Biological Aspects of mTOR in Leukemia


Abstract
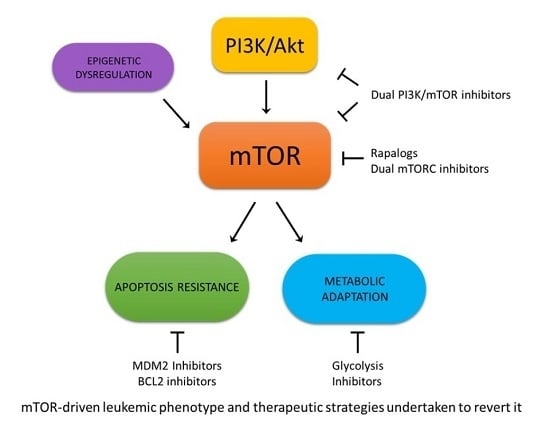
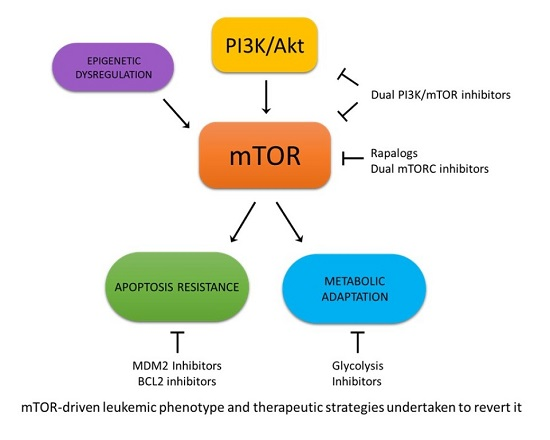
:
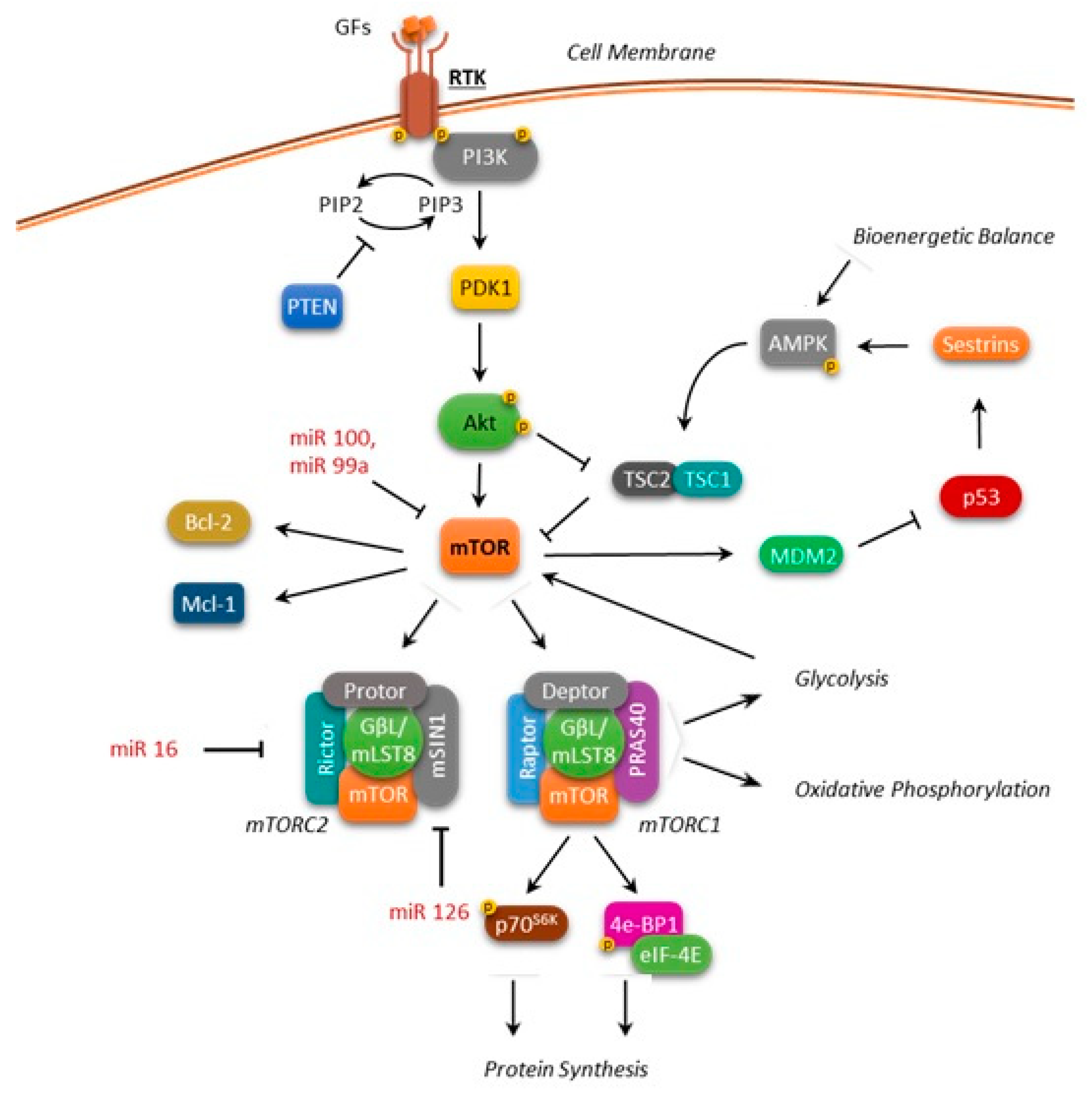
1. mTOR Structure and Function
2. mTOR Deregulation in Leukemias
3. mTOR Involvement in Leukemia Metabolism
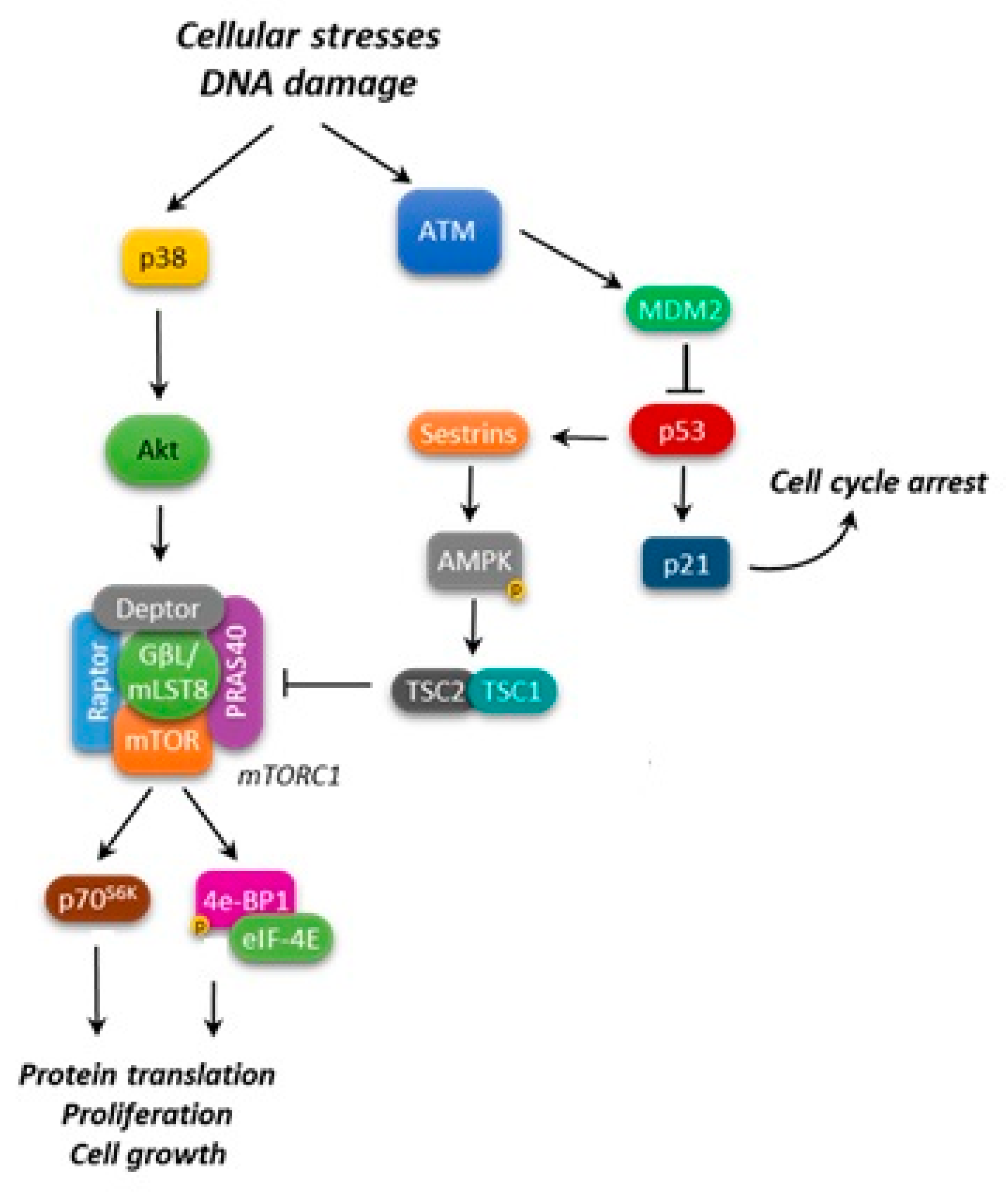
4. mTOR and Apoptosis Regulation in Leukemias
4.1. mTOR and p53
4.2. mTOR and Bcl-2 Family
5. mTOR and MicroRNA Regulation in Leukemias
6. mTOR Axis Inhibition in Leukemias
6.1. Rapamycin and Rapalogs
6.2. Dual Inhibitors
6.3. mTOR Inhibitors in AML
6.4. mTOR Inhibitors in ALL
6.5. mTOR Inhibitors in Other Leukemias
7. Summary
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Memmott, R.M.; Dennis, P.A. Akt-dependent and -independent mechanisms of mTOR regulation in cancer. Cell Signal. 2009, 21, 656–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laplante, M.; Sabatini, D.M. mTOR signaling at a glance. J. Cell Sci. 2009, 122, 3589–3594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarbassov, D.D.; Ali, S.M.; Kim, D.H.; Guertin, D.A.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 2004, 14, 1296–1302. [Google Scholar] [CrossRef] [PubMed]
- Lamming, D.W.; Ye, L.; Katajisto, P.; Goncalves, M.D.; Saitoh, M.; Stevens, D.M.; Davis, J.G.; Salmon, A.B.; Richardson, A.; Ahima, R.S.; et al. Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science 2012, 335, 1638–1643. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Ali, S.M.; Sengupta, S.; Sheen, J.H.; Hsu, P.P.; Bagley, A.F.; Markhard, A.L.; Sabatini, D.M. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell 2006, 22, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Dibble, C.C.; Manning, B.D. Signal integration by mTORC1 coordinates nutrient input with biosynthetic output. Nat. Cell Biol. 2013, 15, 555–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jossé, L.; Xie, J.; Proud, C.G.; Smales, C.M. mTORC1 signalling and eIF4E/4E-BP1 translation initiation factor stoichiometry influence recombinant protein productivity from GS-CHOK1 cells. Biochem. J. 2016, 473, 4651–4664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacinto, E.; Loewith, R.; Schmidt, A.; Lin, S.; Rüegg, M.A.; Hall, A.; Hall, M.N. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat. Cell Biol. 2004, 6, 1122–1128. [Google Scholar] [CrossRef] [PubMed]
- Goncharova, E.A.; Goncharov, D.A.; Li, H.; Pimtong, W.; Lu, S.; Khavin, I.; Krymskaya, V.P. mTORC2 is required for proliferation and survival of TSC2-null cells. Mol. Cell Biol. 2011, 31, 2484–2498. [Google Scholar] [CrossRef] [PubMed]
- Zinzalla, V.; Stracka, D.; Oppliger, W.; Hall, M.N. Activation of mTORC2 by association with the ribosome. Cell 2011, 144, 757–768. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Abrams, S.L.; Fitzgerald, T.L.; Cocco, L.; Martelli, A.M.; Montalto, G.; Cervello, M.; Scalisi, A.; Candido, S.; Libra, M.; et al. Roles of signaling pathways in drug resistance, cancer initiating cells and cancer progression and metastasis. Adv. Biol. Regul. 2015, 57, 75–101. [Google Scholar] [CrossRef] [PubMed]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Hoshii, T.; Tadokoro, Y.; Naka, K.; Ooshio, T.; Muraguchi, T.; Sugiyama, N.; Soga, T.; Araki, K.; Yamamura, K.; Hirao, A. mTORC1 is essential for leukemia propagation but not stem cell self-renewal. J. Clin. Investig. 2012, 122, 2114–2129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beauchamp, E.M.; Platanias, L.C. The evolution of the TOR pathway and its role in cancer. Oncogene 2013, 32, 3923–3932. [Google Scholar] [CrossRef] [PubMed]
- Willems, L.; Tamburini, J.; Chapuis, N.; Lacombe, C.; Mayeux, P.; Bouscary, D. PI3K and mTOR signaling pathways in cancer: New data on targeted therapies. Curr. Oncol. Rep. 2012, 14, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Yang, Y.; Hua, C.; Xu, S.; Zhou, M.; Guo, H.; Wang, N.; Zhao, X.; Huang, L.; Yu, F.; et al. Rictor has a pivotal role in maintaining quiescence as well as stemness of leukemia stem cells in MLL-driven leukemia. Leukemia 2017, 31, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Evangelisti, C.; Chiarini, F.; McCubrey, J.A.; Martelli, A.M. Therapeutic targeting of mTOR in T-cell acute lymphoblastic leukemia: An update. Int. J. Mol. Sci. 2018, 19, 1878. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR signaling in growth, metabolism, and disease. Cell 2017, 169, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Ben-Sahra, I.; Manning, B.D. mTORC1 signaling and the metabolic control of cell growth. Curr. Opin. Cell. Biol. 2017, 45, 72–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathupala, S.P.; Ko, Y.H.; Pedersen, P.L. Hexokinase II: Cancer’s double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene 2006, 25, 4777–4786. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G. AMP-activated/SNF1 protein kinases: Conserved guardians of cellular energy. Nat. Rev. Mol. Cell Biol. 2007, 8, 774–785. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMP-activated protein kinase: A target for drugs both ancient and modern. Chem. Biol. 2012, 19, 1222–1236. [Google Scholar] [CrossRef] [PubMed]
- Pradelli, L.A.; Bénéteau, M.; Chauvin, C.; Jacquin, M.A.; Marchetti, S.; Muñoz-Pinedo, C.; Auberger, P.; Pende, M.; Ricci, J.E. Glycolysis inhibition sensitizes tumor cells to death receptors-induced apoptosis by AMP kinase activation leading to Mcl-1 block in translation. Oncogene 2010, 29, 1641–1652. [Google Scholar] [CrossRef] [PubMed]
- Coloff, J.L.; Macintyre, A.N.; Nichols, A.G.; Liu, T.; Gallo, C.A.; Plas, D.R.; Rathmell, J.C. Akt-dependent glucose metabolism promotes Mcl-1 synthesis to maintain cell survival and resistance to Bcl-2 inhibition. Cancer Res. 2011, 71, 5204–5213. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.L.; Long, Z.J.; Wang, L.X.; Zheng, F.M.; Fang, Z.G.; Yan, M.; Xu, D.F.; Chen, J.J.; Wang, S.W.; Lin, D.J.; et al. Inhibition of mTOR pathway sensitizes acute myeloid leukemia cells to aurora inhibitors by suppression of glycolytic metabolism. Mol. Cancer Res. 2013, 11, 1326–1336. [Google Scholar] [CrossRef] [PubMed]
- Rosilio, C.; Lounnas, N.; Nebout, M.; Imbert, V.; Hagenbeek, T.; Spits, H.; Asnafi, V.; Pontier-Bres, R.; Reverso, J.; Michiels, J.F.; et al. The metabolic perturbators metformin, phenformin and AICAR interfere with the growth and survival of murine PTEN-deficient T cell lymphomas and human T-ALL/T-LL cancer cells. Cancer Lett. 2013, 336, 114–126. [Google Scholar] [CrossRef] [PubMed]
- Estañ, M.C.; Calviño, E.; de Blas, E.; Boyano-Adánez Mdel, C.; Mena, M.L.; Gómez-Gómez, M.; Rial, E.; Aller, P. 2-Deoxy-D-glucose cooperates with arsenic trioxide to induce apoptosis in leukemia cells: Involvement of IGF-1R-regulated Akt/mTOR, MEK/ERK and LKB-1/AMPK signaling pathways. Biochem. Pharmacol. 2012, 84, 1604–1616. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.H.; Pelicano, H.; Zhang, H.; Giles, F.J.; Keating, M.J.; Huang, P. Synergistic effect of targeting mTOR by rapamycin and depleting ATP by inhibition of glycolysis in lymphoma and leukemia cells. Leukemia 2005, 19, 2153–2158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akers, L.J.; Fang, W.; Levy, A.G.; Franklin, A.R.; Huang, P.; Zweidler-McKay, P.A. Targeting glycolysis in leukemia: A novel inhibitor 3-BrOP in combination with rapamycin. Leuk. Res. 2011, 35, 814–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schito, L.; Rey, S.; Konopleva, M. Integration of hypoxic HIF-α signaling in blood cancers. Oncogene 2017, 36, 5331–5340. [Google Scholar] [CrossRef] [PubMed]
- Janku, F.; Yap, T.A.; Meric-Bernstam, F. Targeting the PI3K pathway in cancer: Are we making headway? Nat. Rev. Clin. Oncol. 2018, 15, 273–291. [Google Scholar] [CrossRef] [PubMed]
- Frolova, O.; Samudio, I.; Benito, J.M.; Jacamo, R.; Kornblau, S.M.; Markovic, A.; Schober, W.; Lu, H.; Qiu, Y.H.; Buglio, D.; et al. Regulation of HIF-1α signaling and chemoresistance in acute lymphocytic leukemia under hypoxic conditions of the bone marrow microenvironment. Cancer Biol. Ther. 2012, 13, 858–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braun, M.; Qorraj, M.; Büttner, M.; Klein, F.A.; Saul, D.; Aigner, M.; Huber, W.; Mackensen, A.; Jitschin, R.; Mougiakakos, D. CXCL12 promotes glycolytic reprogramming in acute myeloid leukemia cells via the CXCR4/mTOR axis. Leukemia 2016, 30, 1788–1792. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Wu, L. mTOR up-regulation of PFKFB3 is essential for acute myeloid leukemia cell survival. Biochem. Biophys. Res. Commun. 2017, 483, 897–903. [Google Scholar] [CrossRef] [PubMed]
- Ros, S.; Schulze, A. Balancing glycolytic flux: The role of 6-phosphofructo-2-kinase/fructose 2,6-bisphosphatases in cancer metabolism. Cancer Metab. 2013, 1, 8. [Google Scholar] [CrossRef] [PubMed]
