. Peroxisome proliferator-activated receptor γ coactivator-1α is a central negative regulator of vascular senescence. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 988–998. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.F.; Santana, L.F.; Vermulst, M.; Tomazela, D.M.; Emond, M.J.; MacCoss, M.J.; Gollahon, K.; Martin, G.M.; Loeb, L.A.; Ladiges, W.C.; et al. Overexpression of catalase targeted to mitochondria attenuates murine cardiac aging. Circulation 2009, 119, 2789–2797. [Google Scholar] [CrossRef] [PubMed]
- Patrushev, N.; Seidel-Rogol, B.; Salazar, G. Angiotensin II requires zinc and downregulation of the zinc transporters ZnT3 and ZnT10 to induce senescence of vascular smooth muscle cells. PLoS ONE 2012, 7, e33211. [Google Scholar] [CrossRef] [PubMed]
- Schriner, S.E.; Linford, N.J.; Martin, G.M.; Treuting, P.; Ogburn, C.E.; Emond, M.; Coskun, P.E.; Ladiges, W.; Wolf, N.; Van Remmen, H.; et al. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science 2005, 308, 1909–1911. [Google Scholar] [CrossRef] [PubMed]
- Sikora, E.; Mosieniak, G.; Sliwinska, M.A. Morphological and Functional Characteristic of Senescent Cancer Cells. Curr. Drug Targets 2016, 17, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Roggia, M.F.; Ueta, T. αβ5 Integrin/FAK/PGC-1α Pathway Confers Protective Effects on Retinal Pigment Epithelium. PLoS ONE 2015, 10, e0134870. [Google Scholar] [CrossRef] [PubMed]
- Tsunemi, T.; La Spada, A.R. PGC-1α at the intersection of bioenergetics regulation and neuron function: From Huntington’s disease to Parkinson’s disease and beyond. Prog. Neurobiol. 2012, 97, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Liu, Z.; Feng, Z.; Hao, J.; Shen, W.; Li, X.; Sun, L.; Sharman, E.; Wang, Y.; Wertz, K.; et al. Hydroxytyrosol protects against oxidative damage by simultaneous activation of mitochondrial biogenesis and phase II detoxifying enzyme systems in retinal pigment epithelial cells. J. Nutr. Biochem. 2010, 21, 1089–1098. [Google Scholar] [CrossRef] [PubMed]
- Iacovelli, J.; Rowe, G.C.; Khadka, A.; Diaz-Aguilar, D.; Spencer, C.; Arany, Z.; Saint-Geniez, M. PGC-1α Induces Human RPE Oxidative Metabolism and Antioxidant Capacity. Investig. Ophthalmol. Vis. Sci. 2016, 57, 1038–1051. [Google Scholar] [CrossRef] [PubMed]
- Ueta, T.; Inoue, T.; Yuda, K.; Furukawa, T.; Yanagi, Y.; Tamaki, Y. Intense physiological light upregulates vascular endothelial growth factor and enhances choroidal neovascularization via peroxisome proliferator-activated receptor γ coactivator-1α in mice. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1366–1371. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Jeong, H.; Borovecki, F.; Parkhurst, C.N.; Tanese, N.; Krainc, D. Transcriptional repression of PGC-1α by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell 2006, 127, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Haroutunian, V.; Katsel, P.; Cardozo, C.P.; Ho, L.; Buxbaum, J.D.; Pasinetti, G.M. PGC-1α expression decreases in the Alzheimer disease brain as a function of dementia. Arch. Neurol. 2009, 66, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.H.; Ko, H.S.; Kang, H.; Lee, Y.; Lee, Y.I.; Pletinkova, O.; Troconso, J.C.; Dawson, V.L.; Dawson, T.M. PARIS (ZNF746) repression of PGC-1α contributes to neurodegeneration in Parkinson’s disease. Cell 2011, 144, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Erfurth, U.; Chong, V.; Loewenstein, A.; Larsen, M.; Souied, E.; Schlingemann, R.; Eldem, B.; Mones, J.; Richard, G.; Bandello, F. Guidelines for the management of neovascular age-related macular degeneration by the European Society of Retina Specialists (EURETINA). Br. J. Ophthalmol. 2014, 98, 1144–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sen, N.; Satija, Y.K.; Das, S. PGC-1α, a key modulator of p53, promotes cell survival upon metabolic stress. Mol. Cell 2011, 44, 621–634. [Google Scholar] [CrossRef] [PubMed]
