Pin1 Modulation in Physiological Status and Neurodegeneration. Any Contribution to the Pathogenesis of Type 3 Diabetes?



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Pin1 Characterization: Structure, Regulation and Subcellular Localization
3. Physiological Role of Pin1 in In Vivo Brain Development
3.1. Pin1 Expression during Embryogenesis in the Zebrafish
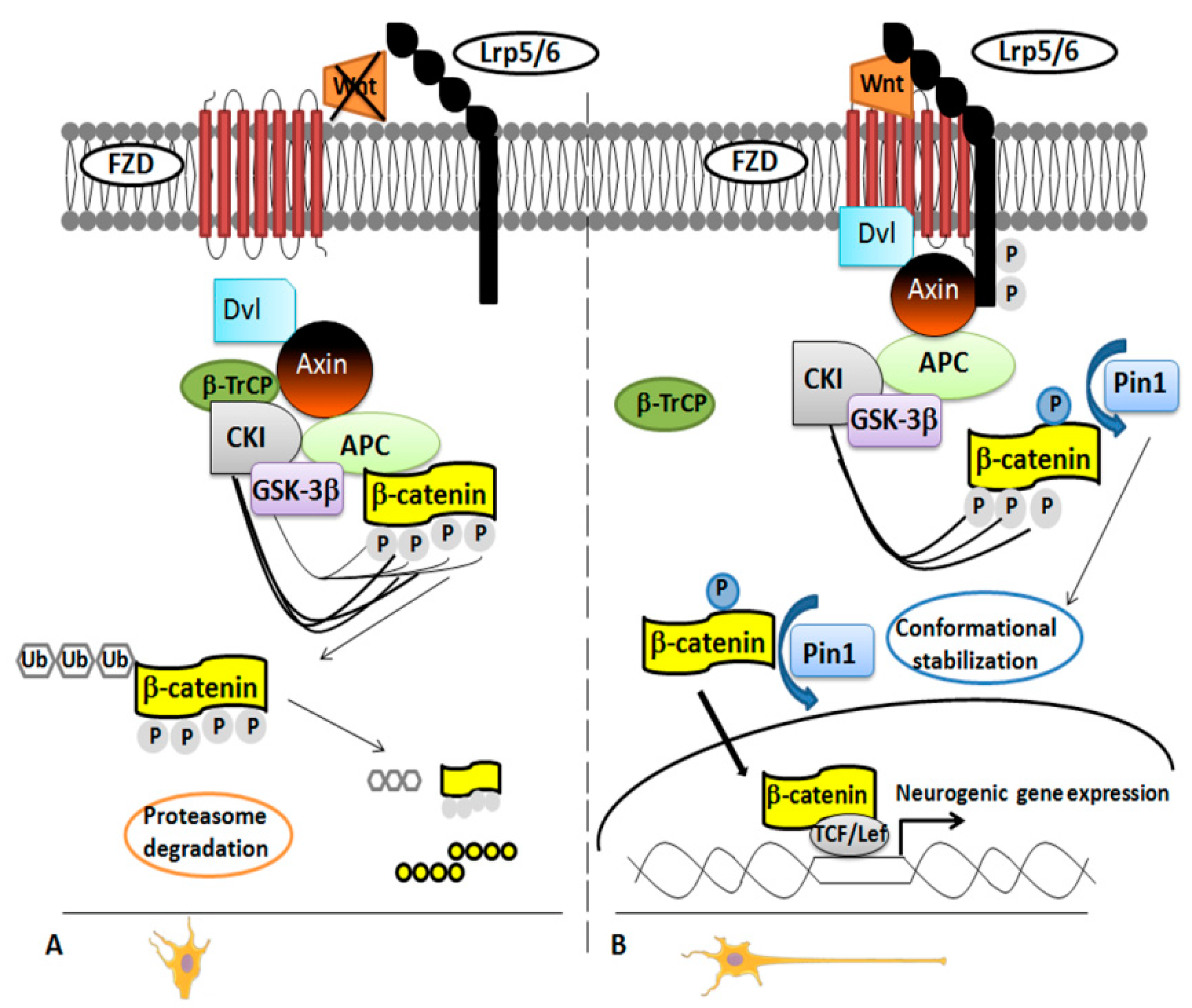
3.2. Pin1 Regulates Neuronal Cortical Differentiation: Modulation of the Wnt/β-Catenin Pathway
3.3. Pin1 Regulation of Axonal Guidance by Modulation of Microtubule Assembly and Buffering Sema-3A Stimulation
4. Role of Pin1 in Neurodegenerative Disorders
4.1. Huntington’s Disease
4.2. Parkinson’s Disease
4.3. Temporal Lobe Epilepsy
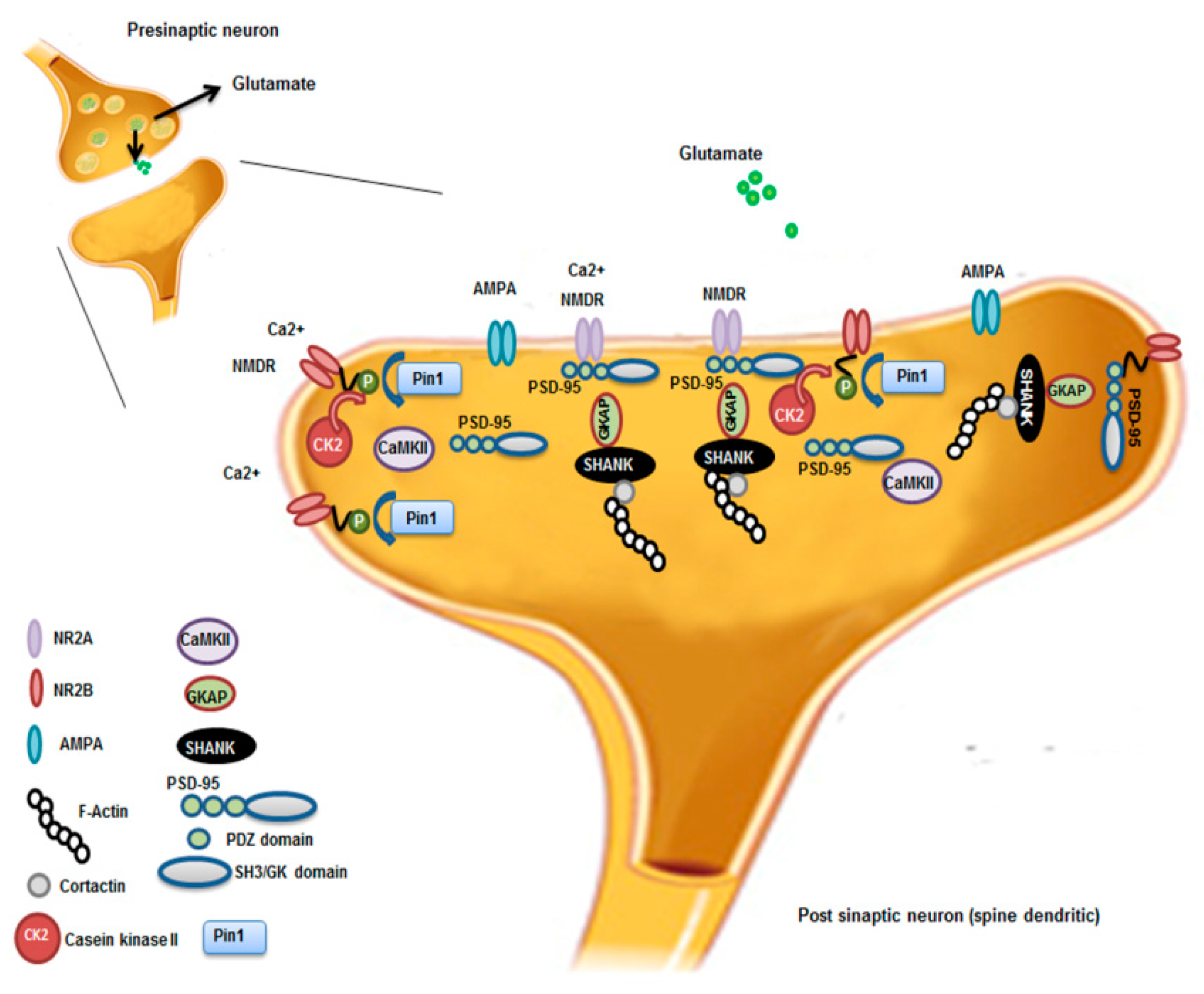
4.4. Pin1 and Alzheimer’s Diseases (AD)
5. Pin1 Links Brain Impaired Glucose Metabolism and Neuronal Degeneration in AD
5.1. Type 3 Diabetes
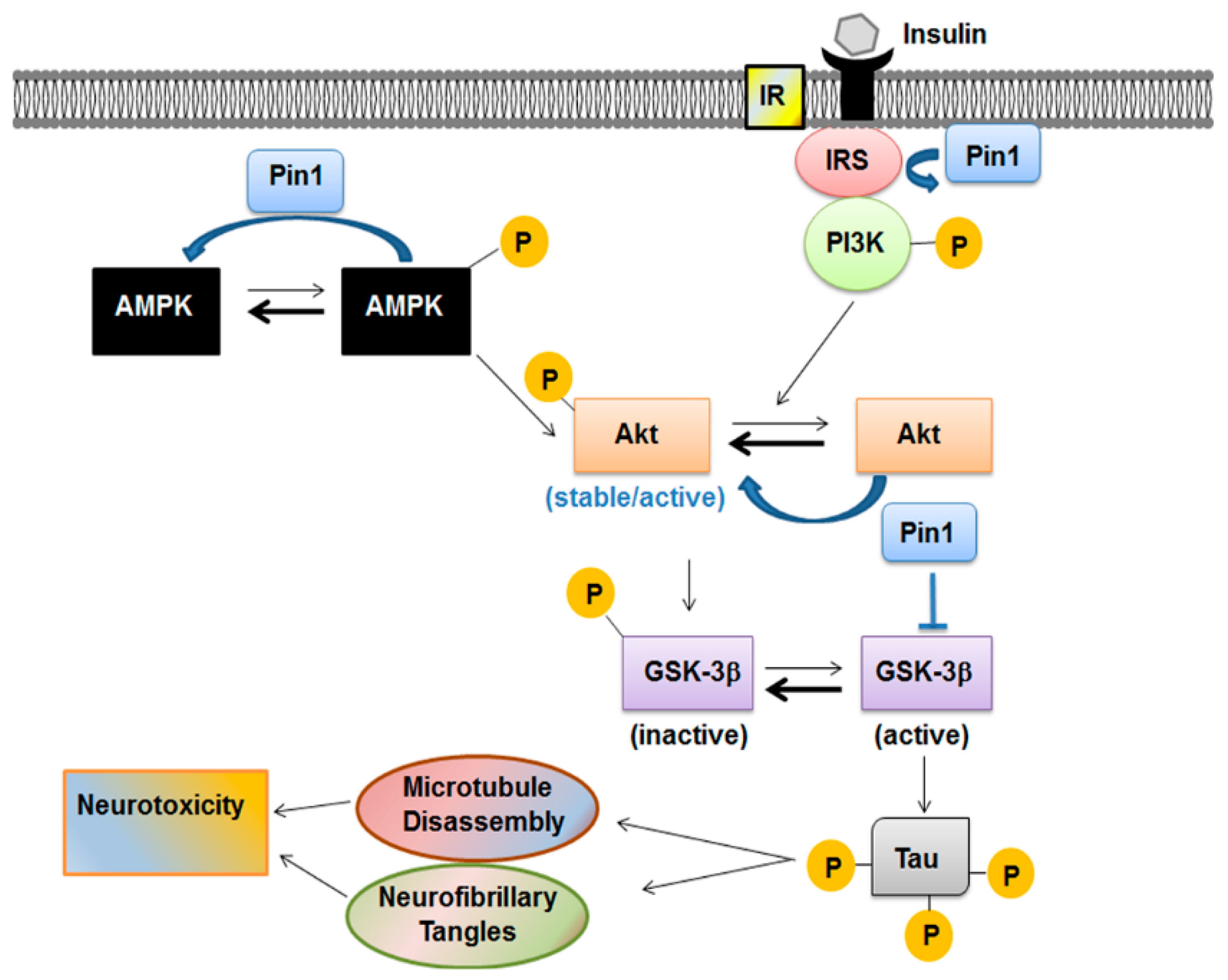
5.2. Pin1 and Insulin Pathways
5.3. Pin1 and GSK-3β(Glycogen Synthase Kinase-3β) Modulation
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Gothel, S.F.; Marahiel, M.A. Peptidyl-prolyl cis-trans isomerases, a superfamily of ubiquitous folding catalysts. Cell. Mol. Life Sci. 1999, 55, 423–436. [Google Scholar] [CrossRef] [PubMed]
- Reimer, U.; Scherer, G.; Drewello, M.; Kruber, S.; Schutkowski, M.; Fischer, G. Side-chain effects on peptidyl-prolyl cis/trans isomerisation. J. Mol. Biol. 1998, 279, 449–460. [Google Scholar] [CrossRef] [PubMed]
- Nakatsu, Y.; Matsunaga, Y.; Yamamotoya, T.; Ueda, K.; Inoue, Y.; Mori, K.; Sakoda, H.; Fujishiro, M.; Ono, H.; Kushiyama, A.; et al. Physiological and Pathogenic Roles of Prolyl Isomerase Pin1 in Metabolic Regulations via Multiple Signal Transduction Pathway Modulations. Int. J. Mol. Sci. 2016, 17, 1495. [Google Scholar] [CrossRef] [PubMed]
- Ibarra, M.S.; Borini Etichetti, C.; di Benedetto, C.; Rosano, G.L.; Margarit, E.; del Sal, G.; Mione, M.; Girardini, J. Dynamic regulation of Pin1 expression and function during zebrafish development. PLoS ONE 2017, 12, e0175939. [Google Scholar] [CrossRef] [PubMed]
- Davies, T.H.; Ning, Y.M.; Sanchez, E.R. A new first step in activation of steroid receptors: Hormone-induced switching of FKBP51 and FKBP52 immunophilins. J. Biol. Chem. 2002, 277, 4597–4600. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Farmer, J.D., Jr.; Lane, W.S.; Friedman, J.; Weissman, I.; Schreiber, S.L. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell 1991, 66, 807–815. [Google Scholar] [CrossRef]
- Lu, K.P.; Finn, G.; Lee, T.H.; Nicholson, L.K. Prolyl cis-trans isomerization as a molecular timer. Nat. Chem. Biol. 2007, 3, 619–629. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.P.; Zhou, X.Z. The prolyl isomerase PIN1: A pivotal new twist in phosphorylation signalling and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 904–916. [Google Scholar] [CrossRef] [PubMed]
- Ranganathan, R.; Lu, K.P.; Hunter, T.; Noel, J.P. Structural and functional analysis of the mitotic rotamase Pin1 suggests substrate recognition is phosphorylation dependent. Cell 1997, 89, 875–886. [Google Scholar] [CrossRef]
- Kim, E.K.; Choi, E.J. Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta 2010, 1802, 396–405. [Google Scholar] [CrossRef] [PubMed]
