Assessment of Genetic Diversity, Population Structure, and Evolutionary Relationship of Uncharacterized Genes in a Novel Germplasm Collection of Diploid and Allotetraploid Gossypium Accessions Using EST and Genomic SSR Markers

 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. SSR Marker Analysis
2.2. Unique Alleles
2.3. Common Alleles
2.4. Analysis of Population Structures
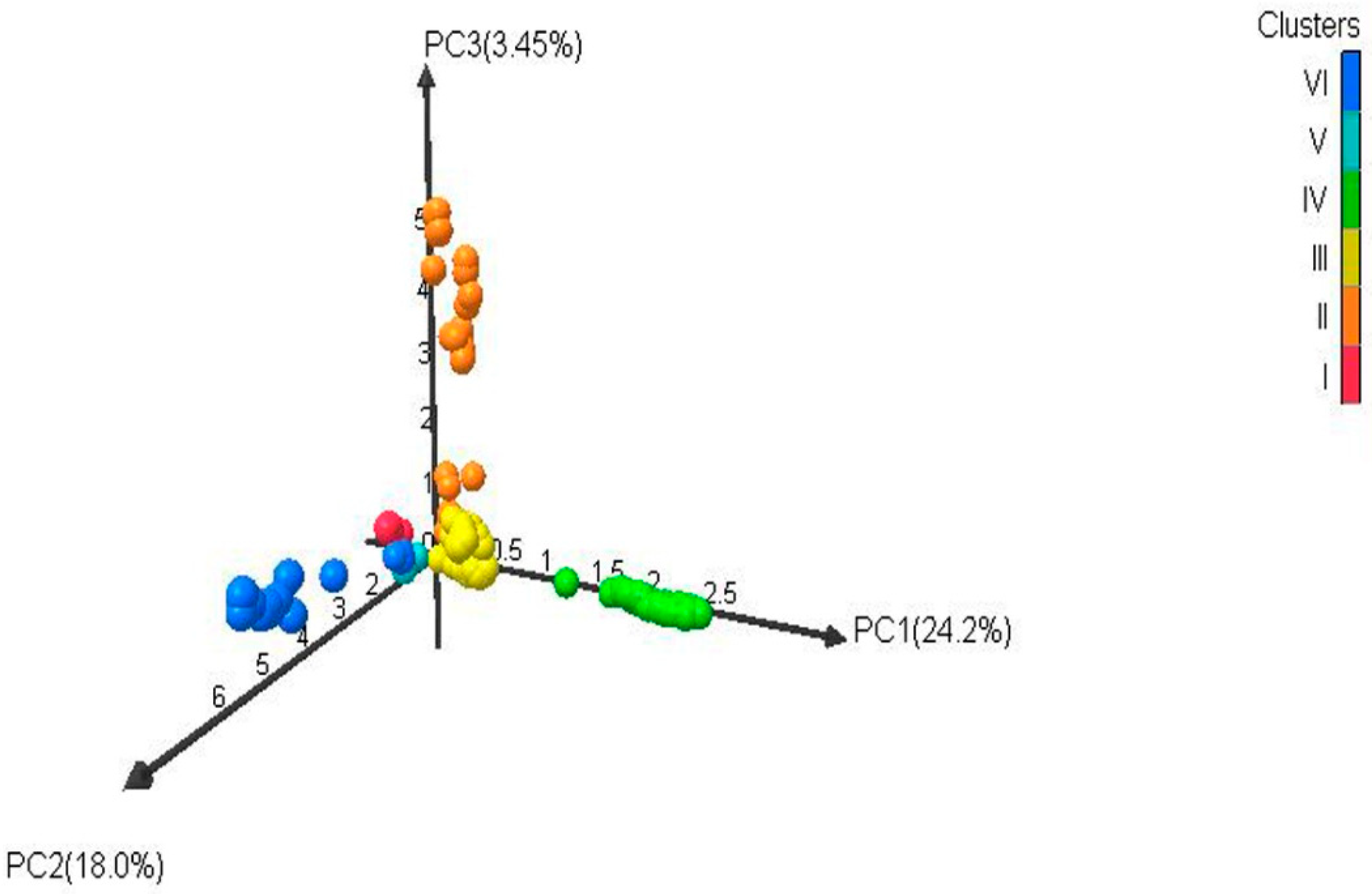
2.5. Genetic Diversity and Cluster Analysis of Phylogenetic Tree
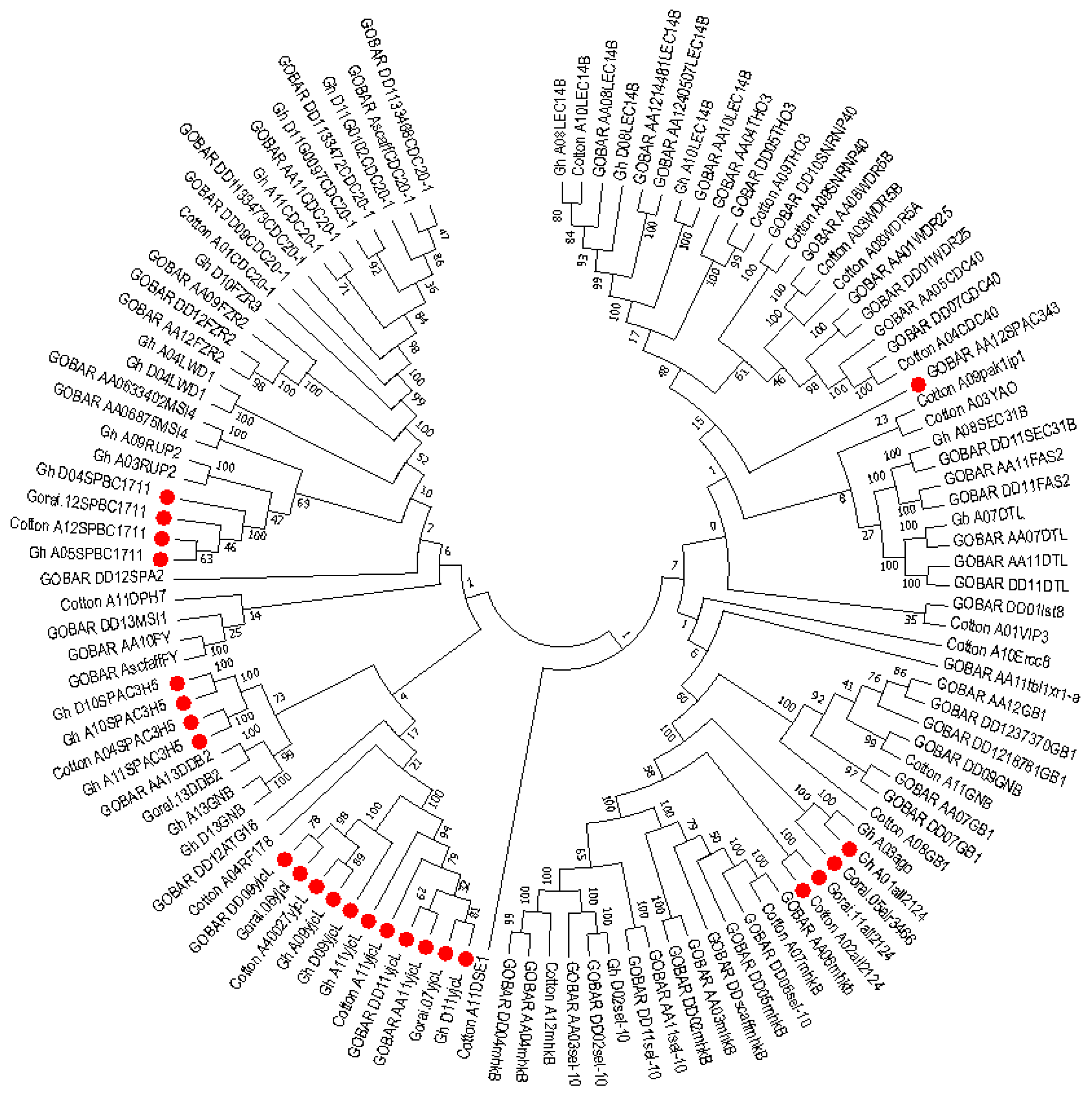
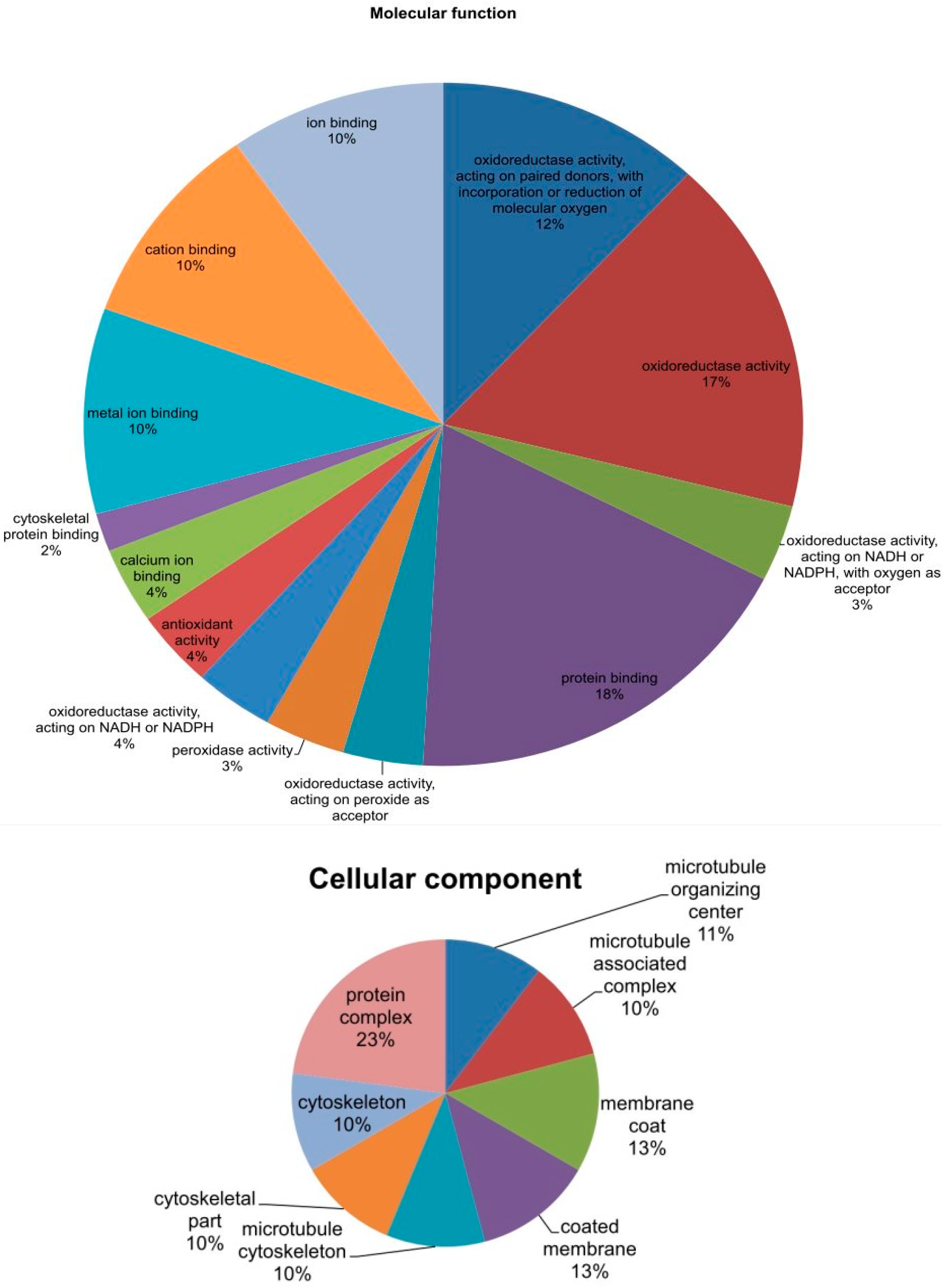
2.6. Phylogenetic Analysis of Mined Genes and Functional Annotation of DUF819 (PF005684)
3. Discussion
4. Materials and Methods
4.1. Plant Material and DNA Extraction
4.2. SSR Marker Selection and Genotyping
4.3. Analysis of Genotypic Data and Genetic Diversity
4.4. Analysis of Genetic Structure
4.5. Gene Mining and Phylogenetic Analysis of DUF819 Proteins
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| PIC | Polymorphism information content |
| MAF | Major Allele frequency |
| GD | Genetic distance |
| Pop | population |
| AMOVA | Analysis of molecular variance |
| PCA | Principal coordinate analysis |
| PCR | Polymerase chain reaction |
| DNA | Deoxyribonucleic acid |
| NPGS | National Plant Germplasm System |
| EST | Expressed sequence tags |
| gSSR | Genomic SSR |
| SSR | Simple sequence repeats |
| RFLP | Restriction fragment polymorphism |
| RAPD | Random amplified polymorphic DNA |
| AFLP | Amplified fragment length polymorphism |
| SNP | Single nucleotide polymorphism |
| G | Gossypium |
| WCGN | Wild cotton Germplasm Nursery of China |
References
- Wendel, J.F.; Brubaker, C.; Alveraz, I.; Cronn, R.; Stewart, J.M. Evolution and natural history of cotton genus. Genet. Genom. 2009, 3, 3–22. [Google Scholar] [CrossRef]
- Wendel, J.F.; Grover, C.E. Taxonomy and Evolution of Cotton Genus, Gossypium; Issue Agronmonogr; Cotton, American Society of Agronomy, Inc.: Madison, WI, USA; Crop Science Society of America, Inc.: Madison, WI, USA; Soil Science Society of America, Inc.: Madison, WI, USA, 2015; Volume 57. [Google Scholar] [CrossRef]
- Gallagher, J.P.; Grover, C.E.; Rex, K.; Moran, M.; Wendel, J.F. A New Species of Cotton from Wake Atoll, Gossypium stephensii (Malvaceae). Syst. Bot. 2017, 42, 115–123. [Google Scholar] [CrossRef]
- Grover, C.E.; Zhu, X.; Grupp, K.K.; Jareczek, J.J.; Gallagher, J.P.; Szadkowski, E.; Seijo, J.G.; Wendel, J.F. Molecular confirmation of species status for the allopolyploid cotton species, Gossypium ekmanianum Wittmack. Genet. Resour. Crop Evol. 2015, 62, 103–114. [Google Scholar] [CrossRef]
- Chandrakanth, K. Wild Crop Relatives: Genomic and Breeding Resources; Springer: Berlin/Heidelberg, Germany, 2011; pp. 109–122. [Google Scholar]
- Krapovickas, A.; Seijo, G. Gossypium ekmanianum (Malvaceae), algodon silvestre de la Republica Dominicana. Bonplandia 2008, 17, 55–63. [Google Scholar]
- Wendel, J.F.; Brubaker, C.L.; Percival, A.E. Genetic diversity in Gossypium hirsutum and the origin of Upland cotton. Am. J. Bot. 1992, 79, 1291–1310. [Google Scholar] [CrossRef]
