The Controversial Role of TGF-β in Neovascular Age-Related Macular Degeneration Pathogenesis


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
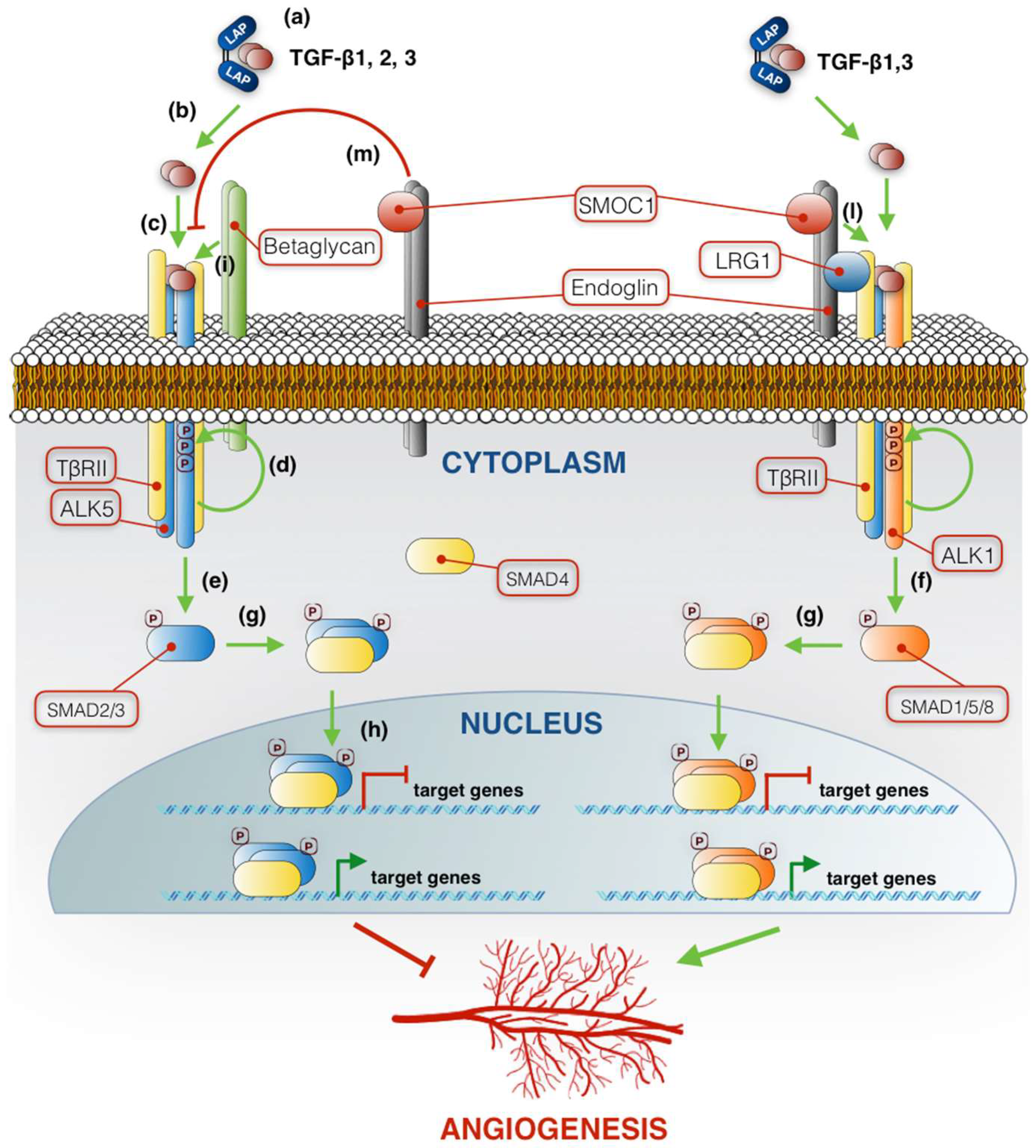
2. TGF-β Synthesis and Signaling Pathway
2.1. TGF-β Family Signaling in Angiogenesis
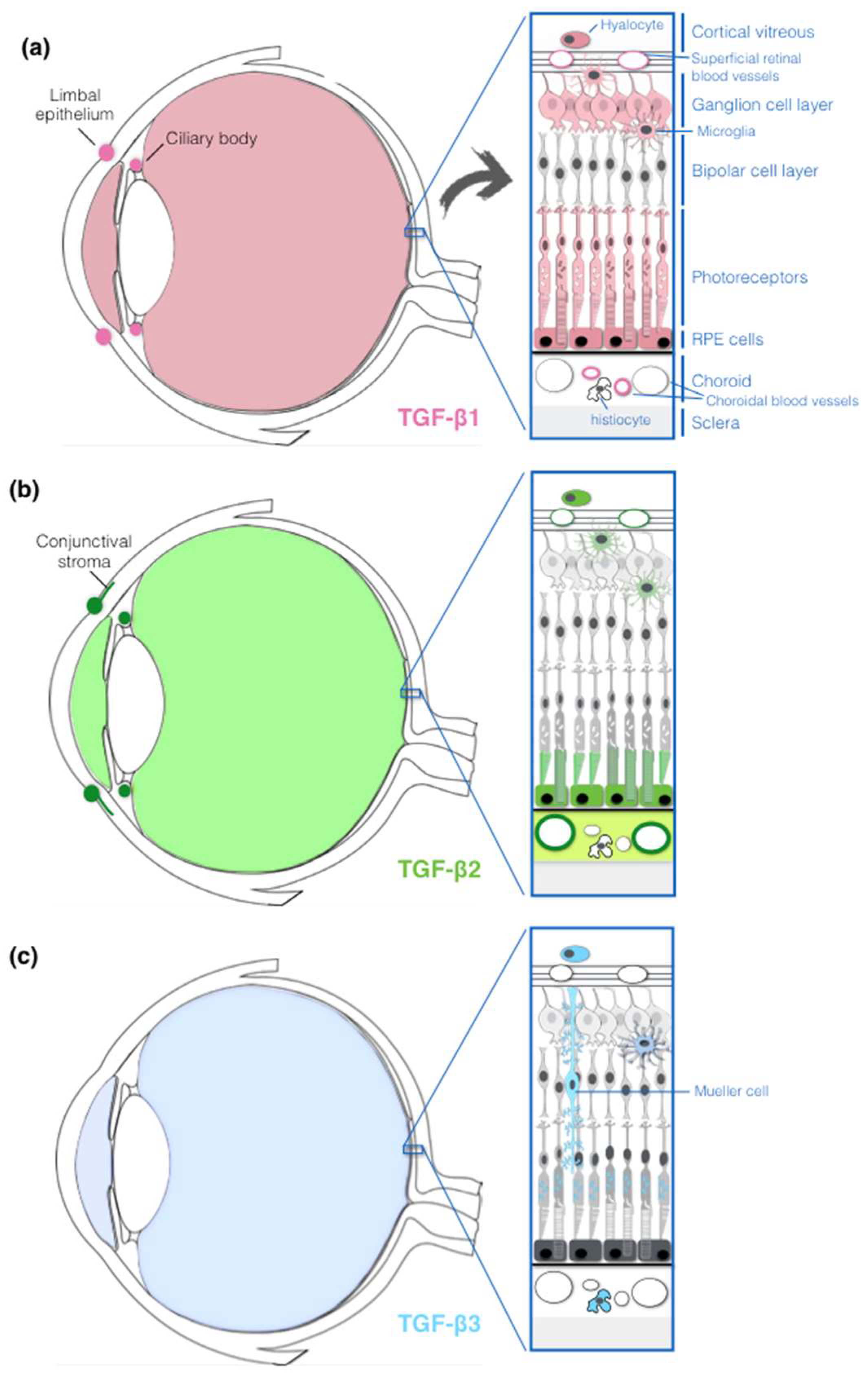
2.2. TGF-β Expression in the Human Eye
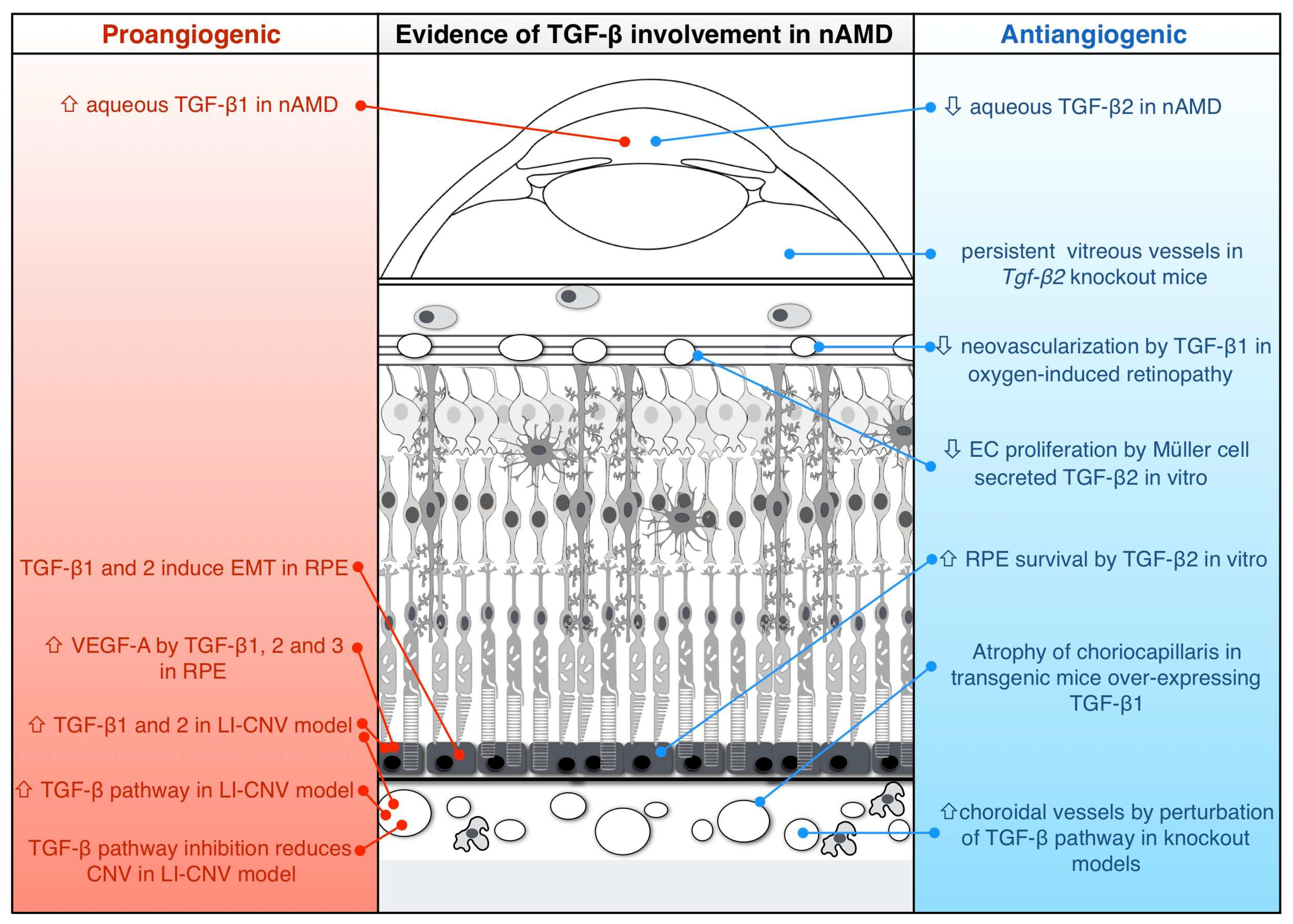
3. Evidence for Proangiogenic Function of TGF-β in nAMD
4. Evidence for Antiangiogenic Function of TGF-β in nAMD
5. TGF-β Signaling in RPE
6. TGF-β and Subretinal Fibrosis
7. TGF-β Signaling in Retinal Neuronal Cells
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| TGF-β | transforming growth factor-beta |
| nAMD | neovascular age-related macular degeneration |
| CNV | choroidal neovascularization |
| LI-CNV | laser-induced CNV |
| TβRII | TGF-β type II receptor |
| ALK | activin receptor-like kinase |
| SMAD | small mother against decapentaplegic |
| VEGF-A | vascular endothelial growth factor-A |
| RPE | retinal pigment epithelium |
| EC | endothelial cell |
| hpRPE | human primary RPE |
References
- Yang, S.; Zhao, J.; Sun, X. Resistance to anti-VEGF therapy in neovascular age-related macular degeneration: A comprehensive review. Drug Des. Devel. Ther. 2016, 10, 1857–1867. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Schor, S.L.; Hinck, A.P. Biological activity differences between TGF-β1 and TGF-β3 correlate with differences in the rigidity and arrangement of their component monomers. Biochemistry 2014, 53, 5737–5749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gentry, L.E.; Nash, B.W. The pro domain of pre-pro-transforming growth factor-β-1 when independently expressed is a functional binding-protein for the mature growth-factor. Biochemistry 1990, 29, 6851–6857. [Google Scholar] [CrossRef] [PubMed]
- Kusakabe, M.; Cheong, P.L.; Nikfar, R.; McLennan, I.S.; Koishi, K. The structure of the TGF-β latency associated peptide region determines the ability of the proprotein convertase furin to cleave TGF-βs. J. Cell. Biochem. 2008, 103, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Poniatowski, L.A.; Wojdasiewicz, P.; Gasik, R.; Szukiewicz, D. Transforming growth factor β family: Insight into the role of growth factors in regulation of fracture healing biology and potential clinical applications. Mediators Inflamm. 2015, 2015, 137823. [Google Scholar] [CrossRef] [PubMed]
- Budi, E.H.; Duan, D.; Derynck, R. Transforming growth factor-β receptors and smads: Regulatory complexity and functional versatility. Trends Cell Biol. 2017, 27, 658–672. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.P.; Seki, T.; Goss, K.A.; Imamura, T.; Yi, Y.; Donahoe, P.K.; Li, L.; Miyazono, K.; ten Dijke, P.; Kim, S.; et al. Activin receptor-like kinase 1 modulates transforming growth factor-β 1 signaling in the regulation of angiogenesis. Proc. Natl. Acad. Sci. USA 2000, 97, 2626–2631. [Google Scholar] [CrossRef] [PubMed]
- Goumans, M.J.; Valdimarsdottir, G.; Itoh, S.; Rosendahl, A.; Sideras, P.; ten Dijke, P. Balancing the activation state of the endothelium via two distinct TGF-β type I receptors. EMBO J. 2002, 21, 1743–1753. [Google Scholar] [CrossRef] [PubMed]
- Hill, C.S. Transcriptional control by the SMADs. Cold Spring Harb. Perspect. Biol. 2016, 8, a022079. [Google Scholar] [CrossRef] [PubMed]
- Goumans, M.J.; Valdimarsdottir, G.; Itoh, S.; Lebrin, F.; Larsson, J.; Mummery, C.; Karlsson, S.; ten Dijke, P. Activin receptor-like kinase (ALK)1 is an antagonistic mediator of lateral TGFβ/ALK5 signaling. Mol. Cell 2003, 12, 817–828. [Google Scholar] [CrossRef]
- Itoh, F.; Itoh, S.; Adachi, T.; Ichikawa, K.; Matsumura, Y.; Takagi, T.; Festing, M.; Watanabe, T.; Weinstein, M.; Karlsson, S.; et al. Smad2/Smad3 in endothelium is indispensable for vascular stability via S1PR1 and N-cadherin expressions. Blood 2012, 119, 5320–5328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walshe, T.E.; Saint-Geniez, M.; Maharaj, A.S.; Sekiyama, E.; Maldonado, A.E.; D’Amore, P.A. TGF-β is required for vascular barrier function, endothelial survival and homeostasis of the adult microvasculature. PLoS ONE 2009, 4, e5149. [Google Scholar] [CrossRef] [PubMed]