- Hay, N.; Sonenberg, N. Upstream and downstream of mTOR. Genes Dev. 2004, 18, 1926–1945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schieke, S.M.; Phillips, D.; McCoy, J.P., Jr.; Aponte, A.M.; Shen, R.F.; Balaban, R.S.; Finkel, T. The mammalian target of rapamycin (mTOR) pathway regulates mitochondrial oxygen consumption and oxidative capacity. J. Biol. Chem. 2006, 281, 27643–27652. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, A.; Schreiber, S.L. Direct control of mitochondrial function by mTOR. Proc. Natl. Acad. Sci. USA 2009, 106, 22229–22232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beesley, A.H.; Firth, M.J.; Ford, J.; Weller, R.E.; Freitas, J.R.; Perera, K.U.; Kees, U.R. Glucocorticoid resistance in T-lineage acute lymphoblastic leukaemia is associated with a proliferative metabolism. Br. J. Cancer 2009, 100, 1926–1936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, A.; Janocha, A.J.; Hill, B.T.; Smith, M.R.; Erzurum, S.C.; Almasan, A. Targeting mTORC1-mediated metabolic addiction overcomes fludarabine resistance in malignant B cells. Mol. Cancer Res. 2014, 12, 1205–1215. [Google Scholar] [CrossRef] [PubMed]
- Allegretti, M.; Ricciardi, M.R.; Licchetta, R.; Mirabilii, S.; Orecchioni, S.; Reggiani, F.; Talarico, G.; Foà, R.; Bertolini, F.; Amadori, S.; et al. The pan-class I phosphatidyl-inositol-3 kinase inhibitor NVP-BKM120 demonstrates anti-leukemic activity in acute myeloid leukemia. Sci. Rep. 2015, 5, 18137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nepstad, I.; Reikvam, H.; Brenner, A.K.; Bruserud, Ø.; Hatfield, K.J. Resistance to the antiproliferative in vitro effect of PI3K-Akt-mTOR inhibition in primary human acute myeloid leukemia cells is associated with altered cell metabolism. Int. J. Mol. Sci. 2018, 19, 382. [Google Scholar] [CrossRef] [PubMed]
- Asnaghi, L.; Calastretti, A.; Bevilacqua, A.; D’Agnano, I.; Gatti, G.; Canti, G.; Delia, D.; Capaccioli, S.; Nicolin, A. Bcl-2 phosphorylation and apoptosis activated by damaged microtubules require mTOR and are regulated by Akt. Oncogene 2004, 23, 5781–5791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, X.; Kinsella, T.J. Mammalian target of rapamycin and S6 kinase 1 positively regulate 6-thioguanine-induced autophagy. Cancer Res. 2008, 68, 2384–2390. [Google Scholar] [CrossRef] [PubMed]
- Castedo, M.; Ferri, K.F.; Kroemer, G. Mammalian target of rapamycin (mTOR): Pro- and anti-apoptotic. Cell Death Differ. 2002, 9, 99–100. [Google Scholar] [CrossRef] [PubMed]
- Hasty, P.; Sharp, Z.D.; Curiel, T.J.; Campisi, J. mTORC1 and p53: Clash of the gods? Cell Cycle 2013, 12, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Zhang, H.; Levine, A.J.; Jin, S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc. Natl. Acad. Sci. USA 2005, 102, 8204–8209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stambolic, V.; MacPherson, D.; Sas, D.; Lin, Y.; Snow, B.; Jang, Y.; Benchimol, S.; Mak, T.W. Regulation of PTEN transcription by p53. Mol. Cell 2001, 8, 317–325. [Google Scholar] [CrossRef]
- Horton, L.E.; Bushell, M.; Barth-Baus, D.; Tilleray, V.J.; Clemens, M.J.; Hensold, J.O. p53 activation results in rapid dephosphorylation of the eIF4E-binding protein 4E-BP1, inhibition of ribosomal protein S6 kinase and inhibition of translation initiation. Oncogene 2002, 21, 5325–5334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Z.; Hu, W.; de Stanchina, E.; Teresky, A.K.; Jin, S.; Lowe, S.; Levine, A.J. The regulation of AMPK beta1, TSC2, and PTEN expression by p53: Stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res. 2007, 67, 3043–3053. [Google Scholar] [CrossRef] [PubMed]
- Budanov, A.V.; Karin, M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell 2008, 134, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Oricchio, E.; Katanayeva, N.; Donaldson, M.C.; Sungalee, S.; Pasion, J.P.; Béguelin, W.; Battistello, E.; Sanghvi, V.R.; Jiang, M.; Jiang, Y.; et al. Genetic and epigenetic inactivation of SESTRIN1 controls mTORC1 and response to EZH2 inhibition in follicular lymphoma. Sci. Transl. Med. 2017, 9, eaak9969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, K.P.; Leong, W.F.; Chau, J.F.; Jia, D.; Zeng, L.; Liu, H.; He, L.; Hao, A.; Zhang, H.; Meek, D.; et al. S6K1 is a multifaceted regulator of Mdm2 that connects nutrient status and DNA damage response. EMBO J. 2010, 29, 2994–3006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Zhang, Y.; Liu, A.; Wang, J.; Li, L.; Chen, X.; Gao, X.; Xue, Y.; Zhang, X.; Liu, Y. Distinct dasatinib-induced mechanisms of apoptotic response and exosome release in imatinib-resistant human chronic myeloid leukemia cells. Int. J. Mol. Sci. 2016, 17, 531. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Simpson, S.E.; Scialla, T.J.; Bagg, A.; Carroll, M. Survival of acute myeloid leukemia cells requires PI3 kinase activation. Blood 2003, 102, 972–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kornblau, S.M.; Womble, M.; Qiu, Y.H.; Jackson, C.E.; Chen, W.; Konopleva, M.; Estey, E.H.; Andreeff, M. Simultaneous activation of multiple signal transduction pathways confers poor prognosis in acute myelogenous leukemia. Blood 2006, 108, 2358–2365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamburini, J.; Chapuis, N.; Bardet, V.; Park, S.; Sujobert, P.; Willems, L.; Ifrah, N.; Dreyfus, F.; Mayeux, P.; Lacombe, C.; et al. Mammalian target of rapamycin (mTOR) inhibition activates phosphatidylinositol 3-kinase/Akt by up-regulating insulin-like growth factor-1 receptor signaling in acute myeloid leukemia: Rationale for therapeutic inhibition of both pathways. Blood 2008, 111, 379–382. [Google Scholar] [CrossRef] [PubMed]
- Kojima, K.; Shimanuki, M.; Shikami, M.; Samudio, I.J.; Ruvolo, V.; Corn, P.; Hanaoka, N.; Konopleva, M.; Andreeff, M.; Nakakuma, H. The dual PI3 kinase/mTOR inhibitor PI-103 prevents p53 induction by Mdm2 inhibition but enhances p53-mediated mitochondrial apoptosis in p53 wild-type AML. Leukemia 2008, 22, 1728–1736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.; Li, Y.; Xiang, B.; Huang, X.O.; Ma, H.B.; Wang, F.F.; Gong, Y.P. Nutlin-3 plus tanshinone IIA exhibits synergetic anti-leukemia effect with imatinib by reactivating p53 and inhibiting the AKT/mTOR pathway in Ph+ ALL. Biochem. J. 2017, 474, 4153–4170. [Google Scholar] [CrossRef] [PubMed]
- Asnaghi, L.; Bruno, P.; Priulla, M.; Nicolin, A. mTOR: A protein kinase switching between life and death. Pharmacol. Res. 2004, 50, 545–549. [Google Scholar] [CrossRef] [PubMed]
- Rahmani, M.; Aust, M.M.; Attkisson, E.; Williams, D.C., Jr.; Ferreira-Gonzalez, A.; Grant, S. Dual inhibition of Bcl-2 and Bcl-xL strikingly enhances PI3K inhibition-induced apoptosis in human myeloid leukemia cells through a GSK3- and Bim-dependent mechanism. Cancer Res. 2013, 73, 1340–1351. [Google Scholar] [CrossRef] [PubMed]
- Wei, G.; Twomey, D.; Lamb, J.; Schlis, K.; Agarwal, J.; Stam, R.W.; Opferman, J.T.; Sallan, S.E.; den Boer, M.L.; Pieters, R.; et al. Gene expression-based chemical genomics identifies rapamycin as a modulator of MCL1 and glucocorticoid resistance. Cancer Cell 2006, 10, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Spender, L.C.; Inman, G.J. Phosphoinositide 3-kinase/AKT/mTORC1/2 signaling determines sensitivity of Burkitt's lymphoma cells to BH3 mimetics. Mol. Cancer Res. 2012, 10, 347–359. [Google Scholar] [CrossRef] [PubMed]
- Iacovelli, S.; Ricciardi, M.R.; Allegretti, M.; Mirabilii, S.; Licchetta, R.; Bergamo, P.; Rinaldo, C.; Zeuner, A.; Foà, R.; Milella, M.; et al. Co-targeting of Bcl-2 and mTOR pathway triggers synergistic apoptosis in BH3 mimetics resistant acute lymphoblastic leukemia. Oncotarget 2015, 6, 32089–32103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahmani, M.; Nkwocha, J.; Hawkins, E.; Pei, X.; Parker, R.E.; Kmieciak, M.; Leverson, J.D.; Sampath, D.; Ferreira-Gonzalez, A.; Grant, S. Cotargeting BCL-2 and PI3K Induces BAX-Dependent Mitochondrial Apoptosis in AML Cells. Cancer Res. 2018, 78, 3075–3086. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Liu, W.; Tsao, T.; Qiu, Y.; Zhao, Y.; Samudio, I.; Sarbassov, D.D.; Kornblau, S.M.; Baggerly, K.A.; Kantarjian, H.M.; et al. High-throughput profiling of signaling networks identifies mechanism-based combination therapy to eliminate microenvironmental resistance in acute myeloid leukemia. Haematologica 2017, 102, 1537–1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calastretti, A.; Bevilacqua, A.; Ceriani, C.; Viganò, S.; Zancai, P.; Capaccioli, S.; Nicolin, A. Damaged microtubules can inactivate BCL-2 by means of the mTOR kinase. Oncogene 2001, 20, 6172–6180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calastretti, A.; Rancati, F.; Ceriani, M.C.; Asnaghi, L.; Canti, G.; Nicolin, A. Rapamycin increases the cellular concentration of the BCL-2 protein and exerts an anti-apoptotic effect. Eur. J. Cancer 2001, 37, 2121–2128. [Google Scholar] [CrossRef]