- Xiong, S.; Patrushev, N.; Forouzandeh, F.; Hilenski, L.; Alexander, R.W. PGC-1α Modulates Telomere Function and DNA Damage in Protecting against Aging-Related Chronic Diseases. Cell Rep. 2015, 12, 1391–1399. [Google Scholar] [CrossRef] [PubMed]
- Blasiak, J.; Szaflik, J.P. DNA damage and repair in age-related macular degeneration. Front. Biosci. (Landmark Ed.) 2011, 16, 1291–1301. [Google Scholar] [CrossRef] [PubMed]
- Hyttinen, J.M.; Blasiak, J.; Niittykoski, M.; Kinnunen, K.; Kauppinen, A.; Salminen, A.; Kaarniranta, K. DNA damage response and autophagy in the degeneration of retinal pigment epithelial cells-Implications for age-related macular degeneration (AMD). Ageing Res. Rev. 2017, 36, 64–77. [Google Scholar] [CrossRef] [PubMed]
- Blasiak, J.; Synowiec, E.; Salminen, A.; Kaarniranta, K. Genetic variability in DNA repair proteins in age-related macular degeneration. Int. J. Mol. Sci. 2012, 13, 13378–13397. [Google Scholar] [CrossRef] [PubMed]
- Kaarniranta, K.; Tokarz, P.; Koskela, A.; Paterno, J.; Blasiak, J. Autophagy regulates death of retinal pigment epithelium cells in age-related macular degeneration. Cell Biol. Toxicol. 2017, 33, 113–128. [Google Scholar] [CrossRef] [PubMed]
- Saint-Geniez, M.; Jiang, A.; Abend, S.; Liu, L.; Sweigard, H.; Connor, K.M.; Arany, Z. PGC-1α regulates normal and pathological angiogenesis in the retina. Am. J. Pathol. 2013, 182, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.; Itin, A.; Alon, T.; Pe’er, J.; Gnessin, H.; Chan-Ling, T.; Keshet, E. Development of retinal vasculature is mediated by hypoxia-induced vascular endothelial growth factor (VEGF) expression by neuroglia. J. Neurosci. 1995, 15, 4738–4747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mimura, T.; Kaji, Y.; Noma, H.; Funatsu, H.; Okamoto, S. The role of SIRT1 in ocular aging. Exp. Eye Res. 2013, 116, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Kauppinen, A.; Paterno, J.J.; Blasiak, J.; Salminen, A.; Kaarniranta, K. Inflammation and its role in age-related macular degeneration. Cell. Mol. Life Sci. 2016, 73, 1765–1786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valle, I.; Alvarez-Barrientos, A.; Arza, E.; Lamas, S.; Monsalve, M. PGC-1α regulates the mitochondrial antioxidant defense system in vascular endothelial cells. Cardiovasc. Res. 2005, 66, 562–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Kanatous, S.B.; Thurmond, F.A.; Gallardo, T.; Isotani, E.; Bassel-Duby, R.; Williams, R.S. Regulation of mitochondrial biogenesis in skeletal muscle by CaMK. Science 2002, 296, 349–352. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 1999, 98, 115–124. [Google Scholar] [CrossRef]
- Eisele, P.S.; Furrer, R.; Beer, M.; Handschin, C. The PGC-1 coactivators promote an anti-inflammatory environment in skeletal muscle in vivo. Biochem. Biophys. Res. Commun. 2015, 464, 692–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golestaneh, N.; Chu, Y.; Cheng, S.K.; Cao, H.; Poliakov, E.; Berinstein, D.M. Repressed SIRT1/PGC-1α pathway and mitochondrial disintegration in iPSC-derived RPE disease model of age-related macular degeneration. J. Transl. Med. 2016, 14, 344. [Google Scholar] [CrossRef] [PubMed]
- SanGiovanni, J.P.; Chen, J.; Sapieha, P.; Aderman, C.M.; Stahl, A.; Clemons, T.E.; Chew, E.Y.; Smith, L.E. DNA sequence variants in PPARGC1A, a gene encoding a coactivator of the omega-3 LCPUFA sensing PPAR-RXR transcription complex, are associated with NV AMD and AMD-associated loci in genes of complement and VEGF signaling pathways. PLoS ONE 2013, 8, e53155. [Google Scholar] [CrossRef] [PubMed]
