Role of mTOR Signaling in Tumor Microenvironment: An Overview

 , , ,
, , ,
Abstract
:
1. Introduction
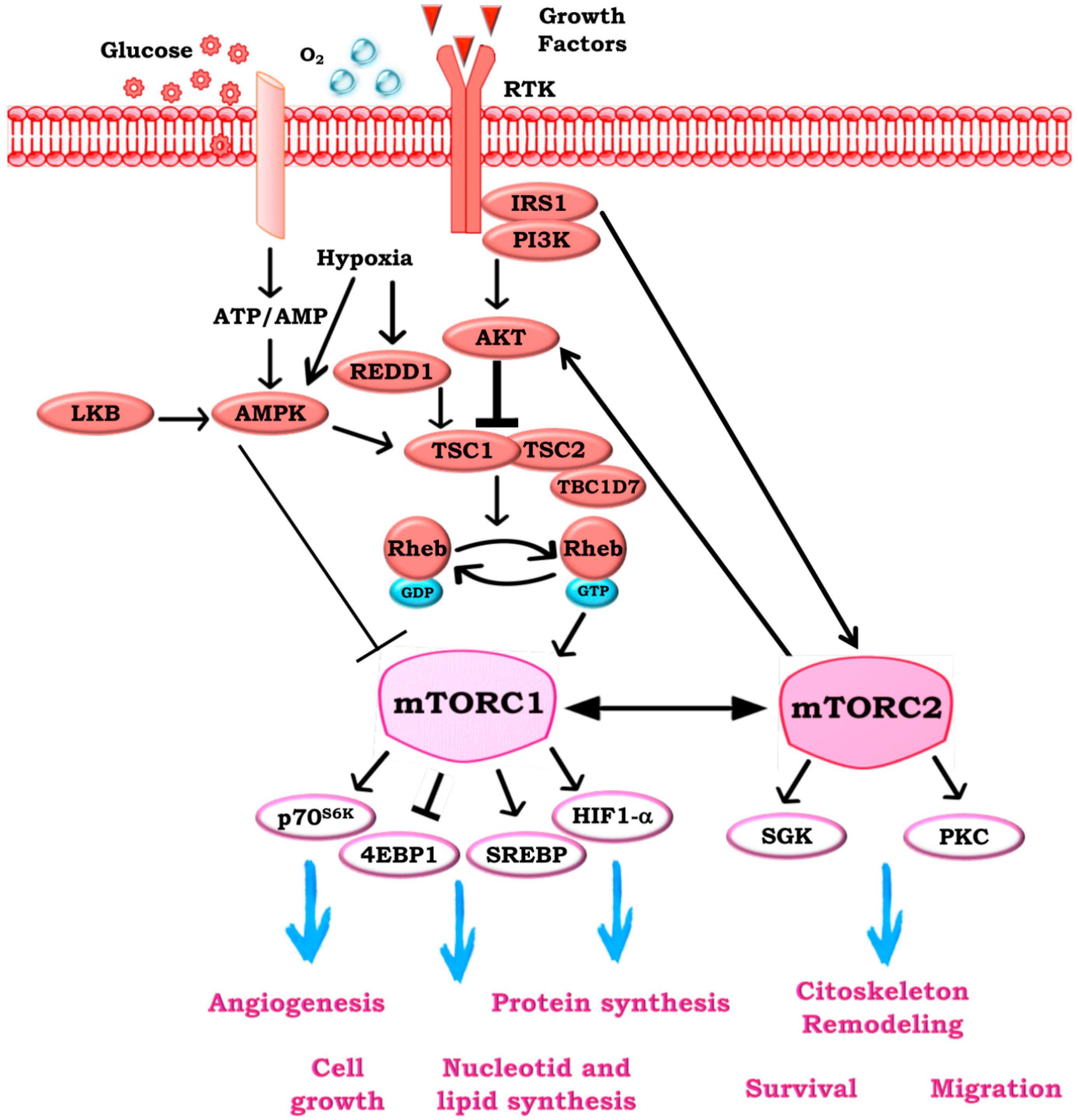
2. mTOR Signaling
3. Tumor Microenvironment
4. Immunoregulatory Functions of mTOR
4.1. T Lymphocytes
4.1.1. CD8+
4.1.2. CD4+
4.1.3. Treg
4.2. TAMs
4.3. MDSCs
5. mTOR and Angiogenesis
6. mTOR in CAFs Regulation
7. Implications in Cancer Immunotherapy
8. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AMPK-β | 5′-AMP activated protein kinase β |
| CAF | cancer-associated fibroblasts |
| CTLA-4 | cytotoxic t-lymphocyte protein 4 |
| DC | dendridic cell |
| DEPTOR | DEP domain-containing mTOR interacting protein |
| ECM | extracellular matrix |
| EGFR | epidermal growth factor receptor |
| FOXO | forkhead box |
| G-CSF | granulocyte colony-stimulating factor |
| HIF | hypoxia-inducible factor |
| Hsp70 | heat shock protein 70 |
| IGF | insulin growth factor |
| IL | interleukin |
| JAK | Janus kinase |
| MDSC | myeloid-derived suppressor cells |
| mLST8 | mammalian lethal with sec13 protein 8 |
| MMP | metalloproteases |
| mSIN1 | mammalian stress-activated protein kinase interacting protein 1 |
| mTOR | mammalian target of rapamycin |
| mTORC | mTOR complex |
| PD-1 | programmed death 1 |
| PDAC | pancreatic ductal adenocarcinoma |
| PDGF | plated-derived growth factor |
| PDK1 | phosphoinositide-dependent kinase-1 |
| PIP2 | phosphatidylinositol 4,5-bisphosphate |
| PIP3 | phosphatidylinositol 3,4,5-trisphosphate |
| PI3K | phosphoinositide 3-kinase |
| PKC | protein kinase C |
| PP2 | protein phosphatase 2 |
| PRAS40 | proline-rich AKT substrate 40 |
| PRR | proline-rich protein |
| Protor | protein observed with rictor |
| PTEN | phosphatase and tensin homolog on chromosome 10 |
| p70S6K1 | p70 ribosomal protein S6 kinase 1 |
| ROS | reactive oxygen species |
| SGK | serum and glucocorticoid kinase |
| SREBP | sterol regulatory element binding proteins |
| STAT | signal transducer and activator of transcription |
| TAM | tumor-associated macrophages |
| TCR | T cell receptor |
| Tel2 | telomere maintenance 2 |
| TGF | trasforming growth factor |
| Th | T helper |
| TLR | tool-like receptor |
| TME | tumor microenvironment |
| Treg | regulatory T cells |
| TSC | tuberous sclerosis complexes |
| TSI | tumor-stroma interactions |
| Tti1 | tel2-interacting protein 1 |
| VEGF | vascular endothelial growth factor |
| VEGFR | VEGF receptor |
| 4EBP-1 | 4E binding protein-1 |
References
- Conciatori, F.; Ciuffreda, L.; Bazzichetto, C.; Falcone, I.; Pilotto, S.; Bria, E.; Cognetti, F.; Milella, M. mTOR Cross-Talk in Cancer and Potential for Combination Therapy. Cancers (Basel) 2018, 10, 23. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 169, 361–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, L.C.; Cook, R.S.; Chen, J. mTORC1 and mTORC2 in cancer and the tumor microenvironment. Oncogene 2017, 36, 2191–2201. [Google Scholar] [CrossRef] [PubMed]
- Schaaf, M.B.; Garg, A.D.; Agostinis, P. Defining the role of the tumor vasculature in antitumor immunity and immunotherapy. Cell Death Dis. 2018, 9, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karar, J.; Maity, A. PI3K/AKT/mTOR Pathway in Angiogenesis. Front. Mol. Neurosci. 2011, 4, 51. [Google Scholar] [CrossRef] [PubMed]
- Laughner, E.; Taghavi, P.; Chiles, K.; Mahon, P.C.; Semenza, G.L. HER2 (neu) signaling increases the rate of hypoxia-inducible factor 1alpha (HIF-1alpha) synthesis: Novel mechanism for HIF-1-mediated vascular endothelial growth factor expression. Mol. Cell Biol. 2001, 21, 3995–4004. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.J.; Albers, M.W.; Shin, T.B.; Ichikawa, K.; Keith, C.T.; Lane, W.S.; Schreiber, S.L. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature 1994, 369, 756–758. [Google Scholar] [CrossRef] [PubMed]
- Chiu, M.I.; Katz, H.; Berlin, V. RAPT1, a mammalian homolog of yeast Tor, interacts with the FKBP12/rapamycin complex. Proc. Natl. Acad. Sci. USA 1994, 91, 12574–12578. [Google Scholar] [CrossRef] [PubMed]