- MacAulay, K.; Doble, B.W.; Patel, S.; Hansotia, T.; Sinclair, E.M.; Drucker, D.J.; Nagy, A.; Woodgett, J.R. Glycogen synthase kinase 3α-specific regulation of murine hepatic glycogen metabolism. Cell Metab. 2007, 6, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Patrick, G.N.; Zukerberg, L.; Nikolic, M.; de la Monte, S.; Dikkes, P.; Tsai, L.H. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature 1999, 402, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Bao, L.; Kimzey, A.; Sauter, G.; Sowadski, J.M.; Lu, K.P.; Wang, D.G. Prevalent overexpression of prolyl isomerase Pin1 in human cancers. Am. J. Pathol. 2004, 164, 1727–1737. [Google Scholar] [CrossRef]
- Angelucci, F.; Hort, J. Prolyl isomerase Pin1 and neurotrophins: A loop that may determine the fate of cells in cancer and neurodegeneration. Ther. Adv. Med. Oncol. 2017, 9, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Talbot, K.; Wang, H.Y.; Kazi, H.; Han, L.Y.; Bakshi, K.P.; Stucky, A.; Fuino, R.L.; Kawaguchi, K.R.; Samoyedny, A.J.; Wilson, R.S.; et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J. Clin. Investig. 2012, 122, 1316–1338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De la Monte, S.M.; Wands, J.R. Alzheimer’s disease is type 3 diabetes-evidence reviewed. J. Diabetes Sci. Technol. 2008, 2, 1101–1113. [Google Scholar] [CrossRef] [PubMed]
- De la Monte, S.M. Insulin Resistance and Neurodegeneration: Progress Towards the Development of New Therapeutics for Alzheimer’s Disease. Drugs 2017, 77, 47–65. [Google Scholar] [CrossRef] [PubMed]
- Verdecia, M.A.; Bowman, M.E.; Lu, K.P.; Hunter, T.; Noel, J.P. Structural basis for phosphoserine-proline recognition by group IV WW domains. Nat. Struct. Biol. 2000, 7, 639–643. [Google Scholar] [CrossRef] [PubMed]
- Bayer, E.; Goettsch, S.; Mueller, J.W.; Griewel, B.; Guiberman, E.; Mayr, L.M.; Bayer, P. Structural analysis of the mitotic regulator hPin1 in solution: Insights into domain architecture and substrate binding. J. Biol. Chem. 2003, 278, 26183–26193. [Google Scholar] [CrossRef] [PubMed]
- Eichner, T.; Kutter, S.; Labeikovsky, W.; Buosi, V.; Kern, D. Molecular Mechanism of Pin1-Tau Recognition and Catalysis. J. Mol. Biol. 2016, 428 Pt A, 1760–1775. [Google Scholar] [CrossRef]
- Labeikovsky, W.; Eisenmesser, E.Z.; Bosco, D.A.; Kern, D. Structure and dynamics of pin1 during catalysis by NMR. J. Mol. Biol. 2007, 367, 1370–1381. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, D.M.; Saxena, K.; Vogtherr, M.; Bernado, P.; Pons, M.; Fiebig, K.M. Peptide binding induces large scale changes in inter-domain mobility in human Pin1. J. Biol. Chem. 2003, 278, 26174–26182. [Google Scholar] [CrossRef] [PubMed]
- Namanja, A.T.; Peng, T.; Zintsmaster, J.S.; Elson, A.C.; Shakour, M.G.; Peng, J.W. Substrate recognition reduces side-chain flexibility for conserved hydrophobic residues in human Pin1. Structure 2007, 15, 313–327. [Google Scholar] [CrossRef] [PubMed]
- Ryo, A.; Liou, Y.C.; Wulf, G.; Nakamura, M.; Lee, S.W.; Lu, K.P. PIN1 is an E2F target gene essential for Neu/Ras-induced transformation of mammary epithelial cells. Mol. Cell. Biol. 2002, 22, 5281–5295. [Google Scholar] [CrossRef] [PubMed]
- Rustighi, A.; Tiberi, L.; Soldano, A.; Napoli, M.; Nuciforo, P.; Rosato, A.; Kaplan, F.; Capobianco, A.; Pece, S.; Di Fiore, P.P.; et al. The prolyl-isomerase Pin1 is a Notch1 target that enhances Notch1 activation in cancer. Nat. Cell Biol. 2009, 11, 133–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, S.L.; Tang, N.L.; Tam, C.W.; Lui, V.W.; Lam, L.C.; Chiu, H.F.; Driver, J.A.; Pastorino, L.; Lu, K.P. A PIN1 polymorphism that prevents its suppression by AP4 associates with delayed onset of Alzheimer’s disease. Neurobiol. Aging 2012, 33, 804–813. [Google Scholar] [CrossRef] [PubMed]
