Substantial Epigenetic Variation Causing Flower Color Chimerism in the Ornamental Tree Prunus mume Revealed by Single Base Resolution Methylome Detection and Transcriptome Sequencing

 ,
,
Abstract
:1. Introduction
2. Results
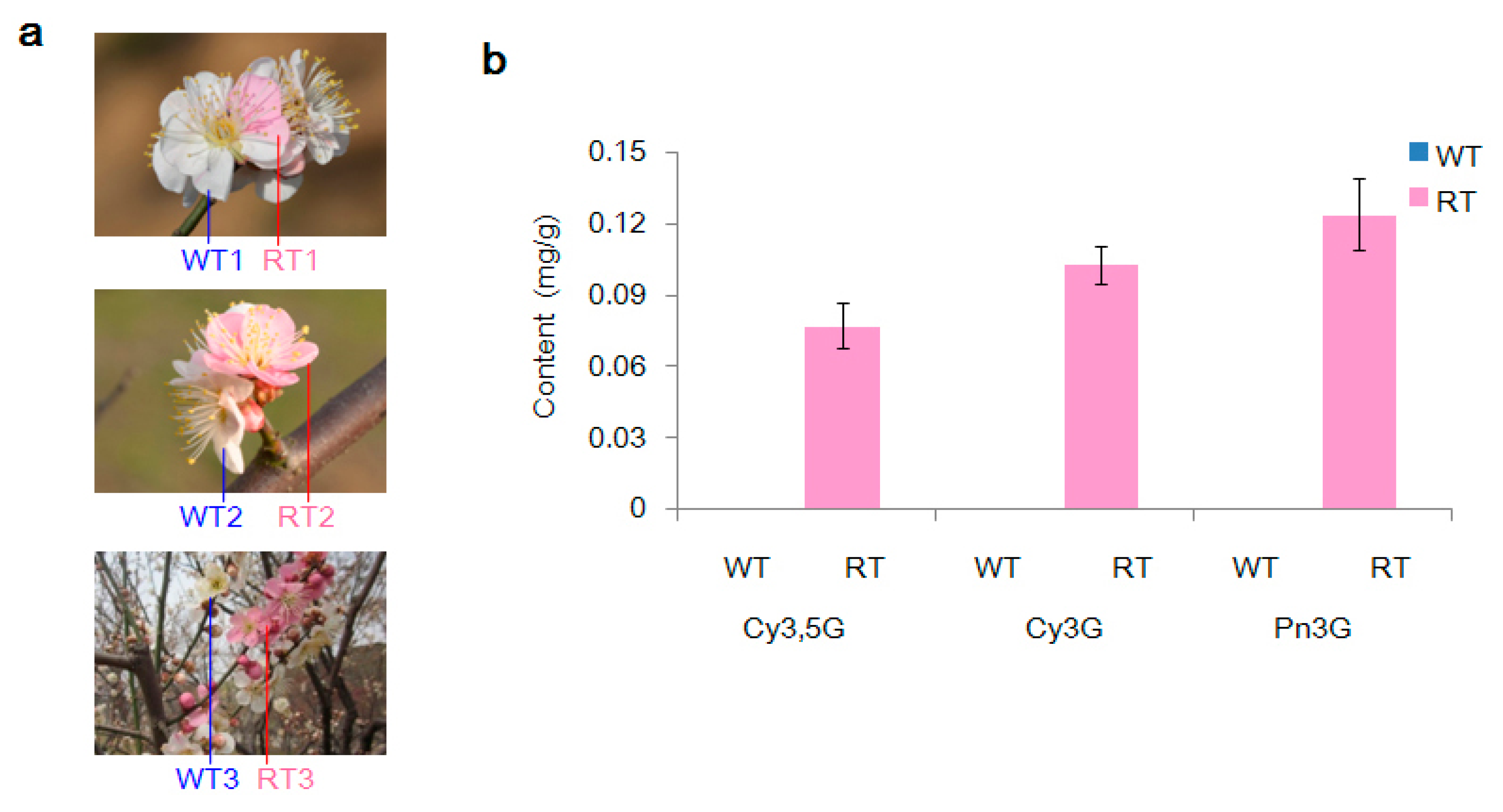
2.1. Variation in Pigmentation in White (WT) and Red (RT) Petal Tissues
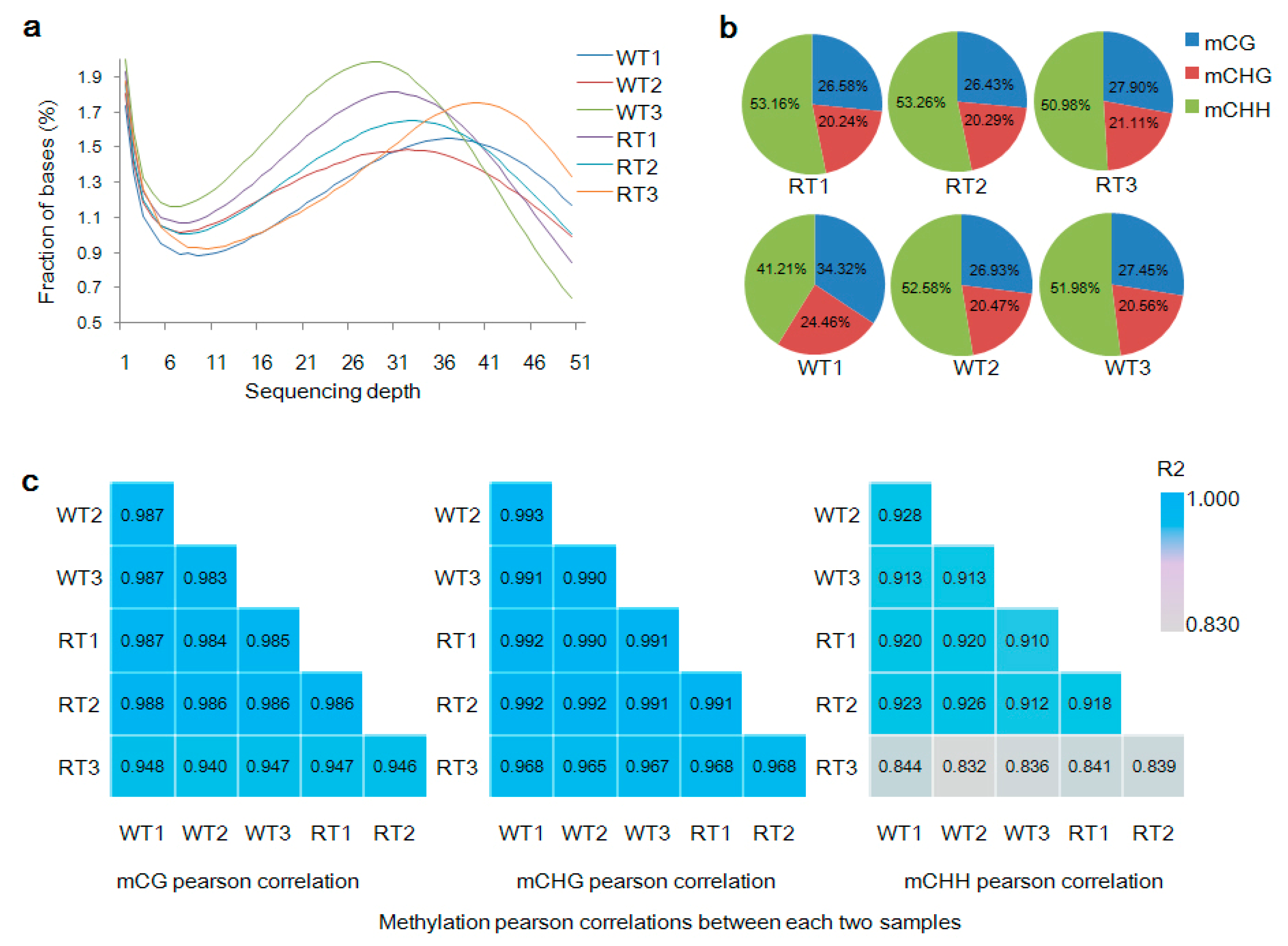
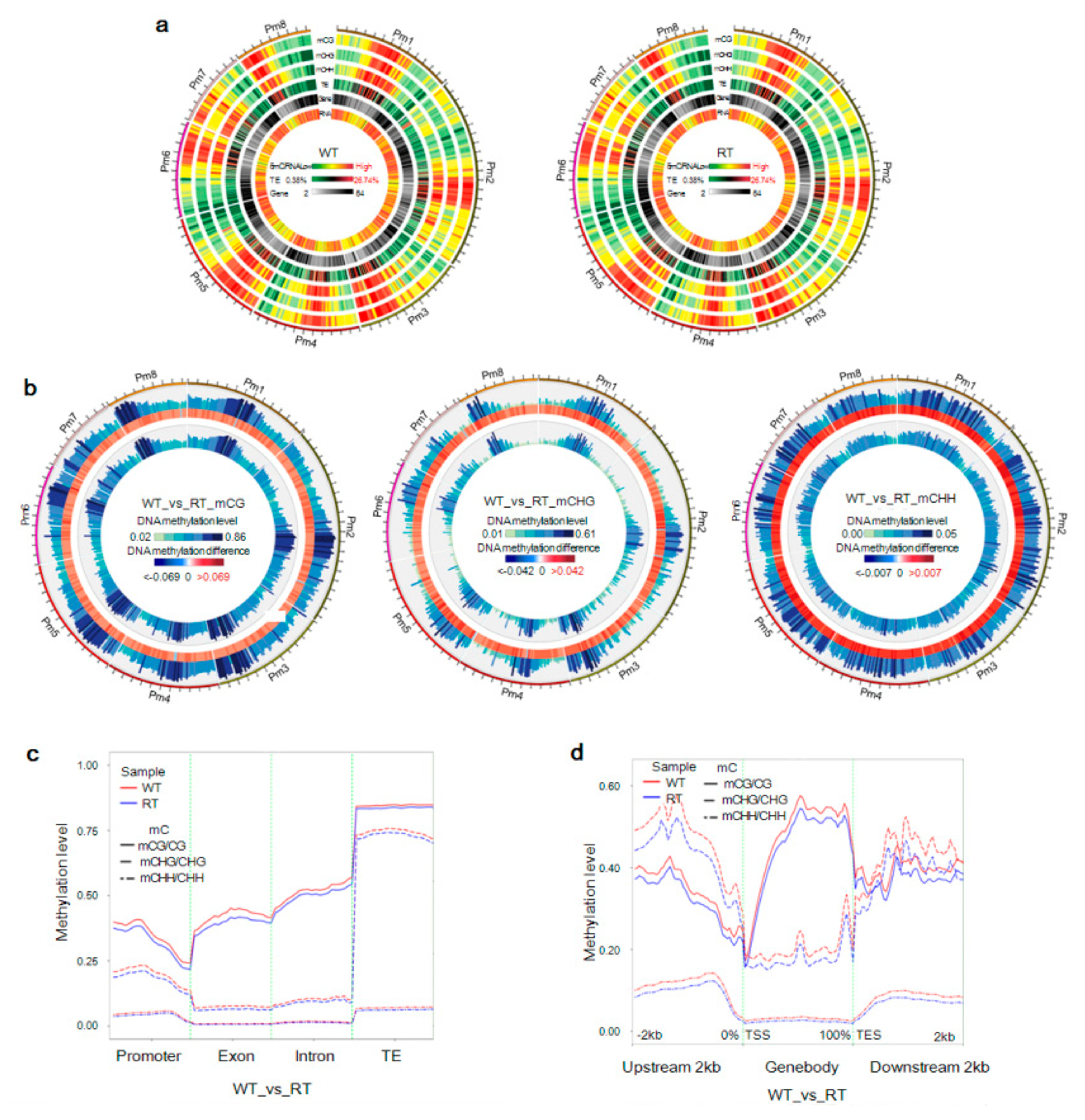
2.2. Genome Methylation Landscape of Prunus mume
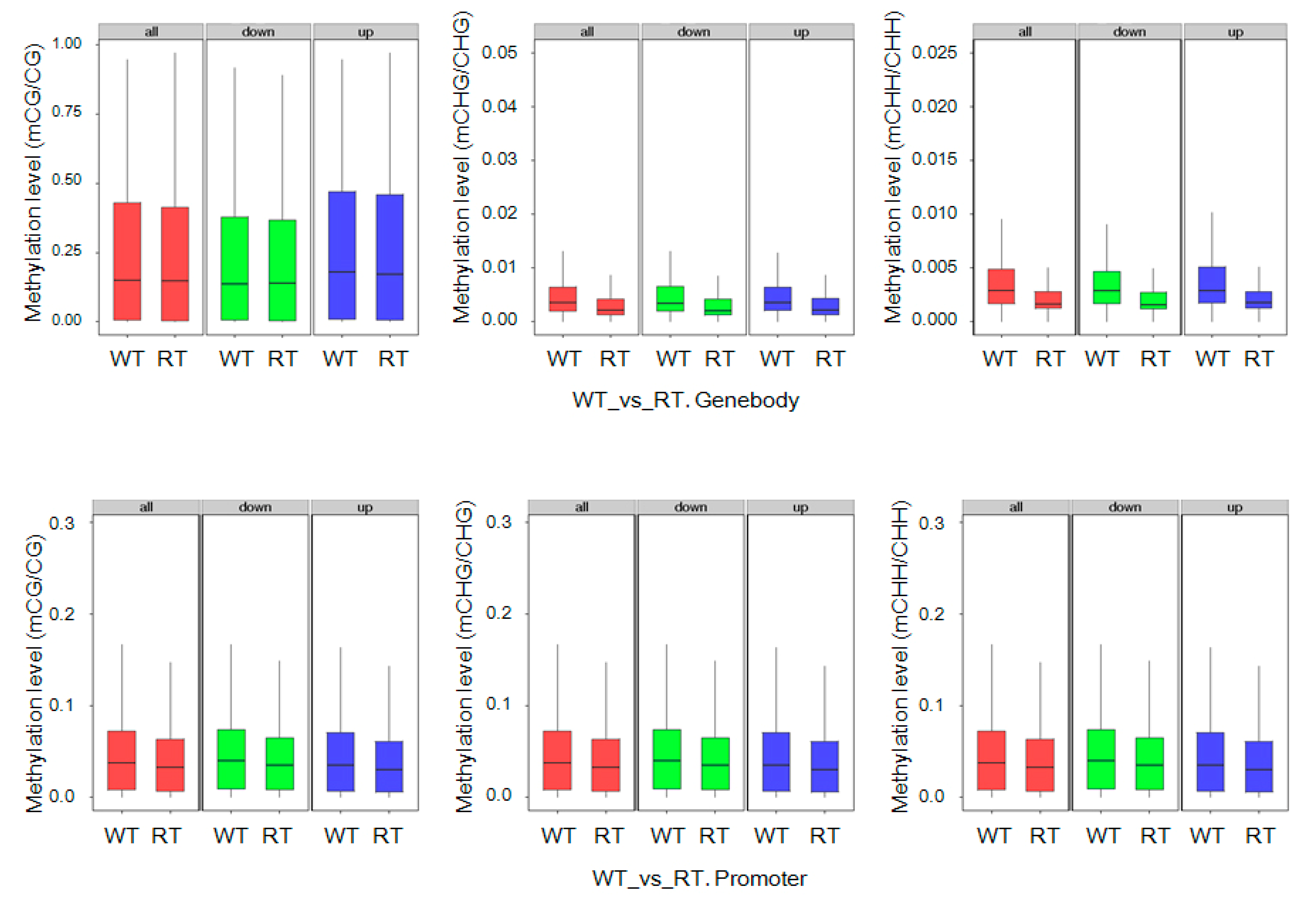
2.3. Levels of mC Variation in Different Colored Petal Tissues
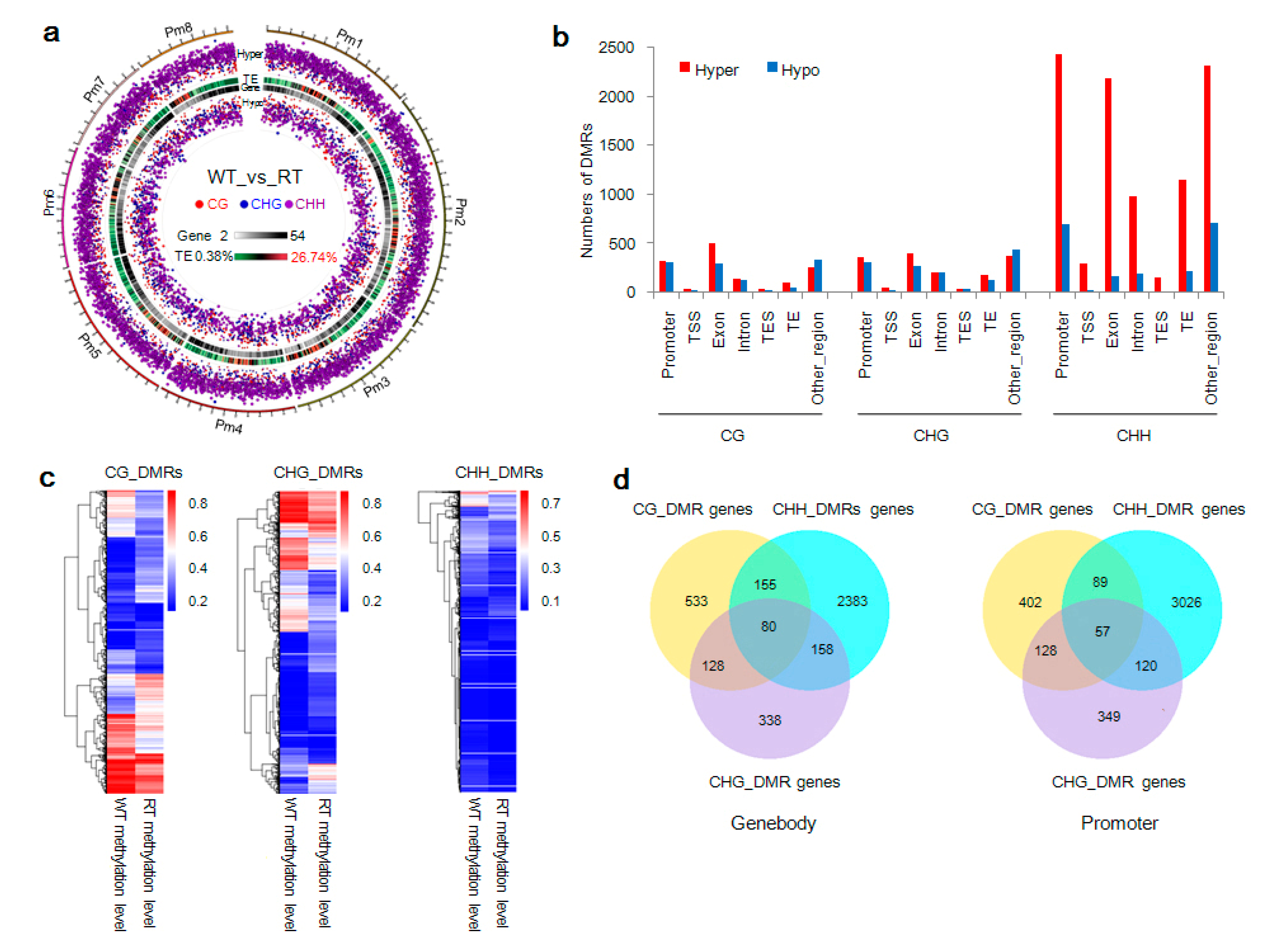
2.4. DMR-Related Genes and Bicolored Flowers on Individual Trees
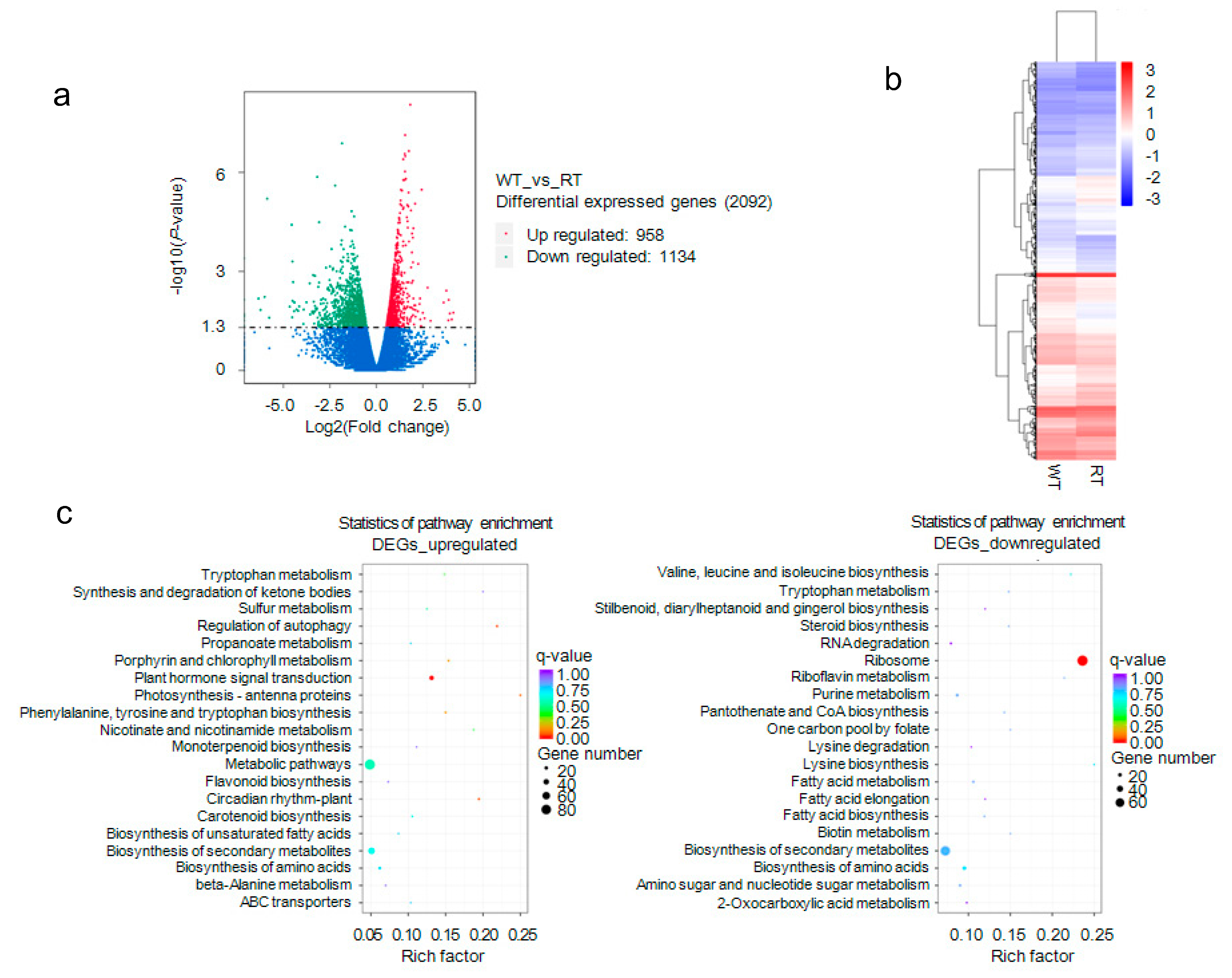
2.5. Analysis of Differential Transcription between WT and RT
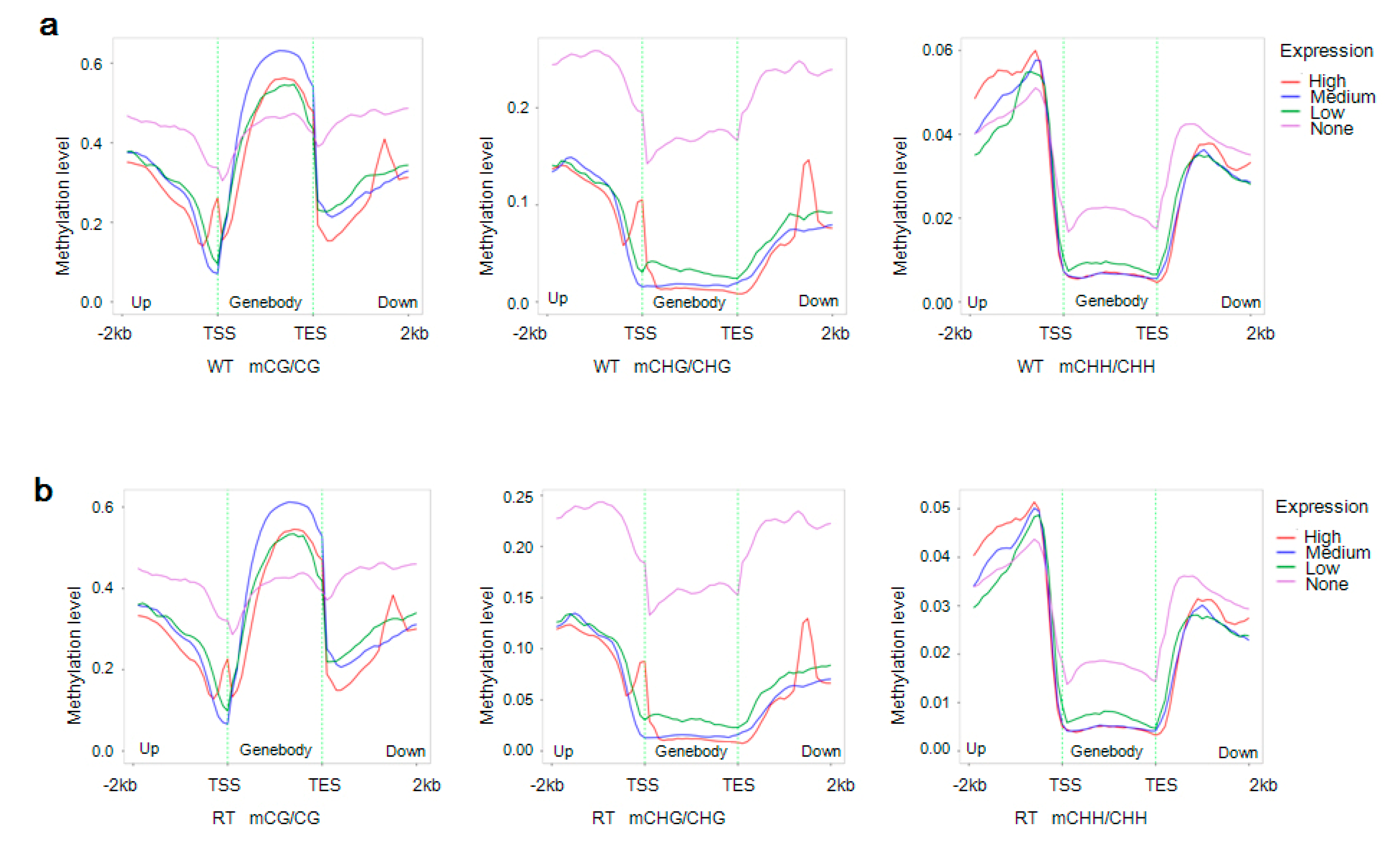
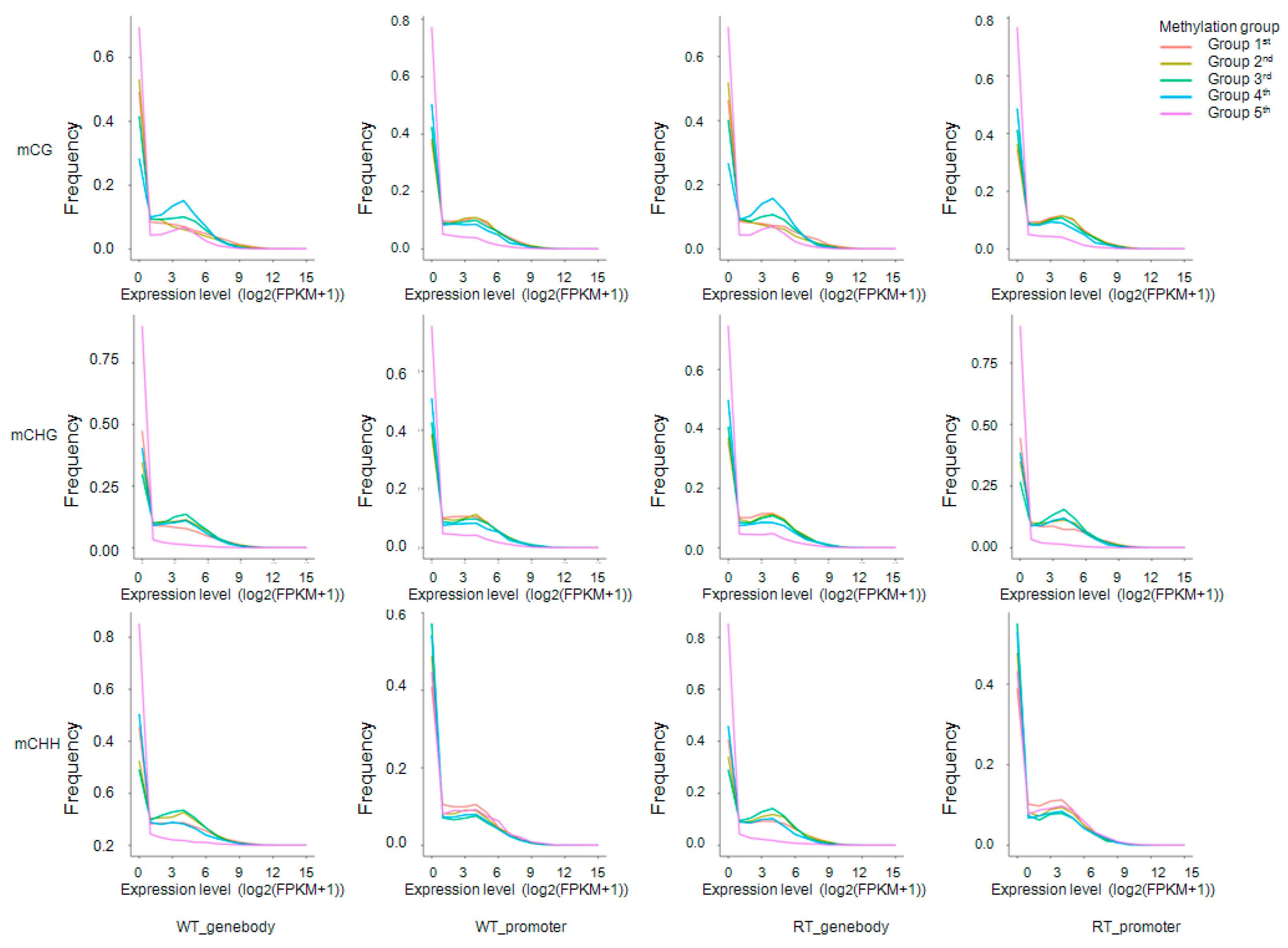