- Wendel, J.F.; Cronn, R.C. Polyploidy and the evolutionary history of cotton. Adv. Agron. 2003, 78, 139–186. [Google Scholar] [Green Version]
- Wang, Q.; Fang, L.; Chen, J.; Hu, Y.; Si, Z.; Wang, S.; Chang, L.; Guo, W.; Zhang, T. Genome-Wide Mining, Characterization, and Development of Microsatellite Markers in Gossypium Species. Sci. Rep. 2015, 5, 10638. [Google Scholar] [CrossRef] [PubMed]
- Wendel, J.F.; Brubaker, C.L. RFLP diversity in Gossypium hirsutum L. and new insights into the domestication of cotton. Am. J. Bot. 1993, 80, 71. [Google Scholar]
- Brubaker, C.L.; Paterson, A.H.; Wendel, J.F. Comparative genetic mapping of allotetraploid cotton and its diploid progenitors. Genome 1999, 42, 184–203. [Google Scholar] [CrossRef]
- Senchina, D.S.; Alvarez, I.; Cronn, R.C.; Liu, B.; Rong, J.; Noyes, R.D.; Paterson, A.H.; Wing, R.A.; Wilkins, T.A.; Wendel, J.F. Rate variation among nuclear genes and the age of polyploidy in Gossypium. Mol. Biol. Evol. 2003, 20, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Campbell, B.T.; Saha, S.; Percy, R.; Frelichowski, J.; Jenkins, J.N.; Park, W.; Mayee, C.D.; Gotmare, V.; Dessauw, D.; Giband, M.; et al. Status of the global cotton germplasm resources. Crop Sci. 2010, 50, 1161–1179. [Google Scholar] [CrossRef]
- Chen, H.; Khan, M.K.R.; Zhou, Z.; Wang, X.; Cai, X.; Ilyas, M.K.; Wang, C.; Wang, Y.; Li, Y.; Liu, F.; et al. A high density SSR genetic map constructed from F2 population of Gossypium hirsutum and Gossypium darwinii. Gene 2015, 574, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Dejoode, D.; Wendel, J. Genetic diversity and origin of the hawaiian-islands cotton, Gossypium tomentosum. Am. J. Bot. 1992, 79, 1311–1319. [Google Scholar] [CrossRef]
- Wendel, J.F.; Rowley, R.; Stewart, J. Genetic diversity in and phylogenetic-relationships of the brazilian endemic cotton, Gossypium mustelinum (Malvaceae). Plant Syst. Evol. 1994, 192, 49–59. [Google Scholar] [CrossRef]
- Wendel, J.F.; Percy, R. Allozyme diversity and introgression in the galapagos-islands endemic Gossypium darwinii and its relationship to continental Gossypium barbadense. Biochem. Syst. Ecol. 1990, 18, 517–528. [Google Scholar] [CrossRef]
- Ulloa, M.; Stewart, J.M.; Garcia-C, E.A.; Goday, A.S.; Gaytan-M, A.; Acosta, N.S. Cotton Genetic Resources in the Western States of Mexico: In Situ Conservation Status and Germplasm Collection for ex situ Preservation. Genet. Resour. Crop Evol. 2006, 53, 653–668. [Google Scholar] [CrossRef]
- Abdurakhmonov, I.Y.; Buriev, Z.T.; Shermatov, S.E.; Abdullaev, A.A.; Urmonov, K.; Kushanov, F.; Egamberdiev, S.S.; Shapulatov, U.; Abdukarimov, A.; Saha, S.; et al. Genetic Diversity in Gossypium genus. In Genetic Diversity in Plants; Caliskan, M., Ed.; InTech: Rijeka, Croatia, 2012; pp. 313–338. [Google Scholar] [CrossRef]
- Van Esbroeck, G.A.; Bowman, D.T. Cotton germplasm diversity and its importance to cultivar development. J. Cotton Sci. 1998, 2, 121–129. [Google Scholar]