- Jacinto, E.; Loewith, R.; Schmidt, A.; Lin, S.; Ruegg, M.A.; Hall, A.; Hall, M.N. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat. Cell Biol. 2004, 6, 1122–1128. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Ali, S.M.; Kim, D.H.; Guertin, D.A.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 2004, 14, 1296–1302. [Google Scholar] [CrossRef] [PubMed]
- Thomanetz, V.; Angliker, N.; Cloetta, D.; Lustenberger, R.M.; Schweighauser, M.; Oliveri, F.; Suzuki, N.; Ruegg, M.A. Ablation of the mTORC2 component rictor in brain or Purkinje cells affects size and neuron morphology. J. Cell Biol. 2013, 201, 293–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Bajraszewski, N.; Wu, E.; Wang, H.; Moseman, A.P.; Dabora, S.L.; Griffin, J.D.; Kwiatkowski, D.J. PDGFRs are critical for PI3K/Akt activation and negatively regulated by mTOR. J. Clin. Investig. 2007, 117, 730–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willems, L.; Tamburini, J.; Chapuis, N.; Lacombe, C.; Mayeux, P.; Bouscary, D. PI3K and mTOR signaling pathways in cancer: New data on targeted therapies. Curr. Oncol. Rep. 2012, 14, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [PubMed]
- Milella, M.; Falcone, I.; Conciatori, F.; Cesta Incani, U.; Del Curatolo, A.; Inzerilli, N.; Nuzzo, C.M.; Vaccaro, V.; Vari, S.; Cognetti, F.; et al. PTEN: Multiple Functions in Human Malignant Tumors. Front. Oncol. 2015, 5, 24. [Google Scholar] [CrossRef] [PubMed]
- Ciuffreda, L.; McCubrey, J.A.; Milella, M. Signaling intermediates (PI3K/PTEN/AKT/mTOR and RAF/MEK/ERK pathways) as therapeutic targets for anti-cancer and anti-angiogenesis treatments. Curr. Signal. Transd. Ther. 2009, 4, 130–143. [Google Scholar] [CrossRef]
- Xue, G.; Zippelius, A.; Wicki, A.; Mandala, M.; Tang, F.; Massi, D.; Hemmings, B.A. Integrated Akt/PKB signaling in immunomodulation and its potential role in cancer immunotherapy. J. Natl. Cancer Inst. 2015, 107, 7. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhao, J.; Zhang, L.; Wei, F.; Lian, Y.; Wu, Y.; Gong, Z.; Zhang, S.; Zhou, J.; Cao, K.; et al. Role of tumor microenvironment in tumorigenesis. J. Cancer 2017, 8, 761–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenny, P.A.; Lee, G.Y.; Bissell, M.J. Targeting the tumor microenvironment. Front. Biosci. 2007, 12, 3468–3474. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The tumor microenvironment at a glance. J. Cell Sci. 2012, 125, 5591–5596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Zhuang, X.; Lin, L.; Yu, P.; Wang, Y.; Shi, Y.; Hu, G.; Sun, Y. New horizons in tumor microenvironment biology: Challenges and opportunities. BMC Med. 2015, 13, 45. [Google Scholar] [CrossRef] [PubMed]
- Zumsteg, A.; Christofori, G. Corrupt policemen: Inflammatory cells promote tumor angiogenesis. Curr. Opin. Oncol. 2009, 21, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, C.; Giannoudis, A.; Lewis, C.E. Mechanisms regulating the recruitment of macrophages into hypoxic areas of tumors and other ischemic tissues. Blood 2004, 104, 2224–2234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrence, T.; Natoli, G. Transcriptional regulation of macrophage polarization: Enabling diversity with identity. Nat. Rev. Immunol. 2011, 11, 750–761. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, B.; Elkord, E. Regulatory T Cells in the Tumor Microenvironment and Cancer Progression: Role and Therapeutic Targeting. Vaccines (Basel) 2016, 4, 28. [Google Scholar] [CrossRef] [PubMed]
- De Palma, M.; Biziato, D.; Petrova, T.V. Microenvironmental regulation of tumour angiogenesis. Nat. Rev. Cancer 2017, 17, 457–474. [Google Scholar] [CrossRef] [PubMed]
- Gattazzo, F.; Urciuolo, A.; Bonaldo, P. Extracellular matrix: A dynamic microenvironment for stem cell niche. Biochim. Biophys. Acta 2014, 1840, 2506–2519. [Google Scholar] [CrossRef] [PubMed]
- Popovic, Z.V.; Sandhoff, R.; Sijmonsma, T.P.; Kaden, S.; Jennemann, R.; Kiss, E.; Tone, E.; Autschbach, F.; Platt, N.; Malle, E.; et al. Sulfated glycosphingolipid as mediator of phagocytosis: SM4s enhances apoptotic cell clearance and modulates macrophage activity. J. Immunol. 2007, 179, 6770–6782. [Google Scholar] [CrossRef] [PubMed]