- Seki, T.; Yun, J.; Oh, S.P. Arterial endothelium-specific activin receptor-like kinase 1 expression suggests its role in arterialization and vascular remodeling. Circ. Res. 2003, 93, 682–689. [Google Scholar] [CrossRef] [PubMed]
- Cunha, S.I.; Pardali, E.; Thorikay, M.; Anderberg, C.; Hawinkels, L.; Goumans, M.-J.; Seehra, J.; Heldin, C.-H.; ten Dijke, P.; Pietras, K. Genetic and pharmacological targeting of activin receptor-like kinase 1 impairs tumor growth and angiogenesis. J. Exp. Med. 2010, 207, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Martín, E.M.; Blanco, F.J.; Roquè, M.; Novensà, L.; Tarocchi, M.; Lang, U.E.; Suzuki, T.; Friedman, S.L.; Botella, L.M.; Bernabéu, C. Vascular injury triggers Krüppel-like factor 6 mobilization and cooperation with specificity protein 1 to promote endothelial activation through upregulation of the activin receptor-like kinase 1 gene. Circ. Res. 2013, 112, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Cheifetz, S.; Hernandez, H.; Laiho, M.; ten Dijke, P.; Iwata, K.K.; Massagué, J. Distinct transforming growth factor-beta (TGF-β) receptor subsets as determinants of cellular responsiveness to three TGF-β isoforms. J. Biol. Chem. 1990, 265, 20533–20538. [Google Scholar] [PubMed]
- Cheifetz, S.; Massagué, J. Isoform-specific transforming growth factor-β binding proteins with membrane attachments sensitive to phosphatidylinositol-specific phospholipase C. J. Biol. Chem. 1991, 266, 20767–20772. [Google Scholar] [PubMed]
- Sankar, S.; Mahooti-Brooks, N.; Centrella, M.; McCarthy, T.L.; Madri, J.A. Expression of transforming growth factor type III receptor in vascular endothelial cells increases their responsiveness to transforming growth factor β 2. J. Biol. Chem. 1995, 270, 13567–13572. [Google Scholar] [CrossRef] [PubMed]
- del Re, E.; Babitt, J.L.; Pirani, A.; Schneyer, A.L.; Lin, H.Y. In the absence of type III receptor, the transforming growth factor (TGF)-β type II-B receptor requires the type I receptor to bind TGF-β2. J. Biol. Chem. 2004, 279, 22765–22772. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.W.; Graulich, W.; Karges, B.; Stahl, S.; Ernst, M.; Ramaswamy, A.; Sedlacek, H.H.; Müller, R.; Adamkiewicz, J. Elevated expression of endoglin, a component of the TGF-β-receptor complex, correlates with proliferation of tumor endothelial cells. Int. J. Cancer 1999, 81, 568–572. [Google Scholar] [CrossRef]
- Torsney, E.; Charlton, R.; Parums, D.; Collis, M.; Arthur, H.M. Inducible expression of human endoglin during inflammation and wound healing in vivo. Inflamm. Res. 2002, 51, 464–470. [Google Scholar] [CrossRef] [PubMed]
- López-Casillas, F.; Cheifetz, S.; Doody, J.; Andres, J.L.; Lane, W.S.; Massague, J. Structure and expression of the membrane proteoglycan betaglycan, a component of the TGF-β receptor system. Cell 1991, 67, 785–795. [Google Scholar] [CrossRef]
- Blanco, F.J.; Santibanez, J.F.; Guerrero-Esteo, M.; Langa, C.; Vary, C.P.H.; Bernabeu, C. Interaction and functional interplay between endoglin and ALK-1, two components of the endothelial transforming growth factor-β receptor complex. J. Cell. Physiol. 2005, 204, 574–584. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Esteo, M.; Sánchez-Elsner, T.; Letamendia, A.; Bernabéu, C. Extracellular and cytoplasmic domains of endoglin interact with the transforming growth factor-β receptors I and II. J. Biol. Chem. 2002, 277, 29197–29209. [Google Scholar] [CrossRef] [PubMed]
- Cheifetz, S.; Bellón, T.; Calés, C.; Vera, S.; Bernabeu, C.; Massagué, J.; Letarte, M. Endoglin is a component of the transforming growth factor-β receptor system in human endothelial cells. J. Biol. Chem. 1992, 267, 19027–19030. [Google Scholar] [PubMed]
- Lastres, P.; Letamendia, A.; Zhang, H.; Rius, C.; Almendro, N.; Raab, U.; Lopez, L.A.; Langa, C.; Fabra, A.; Letarte, M.; et al. Endoglin modulates cellular responses to TGF-β 1. J. Cell Biol. 1996, 133, 1109–1121. [Google Scholar] [CrossRef] [PubMed]
