miRNA-34c Overexpression Causes Dendritic Loss and Memory Decline


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Overexpression of miR-34c Caused Memory Impairment
2.2. Expression Level of miR-34c in the Hippocampus
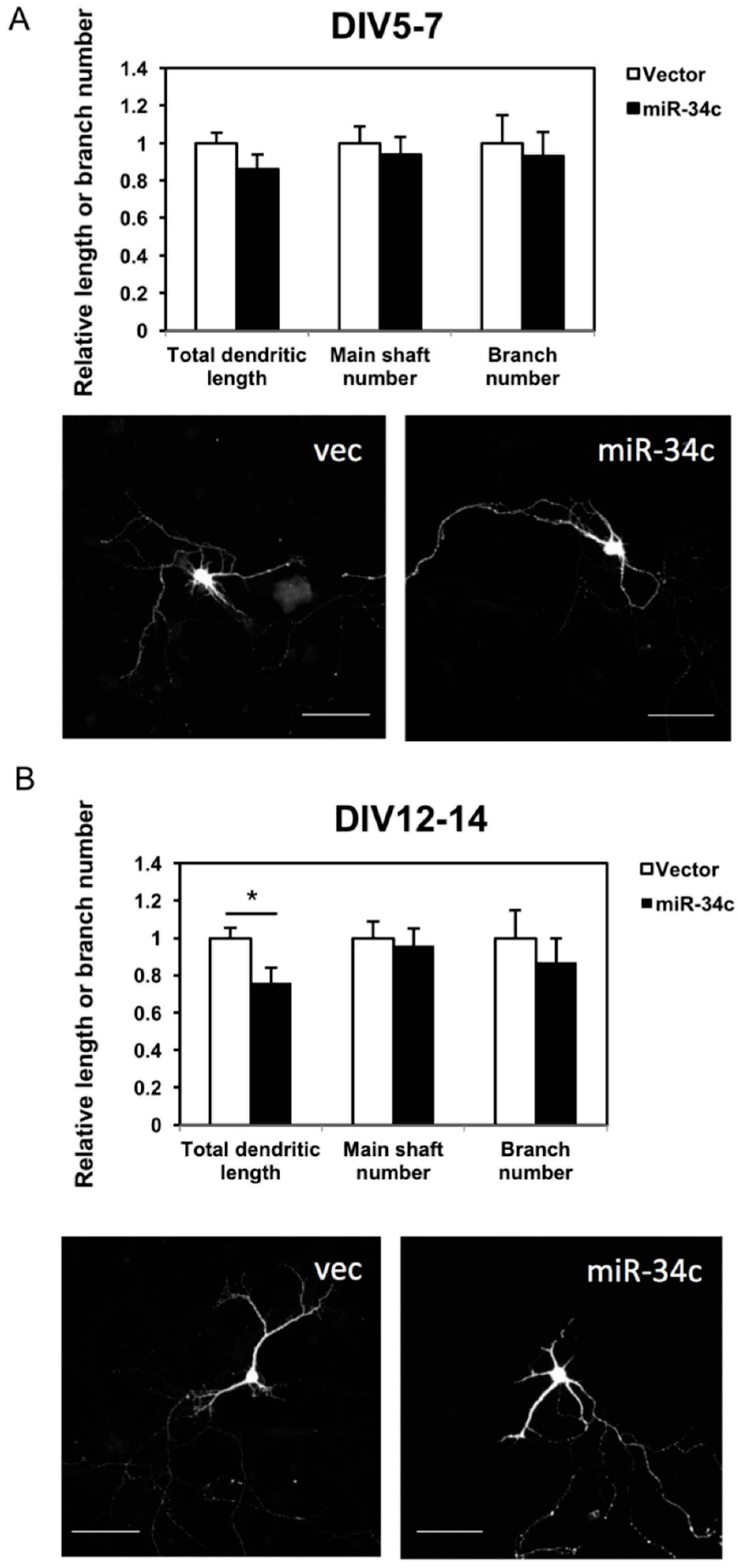
2.3. Overexpression of miR-34c Reduced Dendritic Length and Branch Numbers
2.4. Effects of miR-34c Overexpression on Neurite Protrusion Density
2.5. Effects of miR-34c Overexpression on Protrusion Density in Various Dendritic Areas
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Deoxyribonucleic Acid Constructions
4.3. Lentivirus Production
4.4. Stereotaxic Hippocampal Injections
4.5. Morris Water Maze Test
4.6. Primary Hippocampal Culture
4.7. Transfection
4.8. Immunofluorescence Staining
4.9. Image Analysis
4.10. Reverse Transcription PCR (RT-PCR)
4.11. Statistics
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Forner, S.; Baglietto-Vargas, D.; Martini, A.C.; Trujillo-Estrada, L.; LaFerla, F.M. Synaptic impairment in Alzheimer’s disease: A dysregulated symphony. Trends Neurosci. 2017, 40, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Pillai, R.S.; Bhattacharyya, S.N.; Filipowicz, W. Repression of protein synthesis by mirnas: How many mechanisms? Trends Cell Biol. 2007, 17, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Malmevik, J.; Petri, R.; Klussendorf, T.; Knauff, P.; Akerblom, M.; Johansson, J.; Soneji, S.; Jakobsson, J. Identification of the mirna targetome in hippocampal neurons using rip-seq. Sci. Rep. 2015, 5, 12609. [Google Scholar] [CrossRef] [PubMed]
- Malmevik, J.; Petri, R.; Knauff, P.; Brattas, P.L.; Akerblom, M.; Jakobsson, J. Distinct cognitive effects and underlying transcriptome changes upon inhibition of individual mirnas in hippocampal neurons. Sci. Rep. 2016, 6, 19879. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Rapp, J.; Rainone, S.; Hebert, S.S. MicroRNAs underlying memory deficits in neurodegenerative disorders. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2017, 73, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Gascon, E.; Lynch, K.; Ruan, H.; Almeida, S.; Verheyden, J.M.; Seeley, W.W.; Dickson, D.W.; Petrucelli, L.; Sun, D.; Jiao, J.; et al. Alterations in microRNA-124 and ampa receptors contribute to social behavioral deficits in frontotemporal dementia. Nat. Med. 2014, 20, 1444–1451. [Google Scholar] [CrossRef] [PubMed]
- Zovoilis, A.; Agbemenyah, H.Y.; Agis-Balboa, R.C.; Stilling, R.M.; Edbauer, D.; Rao, P.; Farinelli, L.; Delalle, I.; Schmitt, A.; Falkai, P.; et al. MicroRNA-34c is a novel target to treat dementias. EMBO J. 2011, 30, 4299–4308. [Google Scholar] [CrossRef] [PubMed]
- Bhatnagar, S.; Chertkow, H.; Schipper, H.M.; Yuan, Z.; Shetty, V.; Jenkins, S.; Jones, T.; Wang, E. Increased microRNA-34c abundance in Alzheimer’s disease circulating blood plasma. Front. Mol. Neurosci. 2014, 7, 2. [Google Scholar] [CrossRef] [PubMed]