- Eckerdt, F.; Yuan, J.; Saxena, K.; Martin, B.; Kappel, S.; Lindenau, C.; Kramer, A.; Naumann, S.; Daum, S.; Fischer, G.; et al. Polo-like kinase 1-mediated phosphorylation stabilizes Pin1 by inhibiting its ubiquitination in human cells. J. Biol. Chem. 2005, 280, 36575–36583. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.J.; Zhou, X.Z.; Liou, Y.C.; Noel, J.P.; Lu, K.P. Critical role of WW domain phosphorylation in regulating phosphoserine binding activity and Pin1 function. J. Biol. Chem. 2002, 277, 2381–2384. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.C.; Que, J.; Chen, Y.C.; Lin, J.T.; Liou, Y.C.; Liao, P.C.; Liu, Y.P.; Lee, K.H.; Lin, L.C.; Hsiao, M.; et al. Pin1 acts as a negative regulator of the G2/M transition by interacting with the Aurora-A-Bora complex. J. Cell Sci. 2013, 126 Pt 21, 4862–4872. [Google Scholar] [CrossRef]
- Cho, Y.S.; Park, S.Y.; Kim, D.J.; Lee, S.H.; Woo, K.M.; Lee, K.A.; Lee, Y.J.; Cho, Y.Y.; Shim, J.H. TPA-induced cell transformation provokes a complex formation between Pin1 and 90 kDa ribosomal protein S6 kinase 2. Mol. Cell. Biochem. 2012, 367, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.; Khanal, P.; Kim, J.Y.; Yun, H.J.; Lim, S.C.; Shim, J.H.; Choi, H.S. COT phosphorylates prolyl-isomerase Pin1 to promote tumorigenesis in breast cancer. Mol. Carcinogen. 2015, 54, 440–448. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.H.; Chen, C.H.; Suizu, F.; Huang, P.; Schiene-Fischer, C.; Daum, S.; Zhang, Y.J.; Goate, A.; Chen, R.H.; Zhou, X.Z.; et al. Death-associated protein kinase 1 phosphorylates Pin1 and inhibits its prolyl isomerase activity and cellular function. Mol. Cell 2011, 42, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Rangasamy, V.; Mishra, R.; Sondarva, G.; Das, S.; Lee, T.H.; Bakowska, J.C.; Tzivion, G.; Malter, J.S.; Rana, B.; Lu, K.P.; et al. Mixed-lineage kinase 3 phosphorylates prolyl-isomerase Pin1 to regulate its nuclear translocation and cellular function. Proc. Natl. Acad. Sci. USA 2012, 109, 8149–8154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balastik, M.; Zhou, X.Z.; Alberich-Jorda, M.; Weissova, R.; Ziak, J.; Pazyra-Murphy, M.F.; Cosker, K.E.; Machonova, O.; Kozmikova, I.; Chen, C.H.; et al. Prolyl Isomerase Pin1 Regulates Axon Guidance by Stabilizing CRMP2A Selectively in Distal Axons. Cell Rep. 2015, 13, 812–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baik, S.H.; Fane, M.; Park, J.H.; Cheng, Y.L.; Yang-Wei Fann, D.; Yun, U.J.; Choi, Y.; Park, J.S.; Chai, B.H.; Park, J.S.; et al. Pin1 promotes neuronal death in stroke by stabilizing Notch intracellular domain. Ann. Neurol. 2015, 77, 504–516. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Kosugi, I.; Lee, D.Y.; Hafner, A.; Sinclair, D.A.; Ryo, A.; Lu, K.P. Prolyl isomerase Pin1 regulates neuronal differentiation via beta-catenin. Mol. Cell. Biol. 2012, 32, 2966–2978. [Google Scholar] [CrossRef] [PubMed]