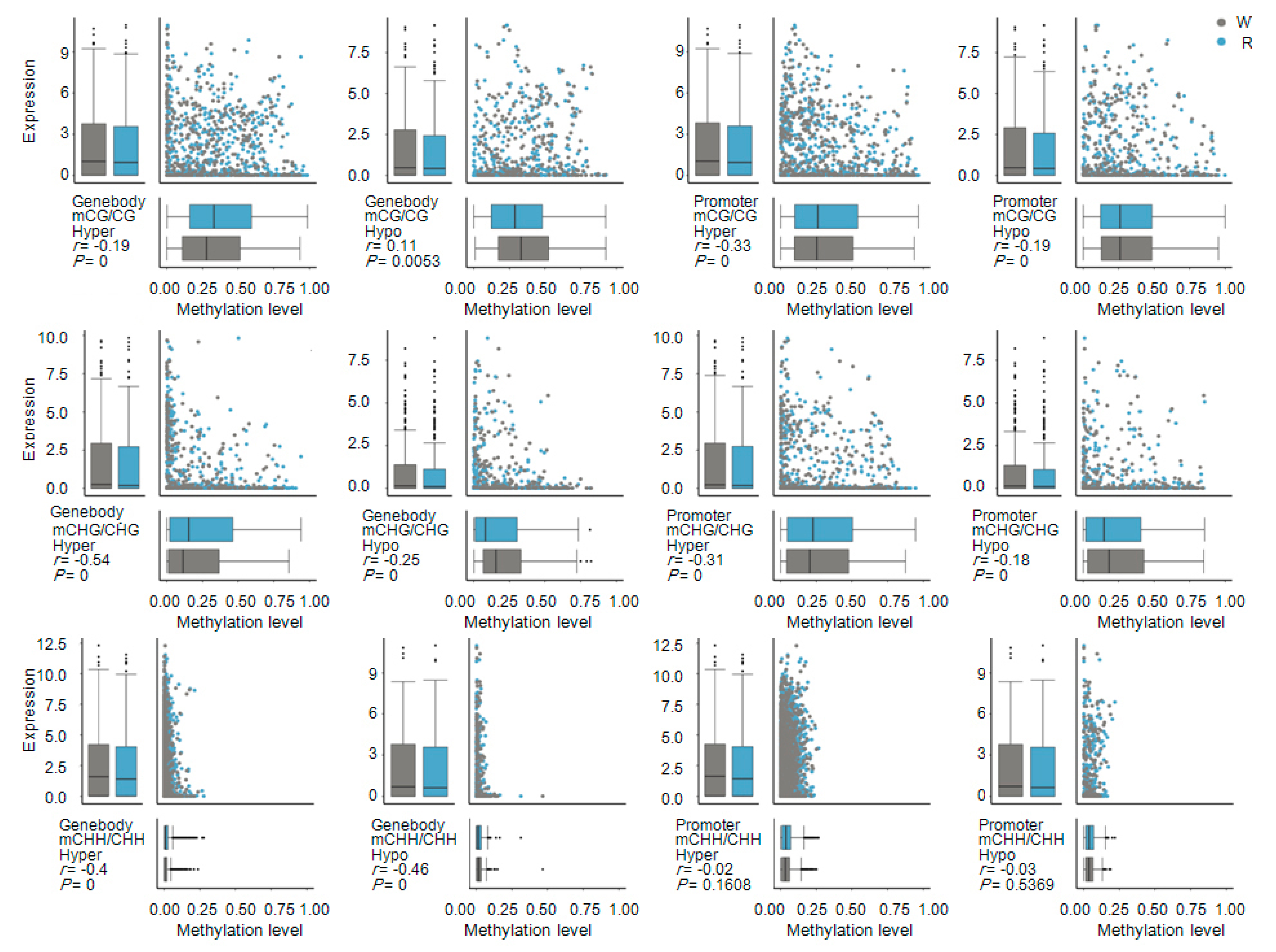
2.6. Correlation between Gene Expression Levels and DNA Methylation
2.7. DEGs with Methylation Modification and DMR-Related Gene Expression
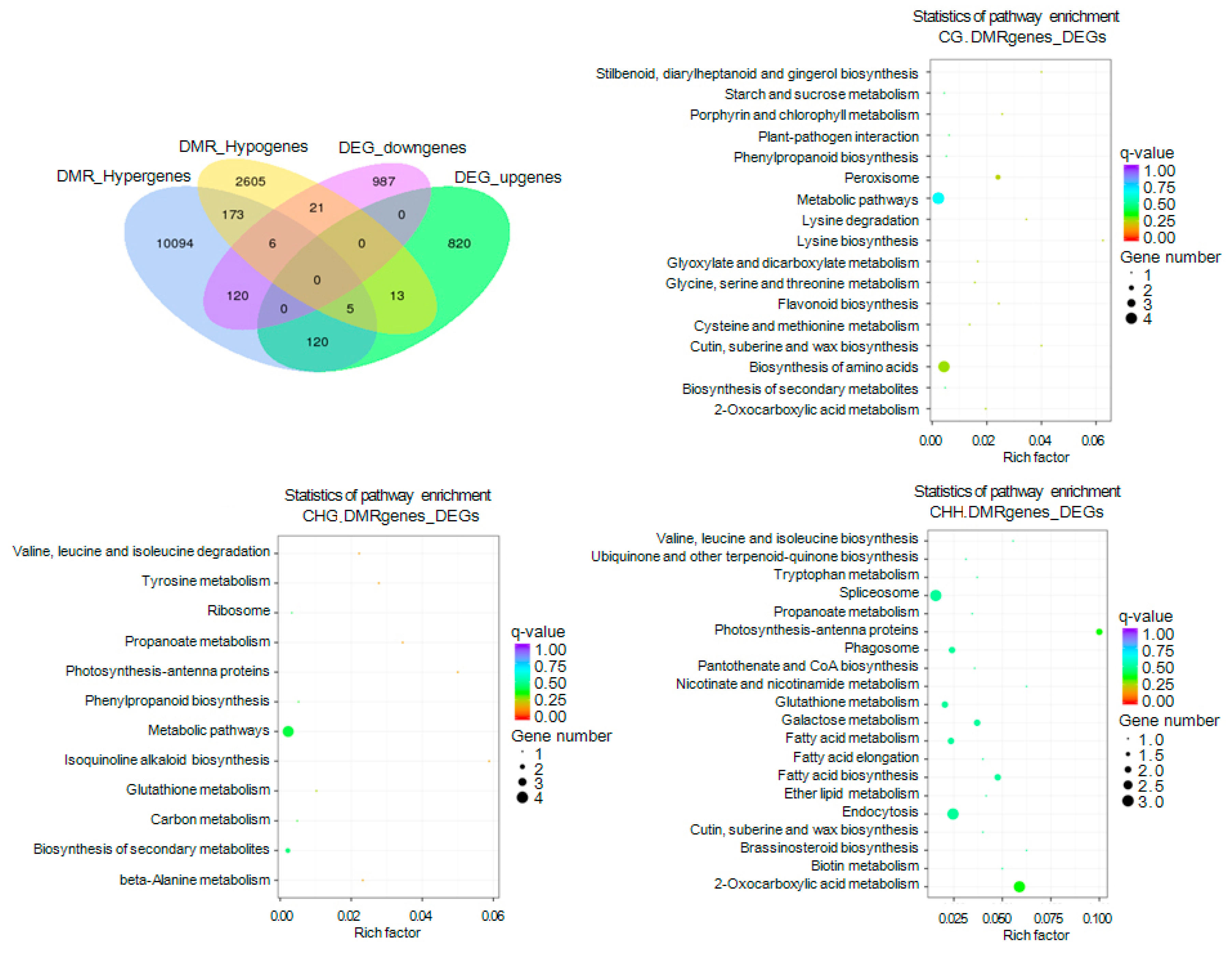
2.8. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) Enrichment of DMR-Related Genes Associated with DEGs
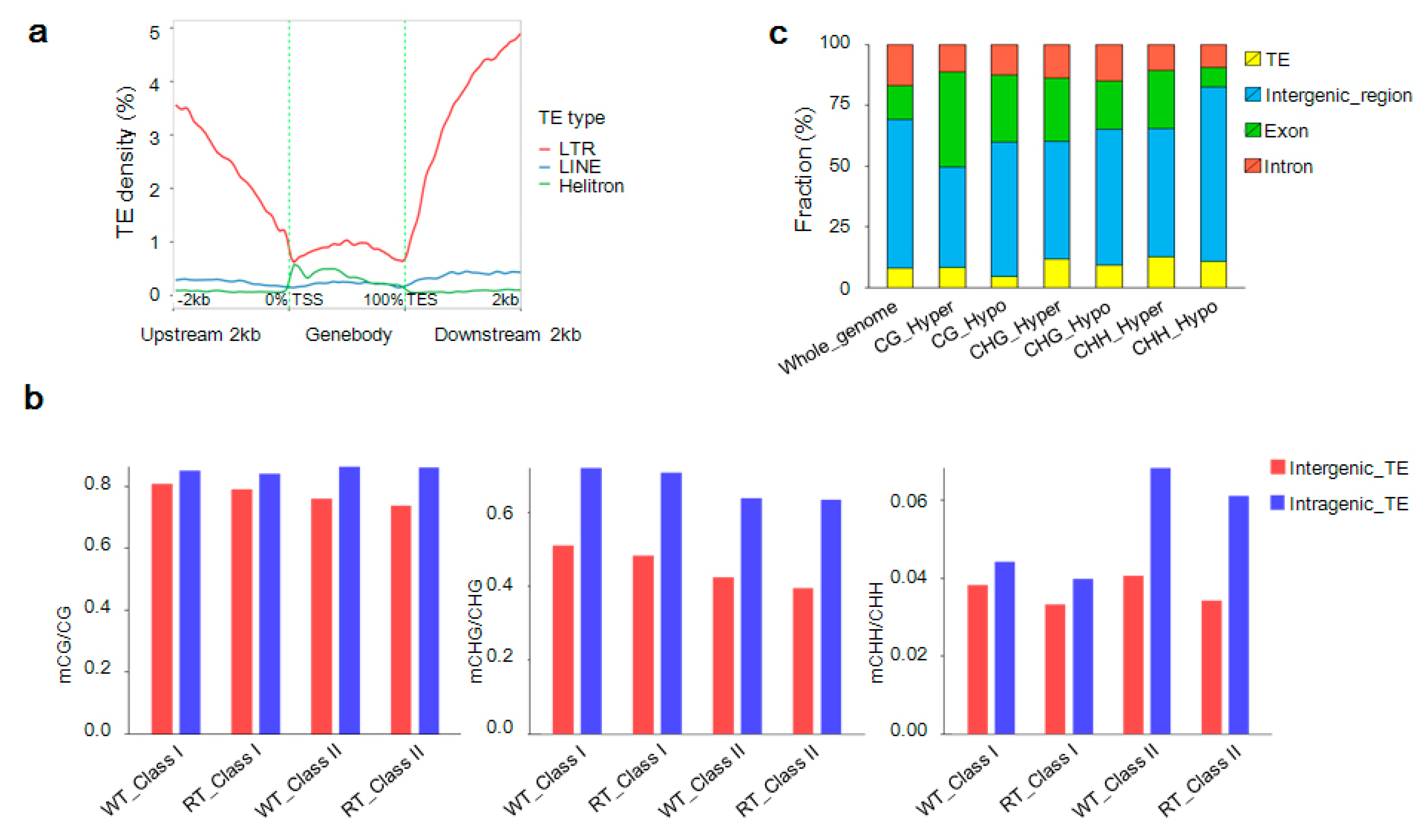
2.9. Detection of Methylated TEs Regulating Candidate Gene Expression
3. Discussion
3.1. Genes within Anthocyanin Regulation Pathway Were Differentially Expressed
3.2. Methylcytosine Modification Affected the Expression of Anthocyanin-Related Genes
3.3. TEs with Methylcytosine Affected the Expression of Anthocyanin-Related Genes
4. Materials and Methods
4.1. Plant Materials
4.2. Qualitative and Quantitative Analysis of Floral Pigments
4.3. Bisulfite Sequencing and DMR Analysis
4.3.1. Extraction of DNA and BS-Seq