- Gerhart-Hines, Z.; Rodgers, J.T.; Bare, O.; Lerin, C.; Kim, S.H.; Mostoslavsky, R.; Alt, F.W.; Wu, Z.; Puigserver, P. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1α. EMBO J. 2007, 26, 1913–1923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Li, S.; Liu, T.; Borjigin, J.; Lin, J.D. Transcriptional coactivator PGC-1α integrates the mammalian clock and energy metabolism. Nature 2007, 447, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Molusky, M.M.; Ma, D.; Buelow, K.; Yin, L.; Lin, J.D. Peroxisomal localization and circadian regulation of ubiquitin-specific protease 2. PLoS ONE 2012, 7, e47970. [Google Scholar] [CrossRef] [PubMed]
- Pfluger, P.T.; Herranz, D.; Velasco-Miguel, S.; Serrano, M.; Tschop, M.H. Sirt1 protects against high-fat diet-induced metabolic damage. Proc. Natl. Acad. Sci. USA 2008, 105, 9793–9798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahin, E.; Colla, S.; Liesa, M.; Moslehi, J.; Muller, F.L.; Guo, M.; Cooper, M.; Kotton, D.; Fabian, A.J.; Walkey, C.; et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature 2011, 470, 359–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitter, S.K.; Song, C.; Qi, X.; Mao, H.; Rao, H.; Akin, D.; Lewin, A.; Grant, M.; Dunn, W., Jr.; Ding, J.; et al. Dysregulated autophagy in the RPE is associated with increased susceptibility to oxidative stress and AMD. Autophagy 2014, 10, 1989–2005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosmidou, C.; Efstathiou, N.E.; Hoang, M.V.; Notomi, S.; Konstantinou, E.K.; Hirano, M.; Takahashi, K.; Maidana, D.E.; Tsoka, P.; Young, L.; et al. Issues with the specificity of immunological reagents for NLRP3: Implications for age-related macular degeneration. Sci. Rep. 2018, 8, 461. [Google Scholar] [CrossRef] [PubMed]
- Bian, Z.M.; Field, M.G.; Elner, S.G.; Kahlenberg, J.M.; Elner, V.M. Distinct CD40L receptors mediate inflammasome activation and secretion of IL-1β and MCP-1 in cultured human retinal pigment epithelial cells. Exp. Eye Res. 2018, 170, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Chu, Y.; Mowery, J.; Konkel, B.; Galli, S.; Theos, A.C.; Golestaneh, N. PGC-1α repression and high fat diet induce age-related macular degeneration-like phenotypes in mice. Dis. Model. Mech. 2018. [Google Scholar] [CrossRef]







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaarniranta, K.; Kajdanek, J.; Morawiec, J.; Pawlowska, E.; Blasiak, J. PGC-1α Protects RPE Cells of the Aging Retina against Oxidative Stress-Induced Degeneration through the Regulation of Senescence and Mitochondrial Quality Control. The Significance for AMD Pathogenesis. Int. J. Mol. Sci. 2018, 19, 2317. https://doi.org/10.3390/ijms19082317
Kaarniranta K, Kajdanek J, Morawiec J, Pawlowska E, Blasiak J. PGC-1α Protects RPE Cells of the Aging Retina against Oxidative Stress-Induced Degeneration through the Regulation of Senescence and Mitochondrial Quality Control. The Significance for AMD Pathogenesis. International Journal of Molecular Sciences. 2018; 19(8):2317. https://doi.org/10.3390/ijms19082317
Chicago/Turabian StyleKaarniranta, Kai, Jakub Kajdanek, Jan Morawiec, Elzbieta Pawlowska, and Janusz Blasiak. 2018. "PGC-1α Protects RPE Cells of the Aging Retina against Oxidative Stress-Induced Degeneration through the Regulation of Senescence and Mitochondrial Quality Control. The Significance for AMD Pathogenesis" International Journal of Molecular Sciences 19, no. 8: 2317. https://doi.org/10.3390/ijms19082317
APA StyleKaarniranta, K., Kajdanek, J., Morawiec, J., Pawlowska, E., & Blasiak, J. (2018). PGC-1α Protects RPE Cells of the Aging Retina against Oxidative Stress-Induced Degeneration through the Regulation of Senescence and Mitochondrial Quality Control. The Significance for AMD Pathogenesis. International Journal of Molecular Sciences, 19(8), 2317. https://doi.org/10.3390/ijms19082317