- Bolek, Y.; El-Zik, K.M.; Pepper, A.E.; Bell, A.A.; Magill, C.W.; Thaxton, P.M.; Reddy, O.U.K. Mapping of verticillium wilt resistance genes in cotton. Plant Sci. 2005, 168, 1581–1590. [Google Scholar] [CrossRef]
- Tyagi, P.; Gore, M.A.; Bowman, D.T.; Campbell, B.T.; Udall, J.A.; Kuraparthy, V. Genetic diversity and population structure in the US Upland cotton (Gossypium hirsutum L.). Theor. Appl. Genet. 2014, 127, 283–295. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, M.A.; Rehman, M.U. Identification of Marker-Trait Associations for Lint Traits in Cotton. Front. Plant Sci. 2017, 8, 86. [Google Scholar] [CrossRef] [PubMed]
- Kalivas, A.; Xanthopoulos, F.; Kehagia, O.; Tsaftaris, A.S. Agronomic characterization, genetic diversity and association analysis of cotton cultivars using simple sequence repeat molecular markers. Genet. Mol. Res. 2011, 10, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; He, S.; Pan, Z.; Du, X. Homologous simple sequence repeats (SSRs) analysis in tetraploid (AD1) and diploid (A2, D5) genomes of Gossypium. Hereditas (Beijing) 2015, 37, 192–203. [Google Scholar]
- Kiflom, W.O.; Xiao, L.; Cai, X.; Wang, X.; Chen, H.; Zhou, Z.; Wang, C.; Wang, Y.; Liu, F.; Wang, K. Genome wide assessment of genetic diversity and fiber quality traits characterization in Gossypium hirsutum races. J. Integr. Agric. 2017, 16, 2402–2412. [Google Scholar] [CrossRef]
- Khan, M.K.R.; Haodong, C.; Zhongli, Z.; Ilyas, M.K.; Xingxing, W.; Cai, X.; Chunying, W.; Fang, L.; Kunbo, W. Genome Wide SSR High Density Genetic Map Construction from an Interspecific Cross of Gossypium hirsutum X Gossypium tomentosum. Front. Plant Sci. 2016, 7, 436. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, M.; Ceasar, S.A.; Duraipandiyan, V.; Dhabi, N.A.A.; Ignacimuthu, S. Assessment of genetic diversity, population structure and relationships in Indian and non-Indian genotypes of finger millet (Eleusine coracana (L.) Gaertn) using genomic SSR markers. SpringerPlus 2016, 5, 120. [Google Scholar] [CrossRef] [PubMed]
- Van Becelaere, G.; Lubbers, E.L.; Paterson, A.H.; Chee, P.W. Pedigree vs. DNA marker-based genetic similarity estimates in cotton. Crop Sci. 2005, 45, 2281–2287. [Google Scholar] [CrossRef]
- Iqbal, M.J.; Aziz, N.; Saeed, N.A.; Zafar, Y. Genetic diversity evaluation of some elite cotton varieties by RAPD analysis. Theor. Appl. Genet. 1997, 94, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Bakht, J.; Iqbal, M.; Shafi, M. Genetic diversity and phylogenetic relationship in different genotypes of cotton for future breeding. Int. Quart. J. Biol. Sci. 2017, 5, 25–29. [Google Scholar]
- Rahman, M.; Yasmin, T.; Tabbasam, N.; Ullah, I.; Asif, M.; Zafar, Y. Studying the extent of genetic diversity among Gossypium arboretum L. genotypes/cultivars using DNA fingerprinting. Genet. Resour. Crop. Evol. 2008, 55, 331–339. [Google Scholar] [CrossRef]
- Shaheen, N.; Pearce, S.R.; Khan, M.A.; Mahmood, T.; Yasmin, G.; Hayat, M.Q. AFLP mediated genetic diversity of Malvaceae species. J. Med. Plant Res. 2010, 4, 148–154. [Google Scholar]