4.3.2. Quality Assessment of Sequencing Data
4.3.3. Reference Genome Preparation and Mapping of Clean Reads
4.3.4. Evaluation of Methylation Level and Distribution
4.3.5. Correlation Analysis and DMR Detection
4.3.6. GO and KEGG Enrichment Analysis of DMR-Related Genes
4.4. Transcriptome Sequencing and Differentially Expressed Gene (DEG) Analysis
4.4.1. RNA Isolation and Sequencing
4.4.2. Mapping and DEG Analysis
4.5. Identification of Transcription Factors and TEs
4.6. Methylation Modification of Gene Expression
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Zhang, Q.X.; Chen, W.B.; Sun, L.D.; Zhao, F.Y.; Huang, B.Q.; Yang, W.R.; Tao, Y.; Wang, J.; Yuan, Z.Q.; Fan, G.Y.; et al. The genome of Prunus mume. Nat. Commun. 2012, 3, 1318. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.Y. Chinese Mei Flowers; Hainan Publishing House: Haikou, China, 1996; pp. 12–58. (In Chinese) [Google Scholar]
- Marcotrigiano, M. Chimeras and variegation: Patterns of deceit. Hortscience 1997, 32, 773–784. [Google Scholar]
- Suzuki, M.; Miyahara, T.; Tokumoto, H.; Hakamatsuka, T.; Goda, Y.; Ozeki, Y.; Nobuhiro Sasaki, N. Transposon-mediated mutation of CYP76AD3 affects betalain synthesis and produces variegated flowers in four o’clock (Mirabilis jalapa). J. Plant Physiol. 2014, 171, 1586–1590. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Chin, Y.; Lin, C.; Chen, P.; To, K. Transforming the snapdragon aurone biosynthetic genes into petunia alters coloration patterns in transgenic flowers. Adv. Biosci. Biotechnol. 2015, 6, 702–722. [Google Scholar] [CrossRef]
- Zhou, H.; Linwang, K.; Wang, H.L.; Gu, C.; Dare, A.P.; Epley, R.V.; He, H.P.; Allan, A.C.; Han, Y.P. Molecular genetics of blood-fleshed peach