- Thorpe, J.R.; Mosaheb, S.; Hashemzadeh-Bonehi, L.; Cairns, N.J.; Kay, J.E.; Morley, S.J.; Rulten, S.L. Shortfalls in the peptidyl-prolyl cis-trans isomerase protein Pin1 in neurons are associated with frontotemporal dementias. Neurobiol. Dis. 2004, 17, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Holzer, M.; Gartner, U.; Stobe, A.; Hartig, W.; Gruschka, H.; Bruckner, M.K.; Arendt, T. Inverse association of Pin1 and tau accumulation in Alzheimer’s disease hippocampus. Acta Neuropathol. 2002, 104, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.J.; Wulf, G.; Zhou, X.Z.; Davies, P.; Lu, K.P. The prolyl isomerase Pin1 restores the function of Alzheimer-associated phosphorylated tau protein. Nature 1999, 399, 784–788. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, P.; Dickson, D.W.; Davies, P. Pin1 colocalization with phosphorylated tau in Alzheimer’s disease and other tauopathies. Neurobiol. Dis. 2003, 14, 251–264. [Google Scholar] [CrossRef]
- Clevers, H. Wnt/β-catenin signaling in development and disease. Cell 2006, 127, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Hirabayashi, Y.; Itoh, Y.; Tabata, H.; Nakajima, K.; Akiyama, T.; Masuyama, N.; Gotoh, Y. The Wnt/β-catenin pathway directs neuronal differentiation of cortical neural precursor cells. Development 2004, 131, 2791–2801. [Google Scholar] [CrossRef] [PubMed]
- Ryo, A.; Nakamura, M.; Wulf, G.; Liou, Y.C.; Lu, K.P. Pin1 regulates turnover and subcellular localization of β-catenin by inhibiting its interaction with APC. Nat. Cell Biol. 2001, 3, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Liou, Y.C.; Sun, A.; Ryo, A.; Zhou, X.Z.; Yu, Z.X.; Huang, H.K.; Uchida, T.; Bronson, R.; Bing, G.; Li, X.; et al. Role of the prolyl isomerase Pin1 in protecting against age-dependent neurodegeneration. Nature 2003, 424, 556–561. [Google Scholar] [CrossRef] [PubMed]
- Culotti, J.G.; Kolodkin, A.L. Functions of netrins and semaphorins in axon guidance. Curr. Opin. Neurobiol. 1996, 6, 81–88. [Google Scholar] [CrossRef]
- Fukata, Y.; Itoh, T.J.; Kimura, T.; Menager, C.; Nishimura, T.; Shiromizu, T.; Watanabe, H.; Inagaki, N.; Iwamatsu, A.; Hotani, H.; et al. CRMP-2 binds to tubulin heterodimers to promote microtubule assembly. Nat. Cell Biol. 2002, 4, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, N.; Chihara, K.; Arimura, N.; Menager, C.; Kawano, Y.; Matsuo, N.; Nishimura, T.; Amano, M.; Kaibuchi, K. CRMP-2 induces axons in cultured hippocampal neurons. Nat. Neurosci. 2001, 4, 781–782. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, T.; Kawano, Y.; Arimura, N.; Kawabata, S.; Kikuchi, A.; Kaibuchi, K. GSK-3beta regulates phosphorylation of CRMP-2 and neuronal polarity. Cell 2005, 120, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Arimura, N.; Menager, C.; Kawano, Y.; Yoshimura, T.; Kawabata, S.; Hattori, A.; Fukata, Y.; Amano, M.; Goshima, Y.; Inagaki, M.; et al. Phosphorylation by Rho kinase regulates CRMP-2 activity in growth cones. Mol. Cell. Biol. 2005, 25, 9973–9984. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, Y.; Cheng, C.; Uchida, Y.; Nakajima, O.; Ohshima, T.; Yagi, T.; Taniguchi, M.; Nakayama, T.; Kishida, R.; Kudo, Y.; et al. Fyn and Cdk5 mediate semaphorin-3A signaling, which is involved in regulation of dendrite orientation in cerebral cortex. Neuron 2002, 35, 907–920. [Google Scholar] [CrossRef]
- Quinn, C.C.; Chen, E.; Kinjo, T.G.; Kelly, G.; Bell, A.W.; Elliott, R.C.; McPherson, P.S.; Hockfield, S. TUC-4b, a novel TUC family variant, regulates neurite outgrowth and associates with vesicles in the growth cone. J. Neurosci. 2003, 23, 2815–2823. [Google Scholar] [CrossRef] [PubMed]