- Hu, W.; Dooley, J.; Chung, S.S.; Chandramohan, D.; Cimmino, L.; Mukherjee, S.; Mason, C.E.; de Strooper, B.; Liston, A.; Park, C.Y. mir29a maintains mouse hematopoietic stem cell self-renewal by regulating Dnmt3a. Blood 2015, 125, 2206–2216. [Google Scholar] [CrossRef] [PubMed]
- Lechman, E.R.; Gentner, B.; van Galen, P.; Giustacchini, A.; Saini, M.; Boccalatte, F.E.; Hiramatsu, H.; Restuccia, U.; Bachi, A.; Voisin, V.; et al. Attenuation of miR-126 activity expands HSC in vivo without exhaustion. Cell Stem Cell 2012, 11, 799–811. [Google Scholar] [CrossRef] [PubMed]
- Metzeler, K.H.; Maharry, K.; Kohlschmidt, J.; Volinia, S.; Mrozek, K.; Becker, H.; Nicolet, D.; Whitman, S.P.; Mendler, J.H.; Schwind, S.; et al. A stem cell-like gene expression signature associates with inferior outcomes and a distinct microRNA expression profile in adults with primary cytogenetically normal acute myeloid leukemia. Leukemia 2013, 27, 2023–2031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lechman, E.R.; Gentner, B.; Ng, S.W.; Schoof, E.M.; van Galen, P.; Kennedy, J.A.; Nucera, S.; Ciceri, F.; Kaufmann, K.B.; Takayama, N.; et al. miR-126 Regulates Distinct Self-Renewal Outcomes in Normal and Malignant Hematopoietic Stem Cells. Cancer Cell 2016, 29, 214–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcais, A.; Blevins, R.; Graumann, J.; Feytout, A.; Dharmalingam, G.; Carroll, T.; Amado, I.F.; Bruno, L.; Lee, K.; Walzer, T.; et al. microRNA-mediated regulation of mTOR complex components facilitates discrimination between activation and anergy in CD4 T cells. J. Exp. Med. 2014, 211, 2281–2295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.J.; Luo, X.Q.; Han, B.W.; Duan, F.T.; Wei, P.P.; Chen, Y.Q. MicroRNA-100/99a, deregulated in acute lymphoblastic leukaemia, suppress proliferation and promote apoptosis by regulating the FKBP51 and IGF1R/mTOR signalling pathways. Br. J. Cancer 2013, 109, 2189–2198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palacios, F.; Abreu, C.; Prieto, D.; Morande, P.; Ruiz, S.; Fernández-Calero, T.; Naya, H.; Libisch, G.; Robello, C.; Landoni, A.I.; et al. Activation of the PI3K/AKT pathway by microRNA-22 results in CLL B-cell proliferation. Leukemia 2015, 29, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.; Yang, Y.; Chen, J.; Li, W.; Li, W.; Zhang, Q.; Mi, Y.; Goswami, R.S.; You, J.Q.; Lin, D.; et al. Regulation of PI3K signaling in T-cell acute lymphoblastic leukemia: A novel PTEN/Ikaros/miR-26b mechanism reveals a critical targetable role for PIK3CD. Leukemia 2017, 31, 2355–2364. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.H.; Wang, S.L.; Zhao, J.T.; Lin, Z.J.; Chen, L.Y.; Su, R.; Xie, S.T.; Carter, B.Z.; Xu, B. miR-150 exerts antileukemia activity in vitro and in vivo through regulating genes in multiple pathways. Cell Death Dis. 2016, 7, e2371. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Gao, L.; Luo, X.; Wang, L.; Gao, X.; Wang, W.; Sun, J.; Dou, L.; Li, J.; Xu, C.; et al. Epigenetic silencing of microRNA-193a contributes to leukemogenesis in t(8;21) acute myeloid leukemia by activating the PTEN/PI3K signal pathway. Blood 2013, 121, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Jia, C.Y.; Li, H.H.; Zhu, X.C.; Dong, Y.W.; Fu, D.; Zhao, Q.L.; Wu, W.; Wu, X.Z. MiR-223 suppresses cell proliferation by targeting IGF-1R. PLoS ONE 2011, 6, e27008. [Google Scholar] [CrossRef] [PubMed]
- Garzon, R.; Heaphy, C.E.; Havelange, V.; Fabbri, M.; Volinia, S.; Tsao, T.; Zanesi, N.; Kornblau, S.M.; Marcucci, G.; Calin, G.A.; et al. MicroRNA 29b functions in acute myeloid leukemia. Blood 2009, 114, 5331–5341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pekarsky, Y.; Santanam, U.; Cimmino, A.; Palamarchuk, A.; Efanov, A.; Maximov, V.; Volinia, S.; Alder, H.; Liu, C.G.; Rassenti, L.; et al. Tcl1 expression in chronic lymphocytic leukemia is regulated by miR-29 and miR-181. Cancer Res. 2006, 66, 11590–11593. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, E.I.; Dollins, C.M.; Crawford, M.; Grant, S.; Nana-Sinkam, S.P.; Richards, K.L.; Hammond, S.M.; Graves, L.M. Lyn kinase-dependent regulation of miR181 and myeloid cell leukemia-1 expression: Implications for drug resistance in myelogenous leukemia. Mol. Pharmacol. 2010, 78, 811–817. [Google Scholar] [CrossRef] [PubMed]