- Ray, B.N.; Lee, N.Y.; How, T.; Blobe, G.C. ALK5 phosphorylation of the endoglin cytoplasmic domain regulates Smad1/5/8 signaling and endothelial cell migration. Carcinogenesis 2010, 31, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Velasco, S.; Alvarez-Muñoz, P.; Pericacho, M.; Dijke, P.t.; Bernabéu, C.; López-Novoa, J.M.; Rodríguez-Barbero, A. L- and S-endoglin differentially modulate TGFβ1 signaling mediated by ALK1 and ALK5 in L6E9 myoblasts. J. Cell Sci. 2008, 121, 913–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebrin, F.; Goumans, M.-J.; Jonker, L.; Carvalho, R.L.C.; Valdimarsdottir, G.; Thorikay, M.; Mummery, C.; Arthur, H.M.; Dijke, P.T. Endoglin promotes endothelial cell proliferation and TGF-β/ALK1 signal transduction. EMBO J. 2004, 23, 4018–4028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awwad, K.; Hu, J.; Shi, L.; Mangels, N.; Abdel Malik, R.; Zippel, N.; Fisslthaler, B.; Eble, J.A.; Pfeilschifter, J.; Popp, R.; et al. Role of secreted modular calcium-binding protein 1 (SMOC1) in transforming growth factor β signalling and angiogenesis. Cardiovasc. Res. 2015, 106, 284–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Abraham, S.; McKenzie, J.A.G.; Jeffs, N.; Swire, M.; Tripathi, V.B.; Luhmann, U.F.O.; Lange, C.A.K.; Zhai, Z.; Arthur, H.M.; et al. LRG1 promotes angiogenesis by modulating endothelial TGF-β signalling. Nature 2013, 499, 306–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bornstein, P.; Sage, E.H. Matricellular proteins: Extracellular modulators of cell function. Curr. Opin. Cell Biol. 2002, 14, 608–616. [Google Scholar] [CrossRef]
- Connor, T.B.; Roberts, A.B.; Sporn, M.B.; Danielpour, D.; Dart, L.L.; Michels, R.G.; de Bustros, S.; Enger, C.; Kato, H.; Lansing, M. Correlation of fibrosis and transforming growth factor-β type 2 levels in the eye. J. Clin. Investig. 1989, 83, 1661–1666. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, B.A.; Flanders, K.C.; Guerin, C.J.; Danielpour, D.; Anderson, D.H. Transforming growth factor β 2 is the predominant isoform in the neural retina, retinal pigment epithelium-choroid and vitreous of the monkey eye. Exp. Eye Res. 1994, 59, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Liang, S.; Yu, W.; Zhao, M.; Huang, L.; Zhao, M.; Li, X. Semaphorin 3A blocks the formation of pathologic choroidal neovascularization induced by transforming growth factor β. Mol. Vis. 2014, 20, 1258–1270. [Google Scholar] [PubMed]
- Jia, Y.; Yue, Y.; Hu, D.N.; Chen, J.L.; Zhou, J.B. Human aqueous humor levels of transforming growth factor-β2: Association with matrix metalloproteinases/tissue inhibitors of matrix metalloproteinases. Biomed. Rep. 2017, 7, 573–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khuu, L.A.; Tayyari, F.; Sivak, J.M.; Flanagan, S.J.; Singer, S.; Brent, M.H.; Huang, D.; Tan, O.; Hudson, C. Aqueous humour concentrations of TGF-β, PLGF and FGF-1 and total retinal blood flow in patients with early non-proliferative diabetic retinopathy. Acta Ophthalmol. 2017, 95, e206–e211. [Google Scholar] [CrossRef] [PubMed]
- Tosi, G.M.; Caldi, E.; Neri, G.; Nuti, E.; Marigliani, D.; Baiocchi, S.; Traversi, C.; Cevenini, G.; Tarantello, A.; Fusco, F.; et al. HTRA1 and TGF-β1 concentrations in the aqueous humor of patients with neovascular age-related macular degeneration. Invest. Ophthalmol. Vis. Sci. 2017, 58, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Tosi, G.M.; Neri, G.; Caldi, E.; Fusco, F.; Bacci, T.; Tarantello, A.; Nuti, E.; Marigliani, D.; Baiocchi, S.; Traversi, C.; et al. TGF-β concentrations and activity are down-regulated in the aqueous humor of patients with neovascular age-related macular degeneration. Sci. Rep. 2018, 8, 8053. [Google Scholar] [CrossRef] [PubMed]
- Kita, T.; Hata, Y.; Arita, R.; Kawahara, S.; Miura, M.; Nakao, S.; Mochizuki, Y.; Enaida, H.; Goto, Y.; Shimokawa, H.; et al. Role of TGF-β in proliferative vitreoretinal diseases and ROCK as a therapeutic target. Proc. Natl. Acad. Sci. USA 2008, 105, 17504–17509. [Google Scholar] [CrossRef] [PubMed]