- Yuasa-Kawada, J.; Suzuki, R.; Kano, F.; Ohkawara, T.; Murata, M.; Noda, M. Axonal morphogenesis controlled by antagonistic roles of two CRMP subtypes in microtubule organization. Eur. J. Neurosci. 2003, 17, 2329–2343. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, E.F.; Strittmatter, S.M. The CRMP family of proteins and their role in Sema3A signaling. Adv. Exp. Med. Biol. 2007, 600, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Goshima, Y.; Nakamura, F.; Strittmatter, P.; Strittmatter, S.M. Collapsin-induced growth cone collapse mediated by an intracellular protein related to UNC-33. Nature 1995, 376, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Arimura, N.; Inagaki, N.; Chihara, K.; Menager, C.; Nakamura, N.; Amano, M.; Iwamatsu, A.; Goshima, Y.; Kaibuchi, K. Phosphorylation of collapsin response mediator protein-2 by Rho-kinase. Evidence for two separate signaling pathways for growth cone collapse. J. Biol. Chem. 2000, 275, 23973–23980. [Google Scholar] [CrossRef] [PubMed]
- Tan, F.; Thiele, C.J.; Li, Z. Collapsin response mediator proteins: Potential diagnostic and prognostic biomarkers in cancers (Review). Oncol. Lett. 2014, 7, 1333–1340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behar, O.; Golden, J.A.; Mashimo, H.; Schoen, F.J.; Fishman, M.C. Semaphorin III is needed for normal patterning and growth of nerves, bones and heart. Nature 1996, 383, 525–528. [Google Scholar] [CrossRef] [PubMed]
- Gu, C.; Rodriguez, E.R.; Reimert, D.V.; Shu, T.; Fritzsch, B.; Richards, L.J.; Kolodkin, A.L.; Ginty, D.D. Neuropilin-1 conveys semaphorin and VEGF signaling during neural and cardiovascular development. Dev. Cell 2003, 5, 45–57. [Google Scholar] [CrossRef]
- Escargueil, A.E.; Larsen, A.K. Mitosis-specific MPM-2 phosphorylation of DNA topoisomerase IIα is regulated directly by protein phosphatase 2A. Biochem. J. 2007, 403, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Grison, A.; Mantovani, F.; Comel, A.; Agostoni, E.; Gustincich, S.; Persichetti, F.; Del Sal, G. Ser46 phosphorylation and prolyl-isomerase Pin1-mediated isomerization of p53 are key events in p53-dependent apoptosis induced by mutant huntingtin. Proc. Natl. Acad. Sci. USA 2011, 108, 17979–17984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryo, A.; Togo, T.; Nakai, T.; Hirai, A.; Nishi, M.; Yamaguchi, A.; Suzuki, K.; Hirayasu, Y.; Kobayashi, H.; Perrem, K.; et al. Prolyl-isomerase Pin1 accumulates in lewy bodies of parkinson disease and facilitates formation of α-synuclein inclusions. J. Biol. Chem. 2006, 281, 4117–4125. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Zhang, Y.; Chen, G.; Xiong, Y.; Wang, X.; Zhu, B. Down-regulation of Pin1 in Temporal Lobe Epilepsy Patients and Mouse Model. Neurochem. Res. 2017, 42, 1211–1218. [Google Scholar] [CrossRef] [PubMed]
- Zita, M.M.; Marchionni, I.; Bottos, E.; Righi, M.; del Sal, G.; Cherubini, E.; Zacchi, P. Post-phosphorylation prolyl isomerisation of gephyrin represents a mechanism to modulate glycine receptors function. EMBO J. 2007, 26, 1761–1771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, M.; Shen, L.; Yin, H.; Pan, Y.M.; Wang, L.; Chen, D.; Xi, Z.Q.; Xiao, Z.; Wang, X.F.; Zhou, S.N. Downregulation of gephyrin in temporal lobe epilepsy neurons in humans and a rat model. Synapse (N. Y.) 2011, 65, 1006–1014. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.A.; Tindale, L.; Cumming, R.C. Age-dependent metabolic dysregulation in cancer and Alzheimer’s disease. Biogerontology 2014, 15, 559–577. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ma, Q.; Zhang, Y.W.; Xu, H. Proteolytic processing of Alzheimer’s beta-amyloid precursor protein. J. Neurochem. 