- Liu, S.; Cantrell, R.G.; McCarty, J.C.J.; Stewart, J.M. Simple sequence repeat-based assessment of genetic diversity in cotton race stock accessions. Crop Sci. 2000, 40, 1459–1469. [Google Scholar] [CrossRef]
- Liu, D.; Guo, X.; Lin, Z.; Nie, Y.; Zhang, X. Genetic diversity of Asian cotton (Gossypium arboretum L.) in china evaluated by microsatellite analysis. Genet. Res. Crop Evol. 2006, 53, 1145–1152. [Google Scholar] [CrossRef]
- Hinze, L.L.; Gazave, E.; Gore, M.A.; Fang, D.D.; Scheffler, B.E.; Yu, J.Z.; Jones, D.C.; Frelichowski, J.; Percy, R.G. Genetic Diversity of the Two Commercial Tetraploid Cotton Species in the Gossypium Diversity Reference Set. J. Hered. 2016, 107, 274–286. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, X.F.; Li, Z.K.; Zhang, G.Y.; Ma, Z.Y. Assessing genetic diversity of cotton cultivars using genomic and newly developed expressed sequence tag-derived microsatellite markers. Genet. Mol. Res. 2011, 10, 1462–1470. [Google Scholar] [CrossRef] [PubMed]
- Dongre, A.; Bhandarkar, M.; Banerjee, S. Genetic diversity in tetraploid and diploid cotton (Gossypium spp.) using ISSR and microsatellite DNA markers. Indian J. Biotechnol. 2007, 6, 349–353. [Google Scholar]
- Liu, B.; Wendel, J.F. Intersimple sequence repeat (ISSR) polymorphisms as a genetic marker system in cotton. Mol. Ecol. Notes 2001, 1, 205–208. [Google Scholar] [CrossRef]
- Meng, K.; Wei, S.J.; Wang, Y.Q.; Zhou, D.U.; Ma, L.; Fang, D.; Yang, W.H.; Ma, Z.Y. Development of a core set of SNP markers for the identification of upland cotton cultivars in China. J. Integr. Agric. 2016, 15, 954–962. [Google Scholar] [CrossRef]
- Park, Y.J.; Lee, J.K.; Kim, N.S. Simple Sequence Repeat Polymorphisms (SSRPs) for Evaluation of Molecular Diversity and Germplasm Classification of Minor Crops. Molecules 2009, 14, 4546–4569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, B.T.; Williams, V.E.; Park, W. Using molecular markers and field performance data to characterize the pee dee cotton germplasm resources. Euphytica 2009, 169, 285–301. [Google Scholar] [CrossRef]
- Kirungu, J.N.K.; Deng, Y.; Cai, X.; Mangwanga, R.O.; Zhou, Z.; Wang, X.; Wang, Y.; Zhang, Z.; Wang, K.; Liu, F. Simple Sequence Repeat (SSR) Genetic Linkage Map of D Genome Diploid Cotton Derived from an Interspecific Cross between Gossypium davidsonii and Gossypium klotzschianum. Int. J. Mol. Sci. 2018, 19, 204. [Google Scholar] [CrossRef] [PubMed]
- Goodacre, N.F.; Gerloff, D.L.; Uetz, P. Protein Domains of Unknown Function Are Essential in Bacteria. mBio 2013, 5, e00744-13. [Google Scholar] [CrossRef] [PubMed]
- Mulder, N.J.; Kersey, P.; Pruess, M.; Apweiler, R. In silico characterization of proteins: UniProt, InterPro and Integr8. Mol. Biotechnol. 2008, 38, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Bateman, A.; Coggill, P.; Finn, R.D. DUFs: Families in search of function. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2010, 66, 1148–1152. [Google Scholar] [CrossRef] [PubMed]