- Pasquale, L.R.; Dorman-Pease, M.E.; Lutty, G.A.; Quigley, H.A.; Jampel, H.D. Immunolocalization of TGF-β 1, TGF-β 2, and TGF-β 3 in the anterior segment of the human eye. Invest. Ophthalmol. Vis. Sci. 1993, 34, 23–30. [Google Scholar] [PubMed]
- Lutty, G.A.; Merges, C.; Threlkeld, A.B.; Crone, S.; McLeod, D.S. Heterogeneity in localization of isoforms of TGF-β in human retina, vitreous, and choroid. Investig. Ophthalmol. Vis. Sci. 1993, 34, 477–487. [Google Scholar]
- Anderson, D.H.; Guerin, C.J.; Hageman, G.S.; Pfeffer, B.A.; Flanders, K.C. Distribution of transforming growth factor-β isoforms in the mammalian retina. J. Neurosci. Res. 1995, 42, 63–79. [Google Scholar] [CrossRef] [PubMed]
- Tanihara, H.; Yoshida, M.; Matsumoto, M.; Yoshimura, N. Identification of transforming growth factor-β expressed in cultured human retinal pigment epithelial cells. Investig. Ophthalmol. Vis. Sci. 1993, 34, 413–419. [Google Scholar]
- Kvanta, A. Expression and secretion of transforming growth factor-β in transformed and nontransformed retinal pigment epithelial cells. Ophthalmic Res. 1994, 26, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Kliffen, M.; Sharma, H.S.; Mooy, C.M.; Kerkvliet, S.; de Jong, P.T.V.M. Increased expression of angiogenic growth factors in age-related maculopathy. Br. J. Ophthalmol. 1997, 81, 154–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagineni, C.N.; Cherukuri, K.S.; Kutty, V.; Detrick, B.; Hooks John, J. Interferon-γ differentially regulates TGF-β1 and TGF-β2 expression in human retinal pigment epithelial cells through JAK-STAT pathway. J. Cell. Physiol. 2007, 210, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, L.; Nazari, H.; Sreekumar, P.G.; Kannan, R.; Dustin, L.; Zhu, D.; Barron, E.; Hinton, D.R. TGF-β2 secretion from RPE decreases with polarization and becomes apically oriented. Cytokine 2015, 71, 394–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanford, L.P.; Ormsby, I.; Gittenberger-de Groot, A.C.; Sariola, H.; Friedman, R.; Boivin, G.P.; Cardell, E.L.; Doetschman, T. TGFβ2 knockout mice have multiple developmental defects that are non-overlapping with other TGFβ knockout phenotypes. Development 1997, 124, 2659–2670. [Google Scholar] [PubMed]
- Saika, S.; Saika, S.; Liu, C.-Y.; Azhar, M.; Sanford, L.P.; Doetschman, T.; Gendron, R.L.; Kao, C.W.C.; Kao, W.W.Y. TGFβ2 in corneal morphogenesis during mouse embryonic development. Dev. Biol. 2001, 240, 419–432. [Google Scholar] [CrossRef] [PubMed]
- Iwanishi, H.; Fujita, N.; Tomoyose, K.; Okada, Y.; Yamanaka, O.; Flanders, K.C.; Saika, S. Inhibition of development of laser-induced choroidal neovascularization with suppression of infiltration of macrophages in Smad3-null mice. Lab. Investig. 2016, 96, 641–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aiello, L.P.; Northrup, J.M.; Keyt, B.A.; Takagi, H.; Iwamoto, M.A. Hypoxic regulation of vascular endothelial growth factor in retinal cells. Arch. Ophthalmol. 1995, 113, 1538–1544. [Google Scholar] [CrossRef] [PubMed]
- Farjood, F.; Vargis, E. Physical disruption of cell–cell contact induces VEGF expression in RPE cells. Mol. Vis. 2017, 23, 431–446. [Google Scholar] [PubMed]
- Nozaki, M.; Raisler, B.J.; Sakurai, E.; Sarma, J.V.; Barnum, S.R.; Lambris, J.D.; Chen, Y.; Zhang, K.; Ambati, B.K.; Baffi, J.Z.; et al. Drusen complement components C3a and C5a promote choroidal neovascularization. Proc. Natl. Acad. Sci. USA 2006, 103, 2328–2333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagineni, C.N.; Kommineni, V.K.; William, A.; Detrick, B.; Hooks, J.J. Regulation of VEGF expression in human retinal cells by cytokines: Implications for the role of inflammation in age-related macular degeneration. J. Cell. Physiol. 2011, 227, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Nagineni, C.N.; Samuel, W.; Nagineni, S.; Pardhasaradhi, K.; Wiggert, B.; Detrick, B.; Hooks, J.J. Transforming growth factor-β induces expression of vascular endothelial growth factor in human retinal pigment epithelial cells: Involvement of mitogen-activated protein kinases. J. Cell. Physiol. 2003, 197, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Bian, Z.