- Xie, B.; Zhou, H.; Zhang, R.; Song, M.; Yu, L.; Wang, L.; Liu, Z.; Zhang, Q.; Cui, D.; Wang, X.; et al. Serum mir-206 and mir-132 as potential circulating biomarkers for mild cognitive impairment. J. Alzheimers Dis. 2015, 45, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Lau, P.; Frigerio, C.S.; de Strooper, B. Variance in the identification of microRNAs deregulated in Alzheimer’s disease and possible role of lincrnas in the pathology: The need of larger datasets. Ageing Res. Rev. 2014, 17, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Landreh, M.; Cao, K.; Abe, M.; Hendriks, G.J.; Kennerdell, J.R.; Zhu, Y.; Wang, L.S.; Bonini, N.M. The microRNA mir-34 modulates ageing and neurodegeneration in drosophila. Nature 2012, 482, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Rokavec, M.; Li, H.; Jiang, L.; Hermeking, H. The p53/mir-34 axis in development and disease. J. Mol. Cell Biol. 2014, 6, 214–230. [Google Scholar] [CrossRef] [PubMed]
- Jauhari, A.; Singh, T.; Singh, P.; Parmar, D.; Yadav, S. Regulation of mir-34 family in neuronal development. Mol. Neurobiol. 2018, 55, 936–945. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Bao, J.; Kim, M.; Yuan, S.; Tang, C.; Zheng, H.; Mastick, G.S.; Xu, C.; Yan, W. Two mirna clusters, mir-34b/c and mir-449, are essential for normal brain development, motile ciliogenesis, and spermatogenesis. Proc. Nat.Acad. Sci. USA 2014, 111, E2851–E2857. [Google Scholar] [CrossRef] [PubMed]
- Agostini, M.; Tucci, P.; Steinert, J.R.; Shalom-Feuerstein, R.; Rouleau, M.; Aberdam, D.; Forsythe, I.D.; Young, K.W.; Ventura, A.; Concepcion, C.P.; et al. MicroRNA-34a regulates neurite outgrowth, spinal morphology, and function. Proc. Nat.Acad. Sci. USA 2011, 108, 21099–21104. [Google Scholar] [CrossRef] [PubMed]
- Soni, K.; Choudhary, A.; Patowary, A.; Singh, A.R.; Bhatia, S.; Sivasubbu, S.; Chandrasekaran, S.; Pillai, B. Mir-34 is maternally inherited in drosophila melanogaster and danio rerio. Nucleic Acids Res. 2013, 41, 4470–4480. [Google Scholar] [CrossRef] [PubMed]
- Fededa, J.P.; Esk, C.; Mierzwa, B.; Stanyte, R.; Yuan, S.; Zheng, H.; Ebnet, K.; Yan, W.; Knoblich, J.A.; Gerlich, D.W. MicroRNA-34/449 controls mitotic spindle orientation during mammalian cortex development. EMBO J. 2016, 35, 2386–2398. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, P.; Zhu, H.; Xu, Y.; Ma, C.; Dai, X.; Huang, L.; Liu, Y.; Zhang, L.; Qin, C. miR-34a, a microRNA up-regulated in a double transgenic mouse model of Alzheimer’s disease, inhibits bcl2 translation. Brain Res. Bull. 2009, 80, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Dickson, J.R.; Kruse, C.; Montagna, D.R.; Finsen, B.; Wolfe, M.S. Alternative polyadenylation and mir-34 family members regulate tau expression. J. Neurochem. 2013, 127, 739–749. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Huang, M.; Lu, M.; Zhu, W.; Shu, Y.; Cao, P.; Liu, P. Regulation of microtubule-associated protein tau (mapt) by mir-34c-5p determines the chemosensitivity of gastric cancer to paclitaxel. Cancer Chemother. Pharmacol. 2013, 71, 1159–1171. [Google Scholar] [CrossRef] [PubMed]
- Uchizono, K. Characteristics of excitatory and inhibitory synapses in the central nervous system of the cat. Nature 1965, 207, 642–643. [Google Scholar] [CrossRef] [PubMed]