- Bertacchini, J.; Heidari, N.; Mediani, L.; Capitani, S.; Shahjahani, M.; Ahmadzadeh, A.; Saki, N. Targeting PI3K/AKT/mTOR network for treatment of leukemia. Cell Mol. Life Sci. 2015, 72, 2337–2347. [Google Scholar] [CrossRef] [PubMed]
- Vézina, C.; Kudelski, A.; Sehgal, S.N. Rapamycin (AY-22,989), a new antifungal antibiotic. I. Taxonomy of the producing streptomycete and isolation of the active principle. J. Antibiot. 1975, 28, 721–726. [Google Scholar] [CrossRef] [PubMed]
- Yatscoff, R.W.; LeGatt, D.F.; Kneteman, N.M. Therapeutic monitoring of rapamycin: A new immunosuppressive drug. Ther. Drug Monit. 1993, 15, 478–482. [Google Scholar] [CrossRef] [PubMed]
- Guertin, D.A.; Sabatini, D.M. Defining the role of mTOR in cancer. Cancer Cell 2007, 12, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Ballou, L.M.; Lin, R.Z. Rapamycin and mTOR kinase inhibitors. J. Chem. Biol. 2008, 1, 27–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feldman, M.E.; Apsel, B.; Uotila, A.; Loewith, R.; Knight, Z.A.; Ruggero, D.; Shokat, K.M. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol. 2009, 7, e1000038. [Google Scholar] [CrossRef] [PubMed]
- Calimeri, T.; Ferreri, A.J.M. m-TOR inhibitors and their potential role in haematological malignancies. Br. J. Haematol. 2017, 177, 684–702. [Google Scholar] [CrossRef] [PubMed]
- Naing, A.; Aghajanian, C.; Raymond, E.; Olmos, D.; Schwartz, G.; Oelmann, E.; Grinsted, L.; Burke, W.; Taylor, R.; Kaye, S.; et al. Safety, tolerability, pharmacokinetics and pharmacodynamics of AZD8055 in advanced solid tumours and lymphoma. Br. J. Cancer 2012, 107, 1093–1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bendell, J.C.; Kelley, R.K.; Shih, K.C.; Grabowsky, J.A.; Bergsland, E.; Jones, S.; Martin, T.; Infante, J.R.; Mischel, P.S.; Matsutani, T.; et al. A phase I dose-escalation study to assess safety, tolerability, pharmacokinetics, and preliminary efficacy of the dual mTORC1/mTORC2 kinase inhibitor CC-223 in patients with advanced solid tumors or multiple myeloma. Cancer 2015, 121, 3481–3490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghobrial, I.M.; Siegel, D.S.; Vij, R.; Berdeja, J.G.; Richardson, P.G.; Neuwirth, R.; Patel, C.G.; Zohren, F.; Wolf, J.L. TAK-228 (formerly MLN0128), an investigational oral dual TORC1/2 inhibitor: A phase I dose escalation study in patients with relapsed or refractory multiple myeloma, non-Hodgkin lymphoma, or Waldenström’s macroglobulinemia. Am. J. Hematol. 2016, 91, 400–405. [Google Scholar] [CrossRef] [PubMed]
- Wallin, J.J.; Edgar, K.A.; Guan, J.; Berry, M.; Prior, W.W.; Lee, L.; Lesnick, J.D.; Lewis, C.; Nonomiya, J.; Pang, J.; et al. GDC-0980 is a novel class I PI3K/mTOR kinase inhibitor with robust activity in cancer models driven by the PI3K pathway. Mol. Cancer Ther. 2011, 10, 2426–2436. [Google Scholar] [CrossRef] [PubMed]
- Wunderle, L.; Badura, S.; Lang, F.; Wolf, A.; Schleyer, E.; Serve, H.; Goekbuget, N.; Pfeifer, H.; Bug, G. Safety and efficacy of BEZ235, a dual PI3-Kinase/mTOR inhibitor, in adult patients with relapsed or refractory acute leukemia: Results of a phase I study. Blood 2013, 122, 2675. [Google Scholar]
- Min, Y.H.; Eom, J.I.; Cheong, J.W.; Maeng, H.O.; Kim, J.Y.; Jeung, H.K.; Lee, S.T.; Lee, M.H.; Hahn, J.S.; Ko, Y.W. Constitutive phosphorylation of Akt/PKB protein in acute myeloid leukemia: Its significance as a prognostic variable. Leukemia 2003, 17, 995–997. [Google Scholar] [CrossRef] [PubMed]
- Reikvam, H.; Tamburini, J.; Skrede, S.; Holdhus, R.; Poulain, L.; Ersvaer, E.; Hatfield, K.J.; Bruserud, Ø. Antileukaemic effect of PI3K-mTOR inhibitors in acute myeloid leukaemia-gene expression profiles reveal CDC25B expression as determinate of pharmacological effect. Br. J. Haematol. 2014, 164, 200–211. [Google Scholar] [CrossRef] [PubMed]
- Boutros, R.; Lobjois, V.; Ducommun, B. CDC25 phosphatases in cancer cells: Key players? Good targets? Nat. Rev. Cancer 2007, 7, 495–507. [Google Scholar] [CrossRef] [PubMed]
- Nepstad, I.; Hatfield, K.J.; Aasebø, E.; Hernandez-Valladares, M.; Brenner, A.K.; Bartaula-Brevik, S.; Berven, F.; Selheim, F.; Skavland, J.; Gjertsen, B.T.; et al. Two acute myeloid leukemia patient subsets are identified based on the constitutive PI3K-Akt-mTOR signaling of their leukemic cells; a functional, proteomic, and transcriptomic comparison. Expert Opin. Ther. Targets 2018, 22, 639–653. [Google Scholar] [CrossRef] [PubMed]