- Nagarsheth, N.; Wicha, M.S.; Zou, W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat. Rev. Immunol. 2017, 17, 559–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landskron, G.; De la Fuente, M.; Thuwajit, P.; Thuwajit, C.; Hermoso, M.A. Chronic inflammation and cytokines in the tumor microenvironment. J. Immunol. Res. 2014, 2014, 149185. [Google Scholar] [CrossRef] [PubMed]
- Zlotnik, A.; Yoshie, O. Chemokines: A new classification system and their role in immunity. Immunity 2000, 12, 121–127. [Google Scholar] [CrossRef]
- Del Bufalo, D.; Ciuffreda, L.; Trisciuoglio, D.; Desideri, M.; Cognetti, F.; Zupi, G.; Milella, M. Antiangiogenic potential of the Mammalian target of rapamycin inhibitor temsirolimus. Cancer Res. 2006, 66, 5549–5554. [Google Scholar] [CrossRef] [PubMed]
- Milella, M.; Falcone, I.; Conciatori, F.; Matteoni, S.; Sacconi, A.; De Luca, T.; Bazzichetto, C.; Corbo, V.; Simbolo, M.; Sperduti, I.; et al. PTEN status is a crucial determinant of the functional outcome of combined MEK and mTOR inhibition in cancer. Sci. Rep. 2017, 7, 43013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De la Iglesia, N.; Konopka, G.; Lim, K.L.; Nutt, C.L.; Bromberg, J.F.; Frank, D.A.; Mischel, P.S.; Louis, D.N.; Bonni, A. Deregulation of a STAT3-interleukin 8 signaling pathway promotes human glioblastoma cell proliferation and invasiveness. J. Neurosci. 2008, 28, 5870–5878. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, P.J.; Coulter, J.; Walker, S.M.; McKechnie, M.; Neisen, J.; McCabe, N.; Kennedy, R.D.; Salto-Tellez, M.; Albanese, C.; Waugh, D.J. Potentiation of inflammatory CXCL8 signalling sustains cell survival in PTEN-deficient prostate carcinoma. Eur. Urol. 2013, 64, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Weichhart, T.; Hengstschlager, M.; Linke, M. Regulation of innate immune cell function by mTOR. Nat. Rev. Immunol. 2015, 15, 599–614. [Google Scholar] [CrossRef] [PubMed]
- Thomson, A.W.; Turnquist, H.R.; Raimondi, G. Immunoregulatory functions of mTOR inhibition. Nat. Rev. Immunol. 2009, 9, 324–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollizzi, K.N.; Patel, C.H.; Sun, I.H.; Oh, M.H.; Waickman, A.T.; Wen, J.; Delgoffe, G.M.; Powell, J.D. mTORC1 and mTORC2 selectively regulate CD8(+) T cell differentiation. J. Clin. Investig. 2015, 125, 2090–2108. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H. mTOR signaling in immune cells and its implications for cancer immunotherapy. Cancer Lett. 2017, 408, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Delgoffe, G.M.; Kole, T.P.; Zheng, Y.; Zarek, P.E.; Matthews, K.L.; Xiao, B.; Worley, P.F.; Kozma, S.C.; Powell, J.D. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity 2009, 30, 832–844. [Google Scholar] [CrossRef] [PubMed]
- Delgoffe, G.M.; Pollizzi, K.N.; Waickman, A.T.; Heikamp, E.; Meyers, D.J.; Horton, M.R.; Xiao, B.; Worley, P.F.; Powell, J.D. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat. Immunol. 2011, 12, 295–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.; Gudapati, P.; Dragovic, S.; Spencer, C.; Joyce, S.; Killeen, N.; Magnuson, M.A.; Boothby, M. Mammalian target of rapamycin protein complex 2 regulates differentiation of Th1 and Th2 cell subsets via distinct signaling pathways. Immunity 2010, 32, 743–753. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.; Jin, H.S.; Lopez, J.; Elly, C.; Kim, G.; Murai, M.; Kronenberg, M.; Liu, Y.C. TSC1 regulates the balance between effector and regulatory T cells. J. Clin. Investig. 2013, 123, 5165–5178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huynh, A.; DuPage, M.; Priyadharshini, B.; Sage, P.T.; Quiros, J.; Borges, C.M.; Townamchai, N.; Gerriets, V.A.; Rathmell, J.C.; Sharpe, A.H.; et al. Control of PI(3) kinase in Treg cells maintains homeostasis and lineage stability. Nat. Immunol. 2015, 16, 188–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shrestha, S.; Yang, K.; Guy, C.; Vogel, P.; Neale, G.; Chi, H. Treg cells require the phosphatase PTEN to restrain TH1 and TFH cell responses. Nat. Immunol. 2015, 16, 178–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byles, V.; Covarrubias, A.J.; Ben-Sahra, I.; Lamming, D.W.; Sabatini, D.M.; Manning, B.D.; Horng, T. The TSC-mTOR pathway regulates macrophage polarization. Nat. Commun. 2013, 4, 2834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, H.; Westerterp, M.; Wang, C.; Zhu, Y.; Ai, D. Macrophage mTORC1 disruption reduces inflammation and insulin resistance in obese mice. Diabetologia 2014, 57, 2393–2404. [Google Scholar] [CrossRef] [PubMed]