- Attardo, A.; Fitzgerald, J.E.; Schnitzer, M.J. Impermanence of dendritic spines in live adult ca1 hippocampus. Nature 2015, 523, 592–596. [Google Scholar] [CrossRef] [PubMed]
- Takashima, S.; Ieshima, A.; Nakamura, H.; Becker, L.E. Dendrites, dementia and the down syndrome. Brain Dev. 1989, 11, 131–133. [Google Scholar] [CrossRef]
- Spires-Jones, T.; Knafo, S. Spines, plasticity, and cognition in Alzheimer’s model mice. Neural Plast. 2012, 2012, 319836. [Google Scholar] [CrossRef] [PubMed]
- DeKosky, S.T.; Scheff, S.W. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: Correlation with cognitive severity. Ann. Neurol. 1990, 27, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Terry, R.D.; Masliah, E.; Salmon, D.P.; Butters, N.; DeTeresa, R.; Hill, R.; Hansen, L.A.; Katzman, R. Physical basis of cognitive alterations in Alzheimer’s disease: Synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 1991, 30, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Alzheimer’s disease is a synaptic failure. Science 2002, 298, 789–791. [Google Scholar] [CrossRef] [PubMed]
- Baloyannis, S.J. Dendritic pathology in Alzheimer’s disease. J. Neurol. Sci. 2009, 283, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Spruston, N. Pyramidal neurons: Dendritic structure and synaptic integration. Nat. Rev. Neurosci. 2008, 9, 206–221. [Google Scholar] [CrossRef] [PubMed]
- Catala, I.; Ferrer, I.; Galofre, E.; Fabregues, I. Decreased numbers of dendritic spines on cortical pyramidal neurons in dementia. A quantitative golgi study on biopsy samples. Hum. Neurobiol. 1988, 6, 255–259. [Google Scholar] [PubMed]
- Einstein, G.; Buranosky, R.; Crain, B.J. Dendritic pathology of granule cells in Alzheimer’s disease is unrelated to neuritic plaques. J. Neurosci. 1994, 14, 5077–5088. [Google Scholar] [CrossRef] [PubMed]
- Androuin, A.; Potier, B.; Nagerl, U.V.; Cattaert, D.; Danglot, L.; Thierry, M.; Youssef, I.; Triller, A.; Duyckaerts, C.; El Hachimi, K.H.; et al. Evidence for altered dendritic spine compartmentalization in Alzheimer’s disease and functional effects in a mouse model. Acta Neuropathol. 2018, 135, 839–854. [Google Scholar] [CrossRef] [PubMed]
- Berry, K.P.; Nedivi, E. Spine dynamics: Are they all the same? Neuron 2017, 96, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.; Lin, A.; Chang, P.; Gan, W.B. Development of long-term dendritic spine stability in diverse regions of cerebral cortex. Neuron 2005, 46, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Kasai, H.; Matsuzaki, M.; Noguchi, J.; Yasumatsu, N.; Nakahara, H. Structure-stability-function relationships of dendritic spines. Trends Neurosci. 2003, 26, 360–368. [Google Scholar] [CrossRef]
- Tada, T.; Sheng, M. Molecular mechanisms of dendritic spine morphogenesis. Curr. Opin. Neurobiol. 2006, 16, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Codocedo, J.F.; Allard, C.; Godoy, J.A.; Varela-Nallar, L.; Inestrosa, N.C. Sirt1 regulates dendritic development in hippocampal neurons. PLoS ONE 2012, 7, e47073. [Google Scholar] [CrossRef] [PubMed]