-M.; Elner, S.G.; Elner, V.M. Regulation of VEGF mRNA expression and protein secretion by TGF-β2 in human retinal pigment epithelial cells. Exp. Eye Res. 2007, 84, 812–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, V.; Lecomte, J.; Hansen, S.; Blacher, S.; Gonzalez, M.-L.A.; Struman, I.; Sounni, N.E.; Rozet, E.; de Tullio, P.; Foidart, J.M.; et al. Laser-induced choroidal neovascularization model to study age-related macular degeneration in mice. Nat. Protoc. 2013, 8, 2197–2211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Ma, W.; Han, S.; Meng, Z.; Zhao, L.; Yin, Y.; Wang, Y.; Li, J. TGF-β participates choroid neovascularization through Smad2/3-VEGF/TNF-alpha signaling in mice with Laser-induced wet age-related macular degeneration. Sci. Rep. 2017, 7, 9672. [Google Scholar] [CrossRef] [PubMed]
- Ogata, N.; Yamamoto, C.; Miyashiro, M.; Yamada, H.; Matsushima, M.; Uyama, M. Expression of transforming growth factor-β mRNA in experimental choroidal neovascularization. Curr. Eye Res. 1997, 16, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Recalde, S.; Zarranz-Ventura, J.; Fernandez-Robredo, P.; Garcia-Gomez, P.J.; Salinas-Alaman, A.; Borras-Cuesta, F.; Dotor, J.; Garcia-Layana, A. Transforming growth factor-β inhibition decreases diode laser-induced choroidal neovascularization development in rats: P17 and P144 peptides. Investig. Ophthalmol. Vis. Sci. 2011, 52, 7090–7097. [Google Scholar] [CrossRef] [PubMed]
- Zarranz-Ventura, J.; Fernandez-Robredo, P.; Recalde, S.; Salinas-Alaman, A.; Borras-Cuesta, F.; Dotor, J.; Garcia-Layana, A. Transforming growth factor-β inhibition reduces progression of early choroidal neovascularization lesions in rats: P17 and P144 peptides. PLoS ONE 2013, 8, e65434. [Google Scholar] [CrossRef] [PubMed]
- Pennesi, M.E.; Neuringer, M.; Courtney, R.J. Animal models of age related macular degeneration. Mol. Aspects Med. 2012, 33, 487–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amin, R.; Puklin, J.E.; Frank, R.N. Growth factor localization in choroidal neovascular membranes of age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 1994, 35, 3178–3188. [Google Scholar]
- Yafai, Y.; Iandiev, I.; Lange, J.; Unterlauft, J.D.; Wiedemann, P.; Bringmann, A.; Reichenbach, A.; Eichler, W. Muller glial cells inhibit proliferation of retinal endothelial cells via TGF-β2 and Smad signaling. Glia 2014, 62, 1476–1485. [Google Scholar] [CrossRef] [PubMed]
- Langmann, T. Microglia activation in retinal degeneration. J. Leukoc. Biol. 2007, 81, 1345–1351. [Google Scholar] [CrossRef] [PubMed]
- Paglinawan, R.; Malipiero, U.; Schlapbach, R.; Frei, K.; Reith, W.; Fontana, A. TGFβ directs gene expression of activated microglia to an anti-inflammatory phenotype strongly focusing on chemokine genes and cell migratory genes. Glia 2003, 44, 219–231. [Google Scholar] [CrossRef] [PubMed]
- Sugino, I.K.; Sun, Q.; Springer, C.; Cheewatrakoolpong, N.; Liu, T.; Li, H.; Zarbin, M.A. Two bioactive molecular weight fractions of a conditioned medium enhance RPE cell survival on age-related macular degeneration and aged Bruch’s membrane. Transl. Vis. Sci. Technol. 2016, 5, 8. [Google Scholar] [CrossRef] [PubMed]
- Ohlmann, A.; Scholz, M.; Koch, M.; Tamm, E.R. Epithelial–mesenchymal transition of the retinal pigment epithelium causes choriocapillaris atrophy. Histochem. Cell Biol. 2016, 146, 769–780. [Google Scholar] [CrossRef] [PubMed]
- Seitz, R.; Weber, G.; Albrecht, S.; Fuchshofer, R.; Tamm, E.R.; Ohlmann, A. Cross-inhibition of norrin and TGF-β signaling modulates development of retinal and choroidal vasculature. Investig. Ophthalmol. Vis. Sci. 2018, 59, 2240–2251. [Google Scholar] [CrossRef] [PubMed]