- Guri, Y.; Nordmann, T.M.; Roszik, J. mTOR at the Transmitting and Receiving Ends in Tumor Immunity. Front. Immunol. 2018, 9, 578. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, L.; Chen, X.; Li, L.; Li, Y.; Ping, Y.; Huang, L.; Yue, D.; Zhang, Z.; Wang, F.; et al. CD39/CD73 upregulation on myeloid-derived suppressor cells via TGF-beta-mTOR-HIF-1 signaling in patients with non-small cell lung cancer. Oncoimmunology 2017, 6, e1320011. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wang, S.; Li, J.; Zhang, W.; Zheng, L.; Yang, C.; Zhu, T.; Rong, R. The mTOR signal regulates myeloid-derived suppressor cells differentiation and immunosuppressive function in acute kidney injury. Cell Death Dis. 2017, 8, e2695. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Chen, S.; Liu, F.; Wu, H.; McHugh, J.; Bergin, I.L.; Gupta, A.; Adams, D.; Guan, J.L. Constitutive Activation of mTORC1 in Endothelial Cells Leads to the Development and Progression of Lymphangiosarcoma through VEGF Autocrine Signaling. Cancer Cell 2015, 28, 758–772. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Amato, K.R.; Song, W.; Youngblood, V.; Lee, K.; Boothby, M.; Brantley-Sieders, D.M.; Chen, J. Regulation of endothelial cell proliferation and vascular assembly through distinct mTORC2 signaling pathways. Mol. Cell. Biol. 2015, 35, 1299–1313. [Google Scholar] [CrossRef] [PubMed]
- Duluc, C.; Moatassim-Billah, S.; Chalabi-Dchar, M.; Perraud, A.; Samain, R.; Breibach, F.; Gayral, M.; Cordelier, P.; Delisle, M.B.; Bousquet-Dubouch, M.P.; et al. Pharmacological targeting of the protein synthesis mTOR/4E-BP1 pathway in cancer-associated fibroblasts abrogates pancreatic tumour chemoresistance. EMBO Mol. Med. 2015, 7, 735–753. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Abu-Eid, R.; Shrimali, R.; Webb, M.; Verma, V.; Doroodchi, A.; Berrong, Z.; Samara, R.; Rodriguez, P.C.; Mkrtichyan, M.; et al. Differential PI3Kdelta Signaling in CD4(+) T-cell Subsets Enables Selective Targeting of T Regulatory Cells to Enhance Cancer Immunotherapy. Cancer Res. 2017, 77, 1892–1904. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Richards, J.A.; Gupta, R.; Aung, P.P.; Emley, A.; Kluger, Y.; Dogra, S.K.; Mahalingam, M.; Wajapeyee, N. PTEN functions as a melanoma tumor suppressor by promoting host immune response. Oncogene 2014, 33, 4632–4642. [Google Scholar] [CrossRef] [PubMed]
- Templeton, A.J.; Dutoit, V.; Cathomas, R.; Rothermundt, C.; Bartschi, D.; Droge, C.; Gautschi, O.; Borner, M.; Fechter, E.; Stenner, F.; et al. Swiss Group for Clinical Cancer, R., Phase 2 trial of single-agent everolimus in chemotherapy-naive patients with castration-resistant prostate cancer (SAKK 08/08). Eur. Urol. 2013, 64, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Abu-Eid, R.; Samara, R.N.; Ozbun, L.; Abdalla, M.Y.; Berzofsky, J.A.; Friedman, K.M.; Mkrtichyan, M.; Khleif, S.N. Selective inhibition of regulatory T cells by targeting the PI3K-Akt pathway. Cancer Immunol. Res. 2014, 2, 1080–1089. [Google Scholar] [CrossRef] [PubMed]
- Schmid, M.C.; Avraamides, C.J.; Dippold, H.C.; Franco, I.; Foubert, P.; Ellies, L.G.; Acevedo, L.M.; Manglicmot, J.R.; Song, X.; Wrasidlo, W.; et al. Receptor tyrosine kinases and TLR/IL1Rs unexpectedly activate myeloid cell PI3kgamma, a single convergent point promoting tumor inflammation and progression. Cancer Cell 2011, 19, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Vinals, F.; Chambard, J.C.; Pouyssegur, J. p70 S6 kinase-mediated protein synthesis is a critical step for vascular endothelial cell proliferation. J. Biol. Chem. 1999, 274, 26776–26782. [Google Scholar] [CrossRef] [PubMed]