- Brenner, A.K.; Andersson Tvedt, T.H.; Bruserud, Ø. The Complexity of Targeting PI3K-Akt-mTOR Signalling in Human Acute Myeloid Leukaemia: The Importance of Leukemic Cell Heterogeneity, Neighbouring Mesenchymal Stem Cells and Immunocompetent Cells. Molecules 2016, 21, 1512. [Google Scholar] [CrossRef] [PubMed]
- Duarte, D.; Hawkins, E.D.; Lo Celso, C. The interplay of leukemia cells and the bone marrow microenvironment. Blood 2018, 131, 1507–1511. [Google Scholar] [CrossRef] [PubMed]
- Reikvam, H.; Nepstad, I.; Bruserud, Ø.; Hatfield, K.J. Pharmacological targeting of the PI3K/mTOR pathway alters the release of angioregulatory mediators both from primary human acute myeloid leukemia cells and their neighboring stromal cells. Oncotarget 2013, 4, 830–843. [Google Scholar] [CrossRef] [PubMed]
- Récher, C.; Beyne-Rauzy, O.; Demur, C.; Chicanne, G.; Dos Santos, C.; Mas, V.M.; Benzaquen, D.; Laurent, G.; Huguet, F.; Payrastre, B. Antileukemic activity of rapamycin in acute myeloid leukemia. Blood 2005, 105, 2527–2534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perl, A.E.; Kasner, M.T.; Tsai, D.E.; Vogl, D.T.; Loren, A.W.; Schuster, S.J.; Porter, D.L.; Stadtmauer, E.A.; Goldstein, S.C.; Frey, N.V.; et al. A phase I study of the mammalian target of rapamycin inhibitor sirolimus and MEC chemotherapy in relapsed and refractory acute myelogenous leukemia. Clin. Cancer Res. 2009, 15, 6732–6739. [Google Scholar] [CrossRef] [PubMed]
- Wei, A.H.; Sadawarte, S.; Catalano, J.; Hills, R.; Avery, S.; Patil, S.S.; Burnett, A.; Spencer, A. Phase Ib study combining the mTOR inhibitor everolimus (RAD001) with low-dose cytarabine in untreated elderly AML. Blood 2010, 116, 3299. [Google Scholar]
- Amadori, S.; Stasi, R.; Martelli, A.M.; Venditti, A.; Meloni, G.; Pane, F.; Martinelli, G.; Lunghi, M.; Pagano, L.; Cilloni, D.; et al. Temsirolimus, an mTOR inhibitor, in combination with lower-dose clofarabine as salvage therapy for older patients with acute myeloid leukaemia: Results of a phase II GIMEMA study (AML-1107). Br. J. Haematol. 2012, 156, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Scholl, S.; Bondeva, T.; Liu, Y.; Clement, J.H.; Höffken, K.; Wetzker, R. Additive effects of PI3-kinase and MAPK activities on NB4 cell granulocyte differentiation: Potential role of phosphatidylinositol 3-kinase gamma. J. Cancer Res. Clin. Oncol. 2008, 134, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Ozpolat, B.; Akar, U.; Steiner, M.; Zorrilla-Calancha, I.; Tirado-Gomez, M.; Colburn, N.; Danilenko, M.; Kornblau, S.; Berestein, G.L. Programmed cell death-4 tumor suppressor protein contributes to retinoic acid-induced terminal granulocytic differentiation of human myeloid leukemia cells. Mol. Cancer Res. 2007, 5, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Messina, M.; Chiaretti, S.; Wang, J.; Fedullo, A.L.; Peragine, N.; Gianfelici, V.; Piciocchi, A.; Brugnoletti, F.; Di Giacomo, F.; Pauselli, S.; et al. Prognostic and therapeutic role of targetable lesions in B-lineage acute lymphoblastic leukemia without recurrent fusion genes. Oncotarget 2016, 7, 13886–13901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, H.; Yang, X.; Liu, T.; Lin, J.; Chen, X.; Gong, Y. The study of resistant mechanisms and reversal in an imatinib resistant Ph+ acute lymphoblastic leukemia cell line. Leuk. Res. 2012, 36, 509–513. [Google Scholar] [CrossRef] [PubMed]
- Gianfelici, V.; Chiaretti, S.; Demeyer, S.; Di Giacomo, F.; Messina, M.; La Starza, R.; Peragine, N.; Paoloni, F.; Geerdens, E.; Pierini, V.; et al. RNA sequencing unravels the genetics of refractory/relapsed T-cell acute lymphoblastic leukemia. Prognostic and therapeutic implications. Haematologica 2016, 101, 941–950. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Ryu, Y.K.; Chen, T.Z.; Hall, C.P.; Webster, D.R.; Kang, M.H. Synergistic activity of rapamycin and dexamethasone in vitro and in vivo in acute lymphoblastic leukemia via cell-cycle arrest and apoptosis. Leuk. Res. 2012, 36, 342–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bressanin, D.; Evangelisti, C.; Ricci, F.; Tabellini, G.; Chiarini, F.; Tazzari, P.L.; Melchionda, F.; Buontempo, F.; Pagliaro, P.; Pession, A.; et al. Harnessing the PI3K/Akt/mTOR pathway in T-cell acute lymphoblastic leukemia: Eliminating activity by targeting at different levels. Oncotarget 2012, 3, 811–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palomero, T.; Sulis, M.L.; Cortina, M.; Real, P.J.; Barnes, K.; Ciofani, M.; Caparros, E.; Buteau, J.; Brown, K.; Perkins, S.L.; et al. Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nat. Med. 2007, 13, 1203–1210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Airiau, K.; Mahon, F.X.; Josselin, M.; Jeanneteau, M.; Belloc, F. PI3K/mTOR pathway inhibitors sensitize chronic myeloid leukemia stem cells to nilotinib and restore the response of progenitors to nilotinib in the presence of stem cell factor. Cell Death Dis. 2013, 4, e827. [Google Scholar] [CrossRef] [PubMed]