- Schlecht, A.; Leimbeck, S.V.; Jägle, H.; Feuchtinger, A.; Tamm, E.R.; Braunger, B.M. Deletion of endothelial transforming growth factor-β signaling leads to choroidal neovascularization. Am. J. Pathol. 2017, 187, 2570–2589. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.S.; Park, J.M.; Kong, T.; Kim, C.; Bae, S.H.; Kim, H.W.; Moon, J. Retinal angiogenesis effects of TGF-β1 and paracrine factors secreted from human placental stem cells in response to a pathological environment. Cell Transplant. 2016, 25, 1145–1157. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.T.; Gao, J.; Cao, S.; Sandhu, N.; Cui, J.Z.; Chou, C.L.; Fang, E.; Matsubara, J.A. Inflammatory mediators induced by amyloid-β in the retina and RPE in vivo: Implications for inflammasome activation in age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2013, 54, 2225–2237. [Google Scholar] [CrossRef] [PubMed]
- Fisichella, V.; Giurdanella, G.; Platania, C.B.M.; Romano, G.L.; Leggio, G.M.; Salomone, S.; Drago, F.; Caraci, F.; Bucolo, C. TGF-β1 prevents rat retinal insult induced by amyloid-β (1–42) oligomers. Eur. J. Pharmacol. 2016, 787, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Platania, C.B.M.; Fisichella, V.; Fidilio, A.; Geraci, F.; Lazzara, F.; Leggio, G.M.; Salomone, S.; Drago, F.; Pignatello, R.; Caraci, F.; et al. Topical ocular delivery of TGF-β1 to the back of the eye: Implications in age-related neurodegenerative diseases. Int. J. Mol. Sci. 2017, 18, E2076. [Google Scholar] [CrossRef] [PubMed]
- Noma, H.; Funatsu, H.; Mimura, T.; Harino, S.; Hori, S. Aqueous humor levels of vasoactive molecules correlate with vitreous levels and macular edema in central retinal vein occlusion. Eur. J. Ophthalmol. 2010, 20, 402–409. [Google Scholar] [CrossRef] [PubMed]
- Hirase, K.; Ikeda, T.; Sotozono, C.; Nishida, K.; Sawa, H.; Kinoshita, S. Transforming growth factor β2 in the vitreous in proliferative diabetic retinopathy. Arch. Ophthalmol. 1998, 116, 738–741. [Google Scholar] [CrossRef] [PubMed]
- Kon, C.H.; Occleston, N.L.; Aylward, G.W.; Khaw, P.T. Expression of vitreous cytokines in proliferative vitreoretinopathy: A prospective study. Investig. Ophthalmol. Vis. Sci. 1999, 40, 705–712. [Google Scholar]
- Hirase, K.; Sugiyama, T.; Ikeda, T.; Sotozono, C.; Yasuhara, T.; Koizumi, K.; Kinoshita, S. Transforming growth factor β(2) increases in subretinal fluid in rhegmatogenous retinal detachment with subretinal strands. Ophthalmologica 2005, 219, 222–225. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Wu, Z.; Wang, F.; Zhang, Z.; Yu, M. Identification of chemokines and growth factors in proliferative diabetic retinopathy vitreous. Biomed. Res. Int. 2014, 2014, 486386. [Google Scholar] [CrossRef] [PubMed]
- Mitsuhiro, M.R.K.H.; Eguchi, S.; Yamashita, H. Regulation mechanisms of retinal pigment epithelial cell migration by the TGF-β superfamily. Acta Ophthal. Scand. 2003, 81, 630–638. [Google Scholar] [CrossRef]
- Kaven, C.; Spraul, C.W.; Zavazava, N.; Lang, G.K.; Lang, G.E. Growth factor combinations modulate human retinal pigment epithelial cell proliferation. Curr. Eye Res. 2000, 20, 480–487. [Google Scholar] [CrossRef]
- Lopez, P.F.; Sippy, B.D.; Lambert, H.M.; Thach, A.B.; Hinton, D.R. Transdifferentiated retinal pigment epithelial cells are immunoreactive for vascular endothelial growth factor in surgically excised age-related macular degeneration-related choroidal neovascular membranes. Investig. Ophthalmol. Vis. Sci. 1996, 37, 855–868. [Google Scholar]
- Hirasawa, M.; Noda, K.; Noda, S.; Suzuki, M.; Ozawa, Y.; Shinoda, K.; Inoue, M.; Ogawa, Y.; Tsubota, K.; Ishida, S. Transcriptional factors associated with epithelial-mesenchymal transition in choroidal neovascularization. Mol. Vis. 2011, 17, 1222–1230. [Google Scholar] [PubMed]
- Kobayashi, Y.; Orita, T.; Yamashiro, C.; Uchi, S.; Hatano, M.; Kobayashi, M.; Tokuda, K.; Yanai, R.; Takeda, A.; Ishibashi, T.; et al. Inhibitional effect of TGF-β2-induced EMT in RPE cells by an RAR-γ agonist. Investig. Ophthalmol. Vis. Sci. 2018, 59, 4003. [Google Scholar]
- Dvashi, Z.; Goldberg, M.; Adir, O.; Shapira, M.; Pollack, A. TGF-β1 induced transdifferentiation of rpe cells is mediated by TAK1. PLoS ONE 2015, 10, e0122229. [Google Scholar] [CrossRef] [PubMed]