2012, 120, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Atchison, F.W.; Capel, B.; Means, A.R. Pin1 regulates the timing of mammalian primordial germ cell proliferation. Development 2003, 130, 3579–3586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atchison, F.W.; Means, A.R. Spermatogonial depletion in adult Pin1-deficient mice. Biol. Reprod. 2003, 69, 1989–1997. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.Y.; Choi, H.K.; Shim, J.H.; Kang, K.W.; Dong, Z.; Choi, H.S. The prolyl isomerase Pin1 interacts with a ribosomal protein S6 kinase to enhance insulin-induced AP-1 activity and cellular transformation. Carcinogenesis 2009, 30, 671–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastorino, L.; Sun, A.; Lu, P.J.; Zhou, X.Z.; Balastik, M.; Finn, G.; Wulf, G.; Lim, J.; Li, S.H.; Li, X.; et al. The prolyl isomerase Pin1 regulates amyloid precursor protein processing and amyloid-beta production. Nature 2006, 440, 528–534. [Google Scholar] [CrossRef] [PubMed]
- Driver, J.A.; Zhou, X.Z.; Lu, K.P. Regulation of protein conformation by Pin1 offers novel disease mechanisms and therapeutic approaches in Alzheimer’s disease. Discov. Med. 2014, 17, 93–99. [Google Scholar] [PubMed]
- Butterfield, D.A.; Abdul, H.M.; Opii, W.; Newman, S.F.; Joshi, G.; Ansari, M.A.; Sultana, R. Pin1 in Alzheimer’s disease. J. Neurochem. 2006, 98, 1697–1706. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.L.; Pastorino, L.; Zhou, X.Z.; Lu, K.P. Prolyl isomerase Pin1 promotes amyloid precursor protein (APP) turnover by inhibiting glycogen synthase kinase-3β (GSK3β) activity: Novel mechanism for Pin1 to protect against Alzheimer disease. J. Biol. Chem. 2012, 287, 6969–6973. [Google Scholar] [CrossRef] [PubMed]
- Ittner, A.; Chua, S.W.; Bertz, J.; Volkerling, A.; van der Hoven, J.; Gladbach, A.; Przybyla, M.; Bi, M.; van Hummel, A.; Stevens, C.H.; et al. Site-specific phosphorylation of tau inhibits amyloid-β toxicity in Alzheimer’s mice. Science (N. Y.) 2016, 354, 904–908. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Huang, N.Q.; Yan, F.; Jin, H.; Zhou, S.Y.; Shi, J.S.; Jin, F. Diabetes mellitus and Alzheimer’s disease: GSK-3β as a potential link. Behav. Brain Res. 2018, 339, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Aplin, A.E.; Gibb, G.M.; Jacobsen, J.S.; Gallo, J.M.; Anderton, B.H. In vitro phosphorylation of the cytoplasmic domain of the amyloid precursor protein by glycogen synthase kinase-3β. J. Neurochem. 1996, 67, 699–707. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Hung, M.C. Physiological regulation of Akt activity and stability. Am. J. Transl. Res. 2010, 2, 19–42. [Google Scholar] [PubMed]
- Brynczka, C.; Labhart, P.; Merrick, B.A. NGF-mediated transcriptional targets of p53 in PC12 neuronal differentiation. BMC Genom. 2007, 8, 139. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Wulf, G.M. The amplifier effect: How Pin1 empowers mutant p53. Breast Cancer Res. BCR 2011, 13, 315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tedeschi, A.; Di Giovanni, S. The non-apoptotic role of p53 in neuronal biology: Enlightening the dark side of the moon. EMBO Rep. 2009, 10, 576–583. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Ren, Z.; Chow, F.E.; Tsai, R.; Liu, T.; Rizzolio, F.; Boffo, S.; Xu, Y.; Huang, S.; Lippa, C.F.; et al. Pathological Role of Peptidyl-Prolyl Isomerase Pin1 in the Disruption of Synaptic Plasticity in Alzheimer’s Disease. Neural Plast. 2017, 2017, 3270725. [Google Scholar] [CrossRef] [PubMed]