- Lamprecht, R.; LeDoux, J. Structural plasticity and memory. Nat. Rev. Neurosci. 2004, 5, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Haditsch, U.; Leone, D.P.; Farinelli, M.; Chrostek-Grashoff, A.; Brakebusch, C.; Mansuy, I.M.; McConnell, S.K.; Palmer, T.D. A central role for the small gtpase rac1 in hippocampal plasticity and spatial learning and memory. Mol. Cell. Neurosci. 2009, 41, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Vadodaria, K.C.; Jessberger, S. Maturation and integration of adult born hippocampal neurons: Signal convergence onto small rho gtpases. Front. Synaptic Neurosci. 2013, 5, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakayama, A.Y.; Harms, M.B.; Luo, L. Small gtpases rac and rho in the maintenance of dendritic spines and branches in hippocampal pyramidal neurons. J. Neurosci. 2000, 20, 5329–5338. [Google Scholar] [CrossRef] [PubMed]
- Hedrick, N.G.; Yasuda, R. Regulation of rho gtpase proteins during spine structural plasticity for the control of local dendritic plasticity. Curr. Opin. Neurobiol. 2017, 45, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, A.Y.; Luo, L. Intracellular signaling pathways that regulate dendritic spine morphogenesis. Hippocampus 2000, 10, 582–586. [Google Scholar] [CrossRef]
- Schumacher, S.; Franke, K. miR-124-regulated RhoG: A conductor of neuronal process complexity. Small GTPases 2013, 4, 42–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castaneda, P.; Munoz, M.; Garcia-Rojo, G.; Ulloa, J.L.; Bravo, J.A.; Marquez, R.; Garcia-Perez, M.A.; Arancibia, D.; Araneda, K.; Rojas, P.S.; et al. Association of n-cadherin levels and downstream effectors of rho gtpases with dendritic spine loss induced by chronic stress in rat hippocampal neurons. J. Neurosci. Res. 2015, 93, 1476–1491. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Jolly, M.K.; Lu, M.; Tsarfaty, I.; Ben-Jacob, E.; Onuchic, J.N. Modeling the transitions between collective and solitary migration phenotypes in cancer metastasis. Sci. Rep. 2015, 5, 17379. [Google Scholar] [CrossRef] [PubMed]
- Bustos, F.J.; Ampuero, E.; Jury, N.; Aguilar, R.; Falahi, F.; Toledo, J.; Ahumada, J.; Lata, J.; Cubillos, P.; Henriquez, B.; et al. Epigenetic editing of the Dlg4/PSD95 gene improves cognition in aged and Alzheimer’s disease mice. Brain 2017, 140, 3252–3268. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kao, Y.-C.; Wang, I.-F.; Tsai, K.-J. miRNA-34c Overexpression Causes Dendritic Loss and Memory Decline. Int. J. Mol. Sci. 2018, 19, 2323. https://doi.org/10.3390/ijms19082323
Kao Y-C, Wang I-F, Tsai K-J. miRNA-34c Overexpression Causes Dendritic Loss and Memory Decline. International Journal of Molecular Sciences. 2018; 19(8):2323. https://doi.org/10.3390/ijms19082323
Chicago/Turabian StyleKao, Yu-Chia, I-Fang Wang, and Kuen-Jer Tsai. 2018. "miRNA-34c Overexpression Causes Dendritic Loss and Memory Decline" International Journal of Molecular Sciences 19, no. 8: 2323. https://doi.org/10.3390/ijms19082323
APA StyleKao, Y. -C., Wang, I. -F., & Tsai, K. -J. (2018). miRNA-34c Overexpression Causes Dendritic Loss and Memory Decline. International Journal of Molecular Sciences, 19(8), 2323. https://doi.org/10.3390/ijms19082323