- Gamulescu, M.A.; Chen, Y.; He, S.; Spee, C.; Jin, M.; Ryan, S.J.; Hinton, D.R. Transforming growth factor β2-induced myofibroblastic differentiation of human retinal pigment epithelial cells: Regulation by extracellular matrix proteins and hepatocyte growth factor. Exp. Eye Res. 2006, 83, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, H.; Wang, F.; Gu, Q.; Xu, X. Snail involves in the transforming growth factor β1-mediated epithelial-mesenchymal transition of retinal pigment epithelial cells. PLoS ONE 2011, 6, e23322. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, P.; Gutteridge, A.; Impey, E.; Storer, R.I.; Owen, R.M.; Whiting, P.J.; Bictash, M.; Benn, C.L. Targeting the cAMP and transforming growth factor-β pathway increases proliferation to promote re-epithelialization of human stem cell-derived retinal pigment epithelium. Stem Cells Transl. Med. 2016, 5, 925–937. [Google Scholar] [CrossRef] [PubMed]
- Sugioka, K.; Kodama, A.; Okada, K.; Iwata, M.; Yoshida, K.; Kusaka, S.; Matsumoto, C.; Kaji, H.; Shimomura, Y. TGF-β2 promotes RPE cell invasion into a collagen gel by mediating urokinase-type plasminogen activator (uPA) expression. Exp. Eye Res. 2013, 115, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Tamiya, S.; Liu, L.; Kaplan, H.J. Epithelial-mesenchymal transition and proliferation of retinal pigment epithelial cells initiated upon loss of cell-cell contact. Investig. Ophthalmol. Vis. Sci. 2010, 51, 2755–2763. [Google Scholar] [CrossRef] [PubMed]
- Grossniklaus, H.E.; Miskala, P.H.; Green, W.R.; Bressler, S.B.; Hawkins, B.S.; Toth, C.; Wilson, D.J.; Bressler, N.M. Histopathologic and ultrastructural features of surgically excised subfoveal choroidal neovascular lesions: Submacular surgery trials report no. 7. Arch. Ophthalmol. 2005, 123, 914–921. [Google Scholar] [CrossRef] [PubMed]
- Daniel, E.; Toth, C.A.; Grunwald, J.E.; Jaffe, G.J.; Martin, D.F.; Fine, S.L.; Huang, J.; Ying, G.-S.; Hagstrom, S.A.; Winter, K.; et al. Risk of scar in the comparison of age-related acular degeneration treatments trials. Ophthalmology 2014, 121, 656–666. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, K.; Kannan, R.; Hinton, D.R. Molecular mechanisms of subretinal fibrosis in age-related macular degeneration. Exp. Eye Res. 2016, 142, 19–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimoto, K.; Nakatsuka, K.; Matsuo, N.; Yoshioka, H. p38 MAPK mediates the expression of type I collagen induced by TGF-β2 in human retinal pigment epithelial cells ARPE-19. Investig. Ophthalmol. Vis. Sci. 2004, 45, 2431–2437. [Google Scholar] [CrossRef]
- Itoh, Y.; Kimoto, K.; Imaizumi, M.; Nakatsuka, K. Inhibition of RhoA/Rho-kinase pathway suppresses the expression of type I collagen induced by TGF-β2 in human retinal pigment epithelial cells. Exp. Eye Res. 2007, 84, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.L.; Fuchshofer, R.; Kook, D.; Kampik, A.; Bloemendal, H.; Welge-Lussen, U. Subtoxic oxidative stress induces senescence in retinal pigment epithelial cells via TGF-β release. Investig. Ophthalmol. Vis. Sci. 2009, 50, 926–935. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Puklin, J.E.; Frank, R.N.; Zhang, N.L. Ulrastructural immunocytochemistry of subretinal neovascular membranes in age-related macular degeneration. Ophthalmology 1992, 99, 1368–1376. [Google Scholar] [CrossRef]
- Nagineni, C.N.; Kutty, V.; Detrick, B.; Hooks, J.J. Expression of PDGF and their receptors in human retinal pigment epithelial cells and fibroblasts: Regulation by TGF-β. J. Cell. Physiol. 2005, 203, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, Z.L. Transforming growth factor-β neutralizing antibodies inhibit subretinal fibrosis in a mouse model. Int. J. Ophthalmol. 2012, 5, 307–311. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Liu, Z.; Zhang, H.; Zhang, Y.; Lin, D. The COX-2-selective antagonist (NS-398) inhibits choroidal neovascularization and subretinal fibrosis. PLoS ONE 2016, 11, e0146808. [Google Scholar] [CrossRef] [PubMed]
- Braunger, B.M.; Pielmeier, S.; Demmer, C.; Landstorfer, V.; Kawall, D.; Abramov, N.; Leibinger, M.; Kleiter, I.; Fischer, D.; Jägle, H.; et al. TGF-β signaling protects retinal neurons from programmed cell death during the development of the mammalian eye. J. Neurosci. 2013, 33, 14246–14258. [Google Scholar] [CrossRef] [PubMed]