- Steen, E.; Terry, B.M.; Rivera, E.J.; Cannon, J.L.; Neely, T.R.; Tavares, R.; Xu, X.J.; Wands, J.R.; de la Monte, S.M. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease—Is this type 3 diabetes? J. Alzheimer’s Dis. 2005, 7, 63–80. [Google Scholar] [CrossRef]
- De la Monte, S.M.; Chen, G.J.; Rivera, E.; Wands, J.R. Neuronal thread protein regulation and interaction with microtubule-associated proteins in SH-Sy5y neuronal cells. Cell. Mol. Life Sci. 2003, 60, 2679–2691. [Google Scholar] [CrossRef] [PubMed]
- Nakatsu, Y.; Mori, K.; Matsunaga, Y.; Yamamotoya, T.; Ueda, K.; Inoue, Y.; Mitsuzaki-Miyoshi, K.; Sakoda, H.; Fujishiro, M.; Yamaguchi, S.; et al. The prolyl isomerase Pin1 increases beta-cell proliferation and enhances insulin secretion. J. Biol. Chem. 2017, 292, 11886–11895. [Google Scholar] [CrossRef] [PubMed]
- Paneni, F.; Costantino, S.; Castello, L.; Battista, R.; Capretti, G.; Chiandotto, S.; D’Amario, D.; Scavone, G.; Villano, A.; Rustighi, A.; et al. Targeting prolyl-isomerase Pin1 prevents mitochondrial oxidative stress and vascular dysfunction: Insights in patients with diabetes. Eur. Heart J. 2015, 36, 817–828. [Google Scholar] [CrossRef] [PubMed]
- Arosio, B.; Bulbarelli, A.; Bastias Candia, S.; Lonati, E.; Mastronardi, L.; Romualdi, P.; Candeletti, S.; Gussago, C.; Galimberti, D.; Scarpini, E.; et al. Pin1 contribution to Alzheimer’s disease: Transcriptional and epigenetic mechanisms in patients with late-onset Alzheimer’s disease. Neuro-Degener. Dis. 2012, 10, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Simon, B.P.; Bennett, D.A.; Schneider, J.A.; Malter, J.S.; Wang, D.S. The significance of Pin1 in the development of Alzheimer’s disease. J. Alzheimer’s Dis. 2007, 11, 13–23. [Google Scholar] [CrossRef]
- Nakatsu, Y.; Iwashita, M.; Sakoda, H.; Ono, H.; Nagata, K.; Matsunaga, Y.; Fukushima, T.; Fujishiro, M.; Kushiyama, A.; Kamata, H.; et al. Prolyl isomerase Pin1 negatively regulates AMP-activated protein kinase (AMPK) by associating with the CBS domain in the gamma subunit. J. Biol. Chem. 2015, 290, 24255–24266. [Google Scholar] [CrossRef] [PubMed]
- Nakatsu, Y.; Sakoda, H.; Kushiyama, A.; Zhang, J.; Ono, H.; Fujishiro, M.; Kikuchi, T.; Fukushima, T.; Yoneda, M.; Ohno, H.; et al. Peptidyl-prolyl cis/trans isomerase NIMA-interacting 1 associates with insulin receptor substrate-1 and enhances insulin actions and adipogenesis. J. Biol. Chem. 2011, 286, 20812–20822. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.L.; Chen, F.Q.; Xu, W.J.; Yue, W.Z.; Yuan, G.; Yang, Y. Early intervention with glucagon-like peptide 1 analog liraglutide prevents tau hyperphosphorylation in diabetic db/db mice. J. Neurochem. 2015, 135, 301–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Nakatsu, Y.; Shinjo, T.; Guo, Y.; Sakoda, H.; Yamamotoya, T.; Otani, Y.; Okubo, H.; Kushiyama, A.; Fujishiro, M.; et al. Par14 protein associates with insulin receptor substrate 1 (IRS-1), thereby enhancing insulin-induced IRS-1 phosphorylation and metabolic actions. J. Biol. Chem. 2013, 288, 20692–20701. [Google Scholar] [CrossRef] [PubMed]