- Dormond, O.; Madsen, J.C.; Briscoe, D.M. The effects of mTOR-Akt interactions on anti-apoptotic signaling in vascular endothelial cells. J. Biol. Chem. 2007, 282, 23679–23686. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, E.T.; Cao, C.; Niermann, K.; Mu, Y.; Zeng, F.; Hallahan, D.E.; Lu, B. Enhanced radiation damage of tumor vasculature by mTOR inhibitors. Oncogene 2005, 24, 5414–5422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heits, N.; Heinze, T.; Bernsmeier, A.; Kerber, J.; Hauser, C.; Becker, T.; Kalthoff, H.; Egberts, J.H.; Braun, F. Influence of mTOR-inhibitors and mycophenolic acid on human cholangiocellular carcinoma and cancer associated fibroblasts. BMC Cancer 2016, 16, 322. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Chi, H. mTOR and lymphocyte metabolism. Curr. Opin. Immunol. 2013, 25, 347–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deberardinis, R.J.; Sayed, N.; Ditsworth, D.; Thompson, C.B. Brick by brick: Metabolism and tumor cell growth. Curr. Opin. Genet. Dev. 2008, 18, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Araki, K.; Ahmed, R. AMPK: A metabolic switch for CD8+ T-cell memory. Eur. J. Immunol. 2013, 43, 878–881. [Google Scholar] [CrossRef] [PubMed]
- Araki, K.; Turner, A.P.; Shaffer, V.O.; Gangappa, S.; Keller, S.A.; Bachmann, M.F.; Larsen, C.P.; Ahmed, R. mTOR regulates memory CD8 T-cell differentiation. Nature 2009, 460, 108–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kidani, Y.; Elsaesser, H.; Hock, M.B.; Vergnes, L.; Williams, K.J.; Argus, J.P.; Marbois, B.N.; Komisopoulou, E.; Wilson, E.B.; Osborne, T.F.; et al. Sterol regulatory element-binding proteins are essential for the metabolic programming of effector T cells and adaptive immunity. Nat. Immunol. 2013, 14, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Kurebayashi, Y.; Nagai, S.; Ikejiri, A.; Ohtani, M.; Ichiyama, K.; Baba, Y.; Yamada, T.; Egami, S.; Hoshii, T.; Hirao, A.; et al. PI3K-Akt-mTORC1-S6K1/2 axis controls Th17 differentiation by regulating Gfi1 expression and nuclear translocation of RORgamma. Cell Rep. 2012, 1, 360–373. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Chi, H. mTOR and metabolic pathways in T cell quiescence and functional activation. Semin. Immunol. 2012, 24, 421–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; Dillon, C.P.; Shi, L.Z.; Milasta, S.; Carter, R.; Finkelstein, D.; McCormick, L.L.; Fitzgerald, P.; Chi, H.; Munger, J.; et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 2011, 35, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.Z.; Wang, R.; Huang, G.; Vogel, P.; Neale, G.; Green, D.R.; Chi, H. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J. Exp. Med. 2011, 208, 1367–1376. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, M.; Stabilini, A.; Migliavacca, B.; Horejs-Hoeck, J.; Kaupper, T.; Roncarolo, M.G. Rapamycin promotes expansion of functional CD4+CD25+FOXP3+ regulatory T cells of both healthy subjects and type 1 diabetic patients. J. Immunol. 2006, 177, 8338–8347. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Yang, K.; Cloer, C.; Neale, G.; Vogel, P.; Chi, H. mTORC1 couples immune signals and metabolic programming to establish T(reg)-cell function. Nature 2013, 499, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Francisco, L.M.; Salinas, V.H.; Brown, K.E.; Vanguri, V.K.; Freeman, G.J.; Kuchroo, V.K.; Sharpe, A.H. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J. Exp. Med. 2009, 206, 3015–3029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef] [PubMed]
- Lastwika, K.J.; Wilson, W.; Li, Q.K.; Norris, J.; Xu, H.; Ghazarian, S.R.; Kitagawa, H.; Kawabata, S.; Taube, J.M.; Yao, S.; et al. Control of PD-L1 Expression by Oncogenic Activation of the AKT-mTOR Pathway in Non-Small Cell Lung Cancer. Cancer Res. 2016, 76, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Mercalli, A.; Calavita, I.; Dugnani, E.; Citro, A.; Cantarelli, E.; Nano, R.; Melzi, R.; Maffi, P.; Secchi, A.; Sordi, V.; et al. Rapamycin unbalances the polarization of human macrophages to M1. Immunology 2013, 140, 179–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shan, M.; Qin, J.; Jin, F.; Han, X.; Guan, H.; Li, X.; Zhang, J.; Zhang, H.; Wang, Y. Autophagy suppresses isoprenaline-induced M2 macrophage polarization via the ROS/ERK and mTOR signaling pathway. Free Radic. Biol. Med. 2017, 110, 432–443. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Ma, T.; Shen, X.N.; Xia, X.F.; Xu, G.D.; Bai, X.L.; Liang, T.B. Macrophage-induced tumor angiogenesis is regulated by the TSC2-mTOR pathway. Cancer Res. 2012, 72, 1363–1372. [Google Scholar] [CrossRef] [PubMed]