- Littler, E. Combinatorial domain hunting: Solving problems in protein expression. Drug Discov. Today 2010, 15, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Hauser, R.; Pech, M.; Kijek, J.; Yamamoto, H.; Titz, B.; Naeve, F.; Tovchigrechko, A.; Yamamoto, K.; Szaflarski, W.; Takeuchi, N.; et al. RsfA (YbeB) proteins are conserved ribosomal silencing factors. PLoS Genet. 2012, 8, e1002815. [Google Scholar] [CrossRef] [PubMed]
- Gruber, H.; Heijde, M.; Heller, W.; Albert, A.; Seidlitz, H.K.; Ulm, R. Negative feedback regulation of UV-B-induced photomorphogenesis and stress acclimation in Arabidopsis. Proc. Natl. Acad. Sci. USA 2010, 107, 20132–20137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Zhao, P.; Li, J.; Zhang, C.; Wang, L.; Ren, Z. Genome-wide analysis of the WD-repeat protein family in Cucumber and Arabidopsis. Mol. Genet. Genom. 2013, 289, 103–124. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Fang, D.; Kohel, R.; Ulloa, M.; Hinze, L.; Percy, R.; Zhang, J.; Chee, P.; Schefer, B.; Jones, D. Development of a core set of SSR markers for the characterization of Gossypium germplasm. Euphytica 2012, 187, 203–213. [Google Scholar] [CrossRef]
- Nei, M.; Tajima, F.; Tateno, Y. Accuracy of estimated phylogenetic trees from molecular data. J. Mol. Evol. 1983, 19, 153–170. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.W.; Cothren, J.T. Cotton: Origin, History, Technology, and Production; John Wiley & Sons: Hoboken, NJ, USA, 1999; p. 43. [Google Scholar]
- Shu, K.; Yang, W. E3 Ubiquitin Ligases: Ubiquitous Actors in Plant Development and Abiotic Stress Responses. Plant Cell Physiol. 2017, 58, 1461–1476. [Google Scholar] [CrossRef] [PubMed]
- Matteo, P.; Edward, M.M.; Michael, J.T.; David, E.; Todd, O.Y. Assigning protein functions by comparative genome analysis: Protein phylogenetic profiles. Proc. Natl. Acad. Sci. USA 1999, 96, 4285–4288. [Google Scholar] [Green Version]
- Tian, T.; Liu, Y.; Yan, H.; You, Q.; Yi, X.; Du, Z.; Xu, W.; Su, Z. agriGO v2.0: A GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res. 2017, 45, W12–W129. [Google Scholar] [CrossRef] [PubMed]
- Dahab, A.A.; Saeed, M.; Mohamed, B.B.; Ashraf, M.A.; Puspito, A.N.; Bajwa, K.S.; Shahid, A.A.; Husnain, T. Genetic diversity assessment of cotton (Gossypium hirsutum L.) genotypes from Pakistan using simple sequence repeat markers. Aust. J. Crop Sci. 2013, 7, 261–267, ISSN 1835-2707. [Google Scholar]
- Bardak, A.; Bolek, Y. Genetic Diversity of Diploid and Tetraploid Cottons Determined By Ssr and Issr Markers. Turk. J. Field Crops 2012, 2, 139–144. [Google Scholar]
- Bertini, C.H.C.D.; Schuster, I.; Sediyama, T.; Barros, E.G.; Moreira, M.A. Characterization and genetic diversity analysis of cotton cultivars using microsatellites. Genet. Mol. Biol. 2006, 29, 321–329. [Google Scholar] [CrossRef] [Green Version]
- Qin, H.; Chen, M.; Yi, X.; Bie, S.; Zhang, C.; Zhang, Y.; Lan, J.; Meng, Y.; Yuan, Y.; Jiao, C. Identification of Associated SSR Markers for Yield Component and Fiber Quality Traits Based on Frame Map and Upland Cotton Collections. PLoS ONE 2015, 10, e0118073. [Google Scholar] [CrossRef] [PubMed]