- Pellicano, F.; Scott, M.T.; Helgason, G.V.; Hopcroft, L.E.; Allan, E.K.; Aspinall-O’Dea, M.; Copland, M.; Pierce, A.; Huntly, B.J.; Whetton, A.D.; et al. The antiproliferative activity of kinase inhibitors in chronic myeloid leukemia cells is mediated by FOXO transcription factors. Stem Cells 2014, 32, 2324–2337. [Google Scholar] [CrossRef] [PubMed]
- Banerjee Mustafi, S.; Chakraborty, P.K.; Raha, S. Modulation of Akt and ERK1/2 pathways by resveratrol in chronic myelogenous leukemia (CML) cells results in the downregulation of Hsp70. PLoS ONE 2010, 5, e8719. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.F.; Zhang, L.; Wu, D.S.; Yuan, X.Y.; Yu, Y.H.; Zhao, X.L.; Chen, F.P.; Zeng, H. PTEN regulates BCRP/ABCG2 and the side population through the PI3K/Akt pathway in chronic myeloid leukemia. PLoS ONE 2014, 9, e88298. [Google Scholar] [CrossRef] [PubMed]
- Boddu, P.; Ferrajoli, A. Prognostic Factors in the Era of Targeted Therapies in CLL. Curr. Hematol. Malig. Rep. 2018, 13, 78–90. [Google Scholar] [CrossRef] [PubMed]
- Kipps, T.J.; Stevenson, F.K.; Wu, C.J.; Croce, C.M.; Packham, G.; Wierda, W.G.; O’Brien, S.; Gribben, J.; Rai, K. Chronic lymphocytic leukaemia. Nat. Rev. Dis. Primers 2017, 3, 16096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thijssen, R.; Ter Burg, J.; Garrick, B.; van Bochove, G.G.; Brown, J.R.; Fernandes, S.M.; Rodríguez, M.S.; Michot, J.M.; Hallek, M.; Eichhorst, B.; et al. Dual TORK/DNA-PK inhibition blocks critical signaling pathways in chronic lymphocytic leukemia. Blood 2016, 128, 574–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
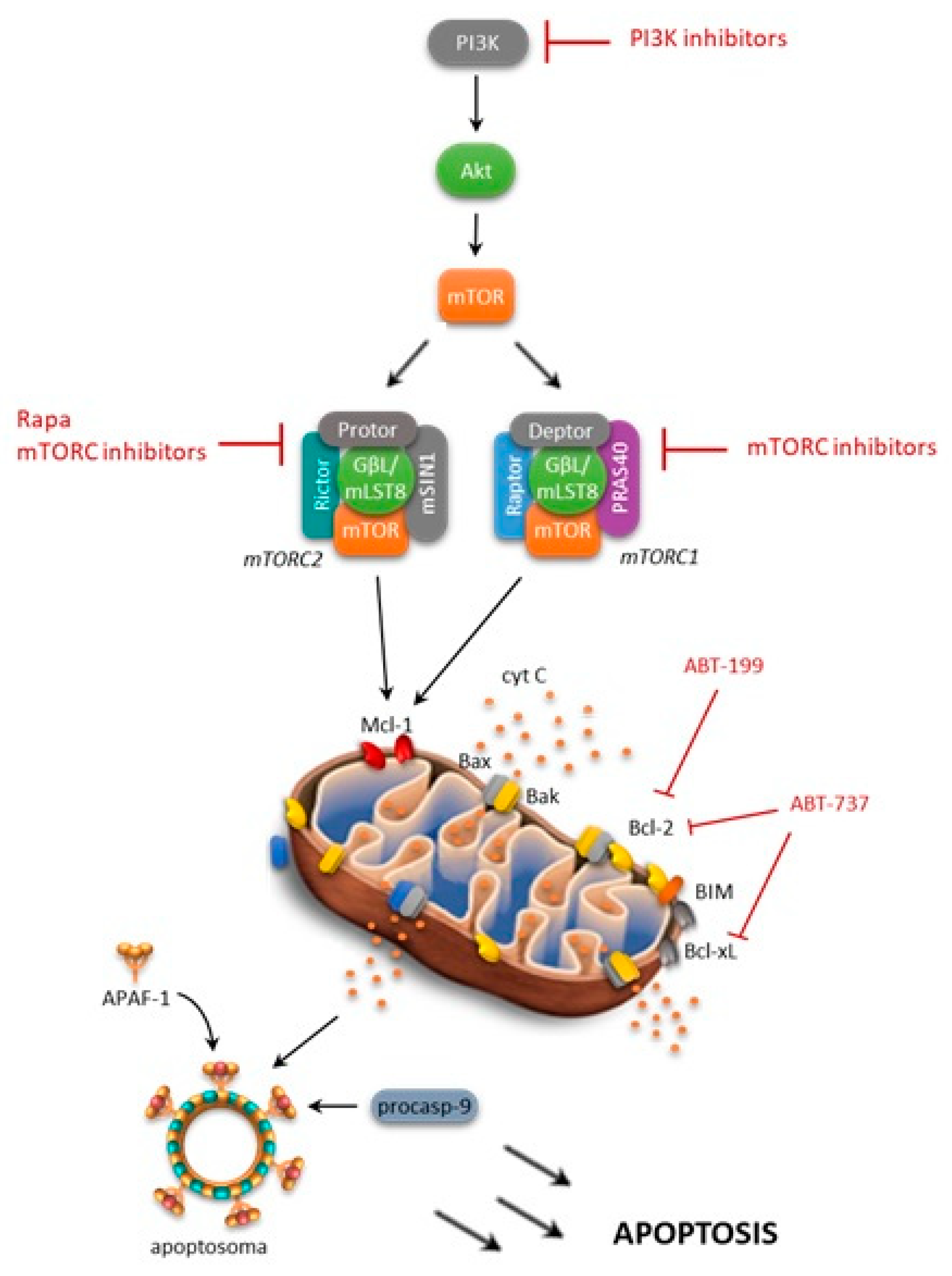{kind=link}
| Inhibitors | Disease | Response | Reference |
|---|---|---|---|
| Rapamycin monotherapy | R/R AML | PR | Recher, et al. [102] |
| Rapamycin + MEC | R/R or untreated secondary AML | CR/PR | Perl, et al. [103] |
| Everolimus + LDAC | Naïve elderly AML | CR/Cri/PR | Wei A.H., et al. [104] |
| Temsirolimus + LDClof | R/R AML | CR/Cri | Amadori, et al. [105] |
| ATRA + LY294002 and PD98059 | APL | Increase granulocyte differentiation | Scholl S., et al. [106] |
| NVPBEZ235 + nilotinib | TKI-resistant BCR-ABL | Increase apoptosis | Airiau K., et al. [114] |
| Rapamycin + dasatinib | CML | Increase apoptosis | Pellicano F., et al. [115] |
| Resveratrol | CML | Inhibits Akt | Banerjee M. S., et al. [116] |
| Inhibitors | Disease | RESPONSE | Reference |
|---|---|---|---|
| CC-115 | R/R CLL | PR | Thijssen, et al. [120] |
| Rapamycin + NVP-BEZ235 | ALL | Increase apoptosis | Messina, et al. [108] |
| RAD001, Torin-2 and CCI-779 | ALL | Increase apoptosis | Bertacchini, et al. [83] |
| Imatinib + mTOR inhibitor | Imatinib-resistant Ph + ALL | Increase apoptosis | Xing H., et al. [109] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mirabilii, S.; Ricciardi, M.R.; Piedimonte, M.; Gianfelici, V.; Bianchi, M.P.; Tafuri, A. Biological Aspects of mTOR in Leukemia. Int. J. Mol. Sci. 2018, 19, 2396. https://doi.org/10.3390/ijms19082396
Mirabilii S, Ricciardi MR, Piedimonte M, Gianfelici V, Bianchi MP, Tafuri A. Biological Aspects of mTOR in Leukemia. International Journal of Molecular Sciences. 2018; 19(8):2396. https://doi.org/10.3390/ijms19082396
Chicago/Turabian StyleMirabilii, Simone, Maria Rosaria Ricciardi, Monica Piedimonte, Valentina Gianfelici, Maria Paola Bianchi, and Agostino Tafuri. 2018. "Biological Aspects of mTOR in Leukemia" International Journal of Molecular Sciences 19, no. 8: 2396. https://doi.org/10.3390/ijms19082396
APA StyleMirabilii, S., Ricciardi, M. R., Piedimonte, M., Gianfelici, V., Bianchi, M. P., & Tafuri, A. (2018). Biological Aspects of mTOR in Leukemia. International Journal of Molecular Sciences, 19(8), 2396. https://doi.org/10.3390/ijms19082396