- Honjo, Y.; Nagineni, C.N.; Larsson, J.; Nandula, S.R.; Hooks, J.J.; Chan, C.C.; Karlsson, S.; Kulkarni, A.B. Neuron-specific TGF-β signaling deficiency results in retinal detachment and cataracts in mice. Biochem. Biophys. Res. Commun. 2007, 352, 418–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beier, M.; Franke, A.; Paunel-Görgülü, A.N.; Scheerer, N.; Dünker, N. Transforming growth factor β mediates apoptosis in the ganglion cell layer during all programmed cell death periods of the developing murine retina. Neurosci. Res. 2006, 56, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Romano, G.L.; Platania, C.B.M.; Drago, F.; Salomone, S.; Ragusa, M.; Barbagallo, C.; Di Pietro, C.; Purrello, M.; Reibaldi, M.; Avitabile, T.; et al. Retinal and circulating miRNAs in age-related macular degeneration: An in vivo animal and human study. Front. Pharmacol. 2017, 8, 168. [Google Scholar] [CrossRef] [PubMed]
- Chae, D.K.; Ban, E.; Yoo, Y.S.; Kim, E.E.; Baik, J.H.; Song, E.J. MIR-27a regulates the TGF-β signaling pathway by targeting SMAD2 and SMAD4 in lung cancer. Mol. Carcinog. 2017, 58, 1992–1998. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Zhu, E.D.; Li, N.; Lu, D.S.; Li, W.; Li, B.S.; Zhao, Y.L.; Mao, X.H.; Guo, G.; Yu, P.W.; et al. Increased miR-146a in gastric cancer directly targets SMAD4 and is involved in modulating cell proliferation and apoptosis. Oncol. Rep. 2012, 27, 559–566. [Google Scholar] [CrossRef] [PubMed]
- Hinck, A.P.; Mueller, T.D.; Springer, T.A. Structural biology and evolution of the TGF-β family. Cold Spring Harb. Perspect. Biol. 2016, 8, a022103. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhu, D.; Sonoda, S.; He, S.; Spee, C.; Ryan, S.J.; Hinton, D.R. Over-expression of BMP4 inhibits experimental choroidal neovascularization by modulating VEGF and MMP-9. Angiogenesis 2012, 15, 213–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyer, L.A.; Pi, X.; Patterson, C. The role of BMPs in endothelial cell function and dysfunction. Trends Endocrinol. Metab. 2014, 25, 472–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larrivée, B.; Prahst, C.; Gordon, E.; del Toro, R.; Mathivet, T.; Duarte, A.; Simons, M.; Eichmann, A. ALK1 signaling inhibits angiogenesis by cooperating with the Notch pathway. Dev. Cell 2012, 22, 489–500. [Google Scholar] [CrossRef] [PubMed]
- Ntumba, K.; Akla, N.; Oh, S.P.; Eichmann, A.; Larrivée, B. BMP9/ALK1 inhibits neovascularization in mouse models of age-related macular degeneration. Oncotarget 2016, 7, 55957–55969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walshe, T.E. TGF-β and microvessel homeostasis. Microvasc. Res. 2010, 80, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Walshe, T.E.; Leach, L.L.; D’Amore, P.A. TGF-β signaling is required for maintenance of retinal ganglion cell differentiation and survival. Neuroscience 2011, 189, 123–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Consortium, T.A.G. Seven new loci associated with age-related macular degeneration. Nat. Genet. 2013, 45, 433–439. [Google Scholar] [CrossRef] [Green Version]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tosi, G.M.; Orlandini, M.; Galvagni, F. The Controversial Role of TGF-β in Neovascular Age-Related Macular Degeneration Pathogenesis. Int. J. Mol. Sci. 2018, 19, 3363. https://doi.org/10.3390/ijms19113363
Tosi GM, Orlandini M, Galvagni F. The Controversial Role of TGF-β in Neovascular Age-Related Macular Degeneration Pathogenesis. International Journal of Molecular Sciences. 2018; 19(11):3363. https://doi.org/10.3390/ijms19113363
Chicago/Turabian StyleTosi, Gian Marco, Maurizio Orlandini, and Federico Galvagni. 2018. "The Controversial Role of TGF-β in Neovascular Age-Related Macular Degeneration Pathogenesis" International Journal of Molecular Sciences 19, no. 11: 3363. https://doi.org/10.3390/ijms19113363
APA StyleTosi, G. M., Orlandini, M., & Galvagni, F. (2018). The Controversial Role of TGF-β in Neovascular Age-Related Macular Degeneration Pathogenesis. International Journal of Molecular Sciences, 19(11), 3363. https://doi.org/10.3390/ijms19113363