- Baytar, A.A.; Erdogan, O.; Frary, A.; Frary, A.; Doganlar, S. Molecular diversity and identification of alleles for Verticillium wilt resistance in elite cotton (Gossypium hirsutum L.) germplasm. Euphytica 2017, 213, 31. [Google Scholar] [CrossRef]
- Iqbal, M.J.; Reddy, O.U.K.; El-Zik, K.M.; Pepper, A.E. A genetic bottleneck in the evolution under domestication of upland cotton Gossypium hirsutum L. examined using DNA fingerprinting. Theor. Appl. Genet. 2001, 103, 547–554. [Google Scholar] [CrossRef]
- Rungis, D.; Llewellyn, D.; Dennis, E.S.; Lyon, B.R. Simple sequence repeat (SSR) markers reveal low levels of polymorphism between cotton (Gossypium hirsutum L.) cultivars. Aust. J. Agric. Res. 2005, 56, 301–307. [Google Scholar] [CrossRef]
- Lacape, J.M.; Dessauw, D.; Rajab, M.; Noyer, J.L.; Hau, B. Microsatellite diversity in tetraploid Gossypium germplasm: Assembling a highly informative genotyping set of cotton SSRs. Mol. Breed. 2007, 19, 45–58. [Google Scholar] [CrossRef]
- Abdurakhmonov, I.Y.; Kohel, R.J.; Yu, J.Z.; Pepper, A.E.; Abdullaev, A.A.; Kushanov, F.N.; Salakhutdinov, L.B.; Buriev, Z.T.; Saha, S.; Scheffler, B.E.; et al. Molecular diversity and association mapping of fiber quality traits in exotic G. hirsutum L. germplasm. Genomics 2008, 92, 478–487. [Google Scholar] [CrossRef] [PubMed]
- Grover, C.E.; Gallagher, J.P.; Jareczek, J.J.; Page, J.T.; Udal, J.A.; Gore, M.A.; Wendel, J.F. Re-evaluating the phylogeny of allopolyploid Gossypium L. Mol. Phylogenet. Evol. 2015, 92, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Noormohammadi, Z.; Farahani, Y.H.A.; Sheidai, M.; Baraki, S.G.; Alishah, O. Genetic diversity analysis in Opal cotton hybrids based on SSR, ISSR, and RAPD markers. Genet. Mol. Res. 2013, 12, 256–269. [Google Scholar] [CrossRef] [PubMed]
- Noormohammadi, Z.; Sheidai, M.; Foroutan, M.; Alishah, O. Networking and Bayesian analyses of genetic affinity in cotton germplasm. Nucleus 2015, 58, 33–45. [Google Scholar] [CrossRef]
- Wu, J.F.; Wang, Y.; Wu, S.H. Two New Clock Proteins, LWD1 and LWD2, Regulate Arabidopsis Photoperiodic Flowering. Plant Physiol. 2008, 148, 948–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biedermann, S.; Hellmann, H. The DDB1a interacting proteins ATCSA-1 and DDB2 are critical factors for UV-B tolerance and genomic integrity in Arabidopsis thaliana. Plant J. 2010, 62, 404–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strohmaier, H.; Spruck, C.H.; Kaiser, P.; Won, K.A.; Sangfelt, O.; Reed, S.I. Human F-box protein hDcd4 targets cyclin E for proteolysis and is mutated in a breast cancer cell line. Nature 2001, 413, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Brown, W.L. Genetic diversity and genetic vulnerability: An appraisal. Econ. Bot. 1983, 37, 4–12. [Google Scholar] [CrossRef]
- Flint-Garcia, S.A.; Anne-Ce line, T.; Yu, J.; Pressoir, G.; Romero, S.M.; Mitchell, S.E.; Doebley, J.; Kresovich, S.; Goodman, M.M.; Buckler, E.S. Maize association population: A high resolution platform for quantitative trait locus dissection. Plant J. 2005, 44, 1054–1064. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Stewart, J.M. Economical and rapid method for extraction cotton genomic DNA. J. Cotton Sci. 2000, 4, 193–201. [Google Scholar]