- Rabiee, A.; Kruger, M.; Ardenkjaer-Larsen, J.; Kahn, C.R.; Emanuelli, B. Distinct signalling properties of insulin receptor substrate (IRS)-1 and IRS-2 in mediating insulin/IGF-1 action. Cell. Signal. 2018, 47, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Park, J.E.; Lee, J.A.; Park, S.G.; Lee, D.H.; Kim, S.J.; Kim, H.J.; Uchida, C.; Uchida, T.; Park, B.C.; Cho, S. A critical step for JNK activation: Isomerization by the prolyl isomerase Pin1. Cell Death Differ. 2012, 19, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Simpson, J.E.; Ince, P.G.; Shaw, P.J.; Heath, P.R.; Raman, R.; Garwood, C.J.; Gelsthorpe, C.; Baxter, L.; Forster, G.; Matthews, F.E.; et al. Microarray analysis of the astrocyte transcriptome in the aging brain: Relationship to Alzheimer’s pathology and APOE genotype. Neurobiol. Aging 2011, 32, 1795–1807. [Google Scholar] [CrossRef] [PubMed]
- Beale, E.G. Insulin signaling and insulin resistance. J. Investig. Med. 2013, 61, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, F.; Lucas, J.J.; Avila, J. GSK3 and tau: Two convergence points in Alzheimer’s disease. J. Alzheimer’s Dis. 2013, 33 (Suppl. 1), S141–S144. [Google Scholar] [CrossRef] [PubMed]
- Takashima, A. GSK-3 is essential in the pathogenesis of Alzheimer’s disease. J. Alzheimer’s Dis. 2006, 9, 309–317. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Poon, H.F.; St Clair, D.; Keller, J.N.; Pierce, W.M.; Klein, J.B.; Markesbery, W.R. Redox proteomics identification of oxidatively modified hippocampal proteins in mild cognitive impairment: Insights into the development of Alzheimer’s disease. Neurobiol. Dis. 2006, 22, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Hall, E.C., 2nd; Lee, S.Y.; Mairuae, N.; Simmons, Z.; Connor, J.R. Expression of the HFE allelic variant H63D in SH-SY5Y cells affects tau phosphorylation at serine residues. Neurobiol. Aging 2011, 32, 1409–1419. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Li, W.; Sultana, R.; You, M.H.; Kondo, A.; Shahpasand, K.; Kim, B.M.; Luo, M.L.; Nechama, M.; Lin, Y.M.; et al. Pin1 cysteine-113 oxidation inhibits its catalytic activity and cellular function in Alzheimer’s disease. Neurobiol. Dis. 2015, 76, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Hall, E.C., 2nd; Lee, S.Y.; Simmons, Z.; Neely, E.B.; Nandar, W.; Connor, J.R. Prolyl-peptidyl isomerase, Pin1, phosphorylation is compromised in association with the expression of the HFE polymorphic allele, H63D. Biochim. Biophys. Acta 2010, 1802, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Min, S.H.; Cho, J.S.; Oh, J.H.; Shim, S.B.; Hwang, D.Y.; Lee, S.H.; Jee, S.W.; Lim, H.J.; Kim, M.Y.; Sheen, Y.Y.; et al. Tau and GSK3β dephosphorylations are required for regulating Pin1 phosphorylation. Neurochem. Res. 2005, 30, 955–961. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bianchi, M.; Manco, M. Pin1 Modulation in Physiological Status and Neurodegeneration. Any Contribution to the Pathogenesis of Type 3 Diabetes? Int. J. Mol. Sci. 2018, 19, 2319. https://doi.org/10.3390/ijms19082319
Bianchi M, Manco M. Pin1 Modulation in Physiological Status and Neurodegeneration. Any Contribution to the Pathogenesis of Type 3 Diabetes? International Journal of Molecular Sciences. 2018; 19(8):2319. https://doi.org/10.3390/ijms19082319
Chicago/Turabian StyleBianchi, Marzia, and Melania Manco. 2018. "Pin1 Modulation in Physiological Status and Neurodegeneration. Any Contribution to the Pathogenesis of Type 3 Diabetes?" International Journal of Molecular Sciences 19, no. 8: 2319. https://doi.org/10.3390/ijms19082319
APA StyleBianchi, M., & Manco, M. (2018). Pin1 Modulation in Physiological Status and Neurodegeneration. Any Contribution to the Pathogenesis of Type 3 Diabetes? International Journal of Molecular Sciences, 19(8), 2319. https://doi.org/10.3390/ijms19082319