- Hallowell, R.W.; Collins, S.L.; Craig, J.M.; Zhang, Y.; Oh, M.; Illei, P.B.; Chan-Li, Y.; Vigeland, C.L.; Mitzner, W.; Scott, A.L.; et al. mTORC2 signalling regulates M2 macrophage differentiation in response to helminth infection and adaptive thermogenesis. Nat. Commun. 2017, 8, 14208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shrivastava, R.; Asif, M.; Singh, V.; Dubey, P.; Ahmad Malik, S.; Lone, M.U.; Tewari, B.N.; Baghel, K.S.; Pal, S.; Nagar, G.K.; et al. M2 polarization of macrophages by Oncostatin M in hypoxic tumor microenvironment is mediated by mTORC2 and promotes tumor growth and metastasis. Cytokine 2018. [Google Scholar] [CrossRef] [PubMed]
- Welte, T.; Kim, I.S.; Tian, L.; Gao, X.; Wang, H.; Li, J.; Holdman, X.B.; Herschkowitz, J.I.; Pond, A.; Xie, G.; et al. Oncogenic mTOR signalling recruits myeloid-derived suppressor cells to promote tumour initiation. Nat. Cell Biol. 2016, 18, 632–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryzhov, S.V.; Pickup, M.W.; Chytil, A.; Gorska, A.E.; Zhang, Q.; Owens, P.; Feoktistov, I.; Moses, H.L.; Novitskiy, S.V. Role of TGF-beta signaling in generation of CD39+CD73+ myeloid cells in tumors. J. Immunol. 2014, 193, 3155–3164. [Google Scholar] [CrossRef] [PubMed]
- Raber, P.L.; Sierra, R.A.; Thevenot, P.T.; Shuzhong, Z.; Wyczechowska, D.D.; Kumai, T.; Celis, E.; Rodriguez, P.C. T cells conditioned with MDSC show an increased anti-tumor activity after adoptive T cell based immunotherapy. Oncotarget 2016, 7, 17565–17578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Hicklin, D.J.; Ellis, L.M. Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J. Clin. Oncol. 2005, 23, 1011–1027. [Google Scholar] [CrossRef] [PubMed]
- Advani, S.H. Targeting mTOR pathway: A new concept in cancer therapy. Indian J. Med. Paediatr. Oncol. 2010, 31, 132–136. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hydroxylation of HIF-1: Oxygen sensing at the molecular level. Physiology (Bethesda) 2004, 19, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Faes, S.; Santoro, T.; Demartines, N.; Dormond, O. Evolving Significance and Future Relevance of Anti-Angiogenic Activity of mTOR Inhibitors in Cancer Therapy. Cancers (Basel) 2017, 9, 152. [Google Scholar] [CrossRef] [PubMed]
- Dodd, K.M.; Tee, A.R. STAT3 and mTOR: Co-operating to drive HIF and angiogenesis. Oncoscience 2015, 2, 913–914. [Google Scholar] [PubMed]
- DeYoung, M.P.; Horak, P.; Sofer, A.; Sgroi, D.; Ellisen, L.W. Hypoxia regulates TSC1/2-mTOR signaling and tumor suppression through REDD1-mediated 14-3-3 shuttling. Genes Dev. 2008, 22, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Squadrito, M.L.; De Palma, M. Macrophage regulation of tumor angiogenesis: Implications for cancer therapy. Mol. Aspects Med. 2011, 32, 123–145. [Google Scholar] [CrossRef] [PubMed]
- Maity, A.; Pore, N.; Lee, J.; Solomon, D.; O’Rourke, D.M. Epidermal growth factor receptor transcriptionally up-regulates vascular endothelial growth factor expression in human glioblastoma cells via a pathway involving phosphatidylinositol 3′-kinase and distinct from that induced by hypoxia. Cancer Res. 2000, 60, 5879–5886. [Google Scholar] [PubMed]
- Pore, N.; Liu, S.; Haas-Kogan, D.A.; O’Rourke, D.M.; Maity, A. PTEN mutation and epidermal growth factor receptor activation regulate vascular endothelial growth factor (VEGF) mRNA expression in human glioblastoma cells by transactivating the proximal VEGF promoter. Cancer Res. 2003, 63, 236–241. [Google Scholar] [PubMed]
- Zundel, W.; Schindler, C.; Haas-Kogan, D.; Koong, A.; Kaper, F.; Chen, E.; Gottschalk, A.R.; Ryan, H.E.; Johnson, R.S.; Jefferson, A.B.; et al. Loss of PTEN facilitates HIF-1-mediated gene expression. Genes Dev. 2000, 14, 391–396. [Google Scholar] [PubMed]