- Zhang, J.; Guo, W.; Zhang, T. Molecular linkage map of allotetraploid cotton (Gossypium hirsutum L. X Gossypium barbadense L.) with a haploid population. Theor. Appl. Genet. 2002, 105, 1166–1174. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Muse, S.V. PowerMarker: An integrated analysis environment for genetic marker analysis. Bioinformatics 2005, 21, 2128–2129. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.A.; Churchill, G.A.; Autrique, J.E.; Tanksley, S.D.; Sorrellis, M.E. Optimizing parental selection for genetic linkage maps. Genome 1993, 36, 181–186. [Google Scholar] [CrossRef] [PubMed]
- XLSTAT. Data Analysis and Statistical Solutions for Microsoft Excell; Addinsoft: Paris, France, 2014. [Google Scholar]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [PubMed]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics 2003, 164, 1567–1587. [Google Scholar] [PubMed]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulationstudy. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Dent, A.E.; Bridgett, M.V. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Liu, K.J.; Goodman, M.; Muse, S.; Smith, J.S.; Buckler, E.; Doebley, J. Genetic structure and diversity among maize inbred lines as inferred from DNA microsatellites. Genetics 2003, 165, 2117–2128. [Google Scholar] [PubMed]
- Wang, K.; Wang, Z.; Li, F.; Ye, W.; Wang, J.; Song, G.; Yue, Z.; Cong, L.; Shang, H.; Zhu, S.; et al. The draft genome of diploid Gossypium raimondii. Nat. Genet. 2012, 44, 1098–1103. [Google Scholar] [CrossRef] [PubMed]
- Magwanga, R.O.; Lu, P.; Nyangasi Kirungu, J.; Diouf, L.; Dong, Q.; Hu, Y.; Cai, X.; Xu, Y.; Hou, Y.; Zhou, Z.; et al. GBS Mapping and Analysis of Genes Conserved between Gossypium tomentosum and Gossypium hirsutum Cotton Cultivars that Respond to Drought Stress at the Seedling Stage of the BC 2 F 2 Generation. Int. J. Mol. Sci. 2018, 19, 1614. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger database. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Priyam, A.; Woodcroft, B.J.; Rai, V.; Munagala, A.; Moghul, I.; Ter, F.; Gibbins, M.A.; Moon, H.K.; Leonard, G.; Rumpf, W.; et al. Sequenceserver: A modern graphical user interface for custom BLAST databases. bioRxiv 2015. [Google Scholar] [CrossRef]
- Saitou, N.; Nei, M. The neighbor joining method: A new method for reconstructing phylogenetic trees. Mol. Bio. Evol. 1987, 4, 406–425. [Google Scholar]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Zuckerkandl, E.; Pauling, L. Evolutionary divergence and convergence in proteins. In Evolving Genes and Proteins; Bryson, V., Vogel, H.J., Eds.; Academic Press: New York, NY, USA, 1965; pp. 97–166. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Panel | Total Alleles | Total Lines | Unique Alleles | Rare Alleles (Freq < 5%) |
|---|---|---|---|---|
| Combined Panel | 382 | 132 | 123 (32.19%) | 108 (28.27%) |