- Fang, J.; Ding, M.; Yang, L.; Liu, L.Z.; Jiang, B.H. PI3K/PTEN/AKT signaling regulates prostate tumor angiogenesis. Cell Signal. 2007, 19, 2487–2497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.M.; Blenis, J. Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol. 2009, 10, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 2005, 121, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, N.A.; Neilson, E.G.; Moses, H.L. Stromal fibroblasts in cancer initiation and progression. Nature 2004, 432, 332–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grum-Schwensen, B.; Klingelhofer, J.; Berg, C.H.; El-Naaman, C.; Grigorian, M.; Lukanidin, E.; Ambartsumian, N. Suppression of tumor development and metastasis formation in mice lacking the S100A4(mts1) gene. Cancer Res. 2005, 65, 3772–3780. [Google Scholar] [CrossRef] [PubMed]
- Apte, M.V.; Pirola, R.C.; Wilson, J.S. Pancreatic stellate cells: A starring role in normal and diseased pancreas. Front. Physiol. 2012, 3, 344. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Donthireddy, L.; Marvel, D.; Condamine, T.; Wang, F.; Lavilla-Alonso, S.; Hashimoto, A.; Vonteddu, P.; Behera, R.; Goins, M.A.; et al. Cancer-Associated Fibroblasts Neutralize the Anti-tumor Effect of CSF1 Receptor Blockade by Inducing PMN-MDSC Infiltration of Tumors. Cancer Cell 2017, 32, 654–668. [Google Scholar] [CrossRef] [PubMed]
- Shan, T.; Chen, S.; Chen, X.; Lin, W.R.; Li, W.; Ma, J.; Wu, T.; Cui, X.; Ji, H.; Li, Y.; et al. Cancer-associated fibroblasts enhance pancreatic cancer cell invasion by remodeling the metabolic conversion mechanism. Oncol. Rep. 2017, 37, 1971–1979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erkan, M. Understanding the stroma of pancreatic cancer: Co-evolution of the microenvironment with epithelial carcinogenesis. J. Pathol. 2013, 231, 4–7. [Google Scholar] [CrossRef] [PubMed]
- Von Ahrens, D.; Bhagat, T.D.; Nagrath, D.; Maitra, A.; Verma, A. The role of stromal cancer-associated fibroblasts in pancreatic cancer. J. Hematol. Oncol. 2017, 10, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumari, N.; Dwarakanath, B.S.; Das, A.; Bhatt, A.N. Role of interleukin-6 in cancer progression and therapeutic resistance. Tumour Biol. 2016, 37, 11553–11572. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gan, G.; Wang, B.; Wu, J.; Cao, Y.; Zhu, D.; Xu, Y.; Wang, X.; Han, H.; Li, X.; et al. Cancer-associated Fibroblasts Promote Irradiated Cancer Cell Recovery Through Autophagy. EBioMedicine 2017, 17, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Bethune, M.T.; Joglekar, A.V. Personalized T cell-mediated cancer immunotherapy: Progress and challenges. Curr. Opin. Biotechnol. 2017, 48, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Pancione, M.; Giordano, G.; Parcesepe, P.; Cerulo, L.; Coppola, L.; Curatolo, A.D.; Conciatori, F.; Milella, M.; Porras, A. Emerging Insight into MAPK Inhibitors and Immunotherapy in Colorectal Cancer. Curr. Med. Chem. 2017, 24, 1383–1402. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, J.S.; Massi, D.; Teng, M.W.L.; Mandala, M. PI3K-AKT-mTOR inhibition in cancer immunotherapy, redux. Semin. Cancer Biol. 2018, 48, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Saunders, R.N.; Metcalfe, M.S.; Nicholson, M.L. Rapamycin in transplantation: A review of the evidence. Kidney Int. 2001, 59, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Parsa, A.T.; Waldron, J.S.; Panner, A.; Crane, C.A.; Parney, I.F.; Barry, J.J.; Cachola, K.E.; Murray, J.C.; Tihan, T.; Jensen, M.C.; et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat. Med. 2007, 13, 84–88. [Google Scholar] [CrossRef] [PubMed]
- George, S.; Miao, D.; Demetri, G.D.; Adeegbe, D.; Rodig, S.J.; Shukla, S.; Lipschitz, M.; Amin-Mansour, A.; Raut, C.P.; Carter, S.L.; et al. Loss of PTEN Is Associated with Resistance to Anti-PD-1 Checkpoint Blockade Therapy in Metastatic Uterine Leiomyosarcoma. Immunity 2017, 46, 197–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, M.; Chen, D.; Lu, B.; Wang, C.; Zhang, J.; Huang, L.; Wang, X.; Timmons, C.L.; Hu, J.; Liu, B.; et al. PTEN loss increases PD-L1 protein expression and affects the correlation between PD-L1 expression and clinical parameters in colorectal cancer. PLoS ONE 2013, 8, e65821. [Google Scholar] [CrossRef] [PubMed]
- Mittendorf, E.A.; Philips, A.V.; Meric-Bernstam, F.; Qiao, N.; Wu, Y.; Harrington, S.; Su, X.; Wang, Y.; Gonzalez-Angulo, A.M.; Akcakanat, A.; et al. PD-L1 expression in triple-negative breast cancer. Cancer Immunol. Res. 2014, 2, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Li, C.; Liu, F.; Zhao, Y.; Liu, J.; Hua, Y.; Liu, J.; Huang, J.; Ge, C. A blockade of PD-L1 produced antitumor and antimetastatic effects in an orthotopic mouse pancreatic cancer model via the PI3K/Akt/mTOR signaling pathway. Onco Targets Ther. 2017, 10, 2115–2126. [Google Scholar] [CrossRef] [PubMed]
- Chapman, N.M.; Chi, H. mTOR signaling, Tregs and immune modulation. Immunotherapy 2014, 6, 1295–1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huijts, C.M.; Santegoets, S.J.; Quiles Del Rey, M.; de Haas, R.R.; Verheul, H.M.; de Gruijl, T.D.; van der Vliet, H.J. Differential effects of inhibitors of the PI3K/mTOR pathway on the expansion and functionality of regulatory T cells. Clin. Immunol. 2016, 168, 47–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marshall, N.A.; Galvin, K.C.; Corcoran, A.M.; Boon, L.; Higgs, R.; Mills, K.H. Immunotherapy with PI3K inhibitor and Toll-like receptor agonist induces IFN-gamma+IL-17+ polyfunctional T cells that mediate rejection of murine tumors. Cancer Res. 2012, 72, 581–591. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Element of TME. | mTORC1/2 Modulation | Effects of Modulation | References |
|---|---|---|---|
| CD8+ | ↓ mTORC1 | ↓ Effector ↑ Memory | [39,40] |
| ↓ mTORC2 | ↓ Memory | [39] | |
| CD4+ | ↑ mTORC1/2 | ↑ Th1, 2, 17 differentiation | [41,42,43] |
| Treg | ↑ mTORC1 | ↑ Differentiation in effector-like T cells | [44] |
| ↑ mTORC2 | ↓ Differentiation | [45,46] | |
| TAM | ↑ mTORC1/2 | ↑ M2 polarization | [47,48] |
| MDSC | ↓↑ mTORC1 | Variable effects | [49,50,51] |
| Endothelial cells | ↑ mTORC1 | ↑ Proliferation | [52,53] |
| CAF | ↓ mTORC1 | ↓ IL-6 secretion | [54] |
| Drug(s) | Target Cell Population | Functional Implication | Potential Therapeutic Benefit | References |
|---|---|---|---|---|
| mTOR/p110β/pan-PI3K inhibitors | CD8+ | ↑ CD8+ infiltration in tumor | ↑ Significant survival benefit | [39,56,57] |
| mTORC1 inhibitor | CD4+ | ↓ number of CD4+ | ↑ Significant survival benefit | [58] |
| mTOR/pan-AKT inhibitors | Treg | ↓ Tregs selectively | ↑ Significant survival benefit | [58,59] |
| PI3K inhibitors | TAM | ↓ TAM recruitment | Variable effects | [60] |
| mTOR inhibitors | MDSC | Variable effects | Variable effects | [49,51] |
| mTOR inhibitors | Endothelial cells | ↓ proliferation, migration and tubular structures formation ↑ apoptosis | ↓Angiogenesis | [33,61,62,63] |
| mTORC1 inhibitor | CAF | ↓ CAF-secreted cytokines | ↓ Of cell migration, invasion, and metastasis | [64] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Conciatori, F.; Bazzichetto, C.; Falcone, I.; Pilotto, S.; Bria, E.; Cognetti, F.; Milella, M.; Ciuffreda, L. Role of mTOR Signaling in Tumor Microenvironment: An Overview. Int. J. Mol. Sci. 2018, 19, 2453. https://doi.org/10.3390/ijms19082453
Conciatori F, Bazzichetto C, Falcone I, Pilotto S, Bria E, Cognetti F, Milella M, Ciuffreda L. Role of mTOR Signaling in Tumor Microenvironment: An Overview. International Journal of Molecular Sciences. 2018; 19(8):2453. https://doi.org/10.3390/ijms19082453
Chicago/Turabian StyleConciatori, Fabiana, Chiara Bazzichetto, Italia Falcone, Sara Pilotto, Emilio Bria, Francesco Cognetti, Michele Milella, and Ludovica Ciuffreda. 2018. "Role of mTOR Signaling in Tumor Microenvironment: An Overview" International Journal of Molecular Sciences 19, no. 8: 2453. https://doi.org/10.3390/ijms19082453
APA StyleConciatori, F., Bazzichetto, C., Falcone, I., Pilotto, S., Bria, E., Cognetti, F., Milella, M., & Ciuffreda, L. (2018). Role of mTOR Signaling in Tumor Microenvironment: An Overview. International Journal of Molecular Sciences, 19(8), 2453. https://doi.org/10.3390/ijms19082453