| Gossypium barbadense | 258 | 20 | 19 (7.36%) | 12 (4.65%) |
| Gossypium darwinii | 309 | 59 | 18 (5.83%) | 57 (18.44%) |
| Gossypium tomentosum | 205 | 32 | 13 (6.34%) | 15 (7.31%) |
| Gossypium hirsutum | 143 | 2 | 25 (17.48%) | 22 (15.38%) |
| Gossypium ekmanianum | 177 | 10 | 31 (17.51%) | 2 (1.13%) |
| Gossypium stephensii | 125 | 4 | 4 (3.2%) | 0 |
| Gossypium klotzschianum | 90 | 5 | 13 (14.44%) | 0 |
| Source of Variation | df | Sum of Squares | Mean Squares | Estimated Variation | Percentage of Variation |
|---|---|---|---|---|---|
| Among Pops | 5 | 4201.563 | 840.313 | 38.134 ** | 49% |
| Within Pops | 126 | 4932.945 | 39.150 | 39.150 | 51% |
| Total | 131 | 9134.508 | 77.284 | 100% |
| Populations with Origin | Pop1 (Santa Cruz) | Pop2 (Hawaiian) | Pop3 (Isabella) | Pop4 (Dominican Republic) | Pop5 (San Cristobal) |
|---|---|---|---|---|---|
| Pop2 (Hawaiian) | 0.869 | ||||
| Pop3 (Isabella) | 0.697 | 0.651 | |||
| Pop4 (Dominican Republic) | 0.757 | 0.689 | 0.544 | ||
| Pop5 (San Cristobal) | 0.810 | 0.749 | 0.301 | 0.638 | |
| Pop6 (Santa Cruz) | 0.813 | 0.752 ** | 0.413 | 0.626 | 0.517 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ditta, A.; Zhou, Z.; Cai, X.; Wang, X.; Okubazghi, K.W.; Shehzad, M.; Xu, Y.; Hou, Y.; Sajid Iqbal, M.; Khan, M.K.R.; et al. Assessment of Genetic Diversity, Population Structure, and Evolutionary Relationship of Uncharacterized Genes in a Novel Germplasm Collection of Diploid and Allotetraploid Gossypium Accessions Using EST and Genomic SSR Markers. Int. J. Mol. Sci. 2018, 19, 2401. https://doi.org/10.3390/ijms19082401
Ditta A, Zhou Z, Cai X, Wang X, Okubazghi KW, Shehzad M, Xu Y, Hou Y, Sajid Iqbal M, Khan MKR, et al. Assessment of Genetic Diversity, Population Structure, and Evolutionary Relationship of Uncharacterized Genes in a Novel Germplasm Collection of Diploid and Allotetraploid Gossypium Accessions Using EST and Genomic SSR Markers. International Journal of Molecular Sciences. 2018; 19(8):2401. https://doi.org/10.3390/ijms19082401
Chicago/Turabian StyleDitta, Allah, Zhongli Zhou, Xiaoyan Cai, Xingxing Wang, Kiflom Weldu Okubazghi, Muhammad Shehzad, Yanchao Xu, Yuqing Hou, Muhammad Sajid Iqbal, Muhammad Kashif Riaz Khan, and et al. 2018. "Assessment of Genetic Diversity, Population Structure, and Evolutionary Relationship of Uncharacterized Genes in a Novel Germplasm Collection of Diploid and Allotetraploid Gossypium Accessions Using EST and Genomic SSR Markers" International Journal of Molecular Sciences 19, no. 8: 2401. https://doi.org/10.3390/ijms19082401
APA StyleDitta, A., Zhou, Z., Cai, X., Wang, X., Okubazghi, K. W., Shehzad, M., Xu, Y., Hou, Y., Sajid Iqbal, M., Khan, M. K. R., Wang, K., & Liu, F. (2018). Assessment of Genetic Diversity, Population Structure, and Evolutionary Relationship of Uncharacterized Genes in a Novel Germplasm Collection of Diploid and Allotetraploid Gossypium Accessions Using EST and Genomic SSR Markers. International Journal of Molecular Sciences, 19(8), 2401. https://doi.org/10.3390/ijms19082401