reveals activation of anthocyanin biosynthesis by NAC transcription factors. Plant J. 2015, 82, 105–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Gong, Q.; Ni, X.; Zhou, Y.; Gao, Z. UFGT: The key enzyme associated with the petals variegation in Japanese apricot. Front. Plant Sci. 2017, 8, 108. [Google Scholar] [CrossRef] [PubMed]
- McClintock, B. Chromosome organization and gene expression. Cold Spring Harb. Symp. Quant. Biol. 1951, 16, 13–47. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, Y.; Hisatomi, Y.; Iida, S. Somatic mutations caused by excision of the transposable element, Tpn1, from the DFR gene for pigmentation in sub-epidermal layer of periclinally chimeric flowers of Japanese morning glory and their germinal transmission to their progeny. Theor. Appl. Genet. 1996, 92, 499–504. [Google Scholar] [CrossRef] [PubMed]
- Abe, Y.; Hoshino, A.; Iida, S. Appearance of flower variegation in the mutable speckled line of the Japanese morning glory is controlled by two genetic elements. Genes Genet. Syst. 1997, 72, 57–62. [Google Scholar] [CrossRef]
- Iida, S.; Morita, Y.; Choi, J.; Park, K.; Hoshino, A. Genetics and epigenetics in flower pigmentation associated with transposable elements in morning glories. Adv. Biophys. 2004, 38, 141–159. [Google Scholar] [CrossRef]
- Fukada-Tanaka, S.; Inagaki, Y.; Yamaguchi, T.; Saito, N.; Iida, S. Colour-enhancing protein in blue petals. Nature 2000, 407, 581. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Fukadatanaka, S.; Inagaki, Y.; Saito, N.; Yonekurasakakibara, K.; Tanaka, Y.; Kusumi, T.; Iida, S. Genes en-coding the vacuolar Na+/H+ exchanger and flower coloration. Plant Cell Physiol. 2001, 42, 451–461. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Galli, M.; Crawford, N.M. Engineering variegated floral patterns in tobacco plants using the Arabidopsis transposable element Tag1. Plant Cell Physiol. 2001, 42, 419–423. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Goto-Yamamoto, N.; Hirochika, H. Retrotransposon-induced mutations in grape skin color. Science 2004, 304, 982. [Google Scholar] [CrossRef] [PubMed]
- Zabala, G.; Vodkin, L. A putative autonomous 20.5 kb-CACTA transposon insertion in an F3'H allele identifies a new CACTA transposon subfamily in Glycine max. BMC Plant Biol. 2008, 8, 124. [Google Scholar] [CrossRef] [PubMed]
- Nishizaki, Y.; Matsuba, Y.; Okamoto, E.; Okamura, M.; Yoshihiro Ozeki, Y.; Sasaki, N. Structure of the acyl-glucose-dependent anthocyanin 5-O-glucosyltransferase gene in carnations and its disruption by transposable elements in some varieties. Mol. Genet. Genom. 2011, 286, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Fukada-Tanaka, S.; Hoshino, A.; Hisatomi, Y.; Habu, Y.; Hasebe, M.; Iida, S. Identification of new chalcone synthase genes for flower pigmentation in the Japanese and common morning glories. Plant Cell Physiol. 1997, 38, 754–758. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.N.; Mao, Y.; Liu, H.L.; Yu, F.X.; Li, S.X.; Yin, T.M. Transcriptome analysis of differentially expressed genes relevant to variegation in peach flowers. PLoS ONE 2014, 9, e90842. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.G.; Jiang, W.; Mantri, N.; Bao, X.Q.; Chen, S.L.; Tao, Z.M. Transciptome analysis reveals flavonoid biosynthesis regulation and simple sequence repeats in yam (Dioscorea alata L.) tubers. BMC Genom. 2015, 16, 346. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cheng, Y.; Ya, H.; Xu, S.; Han, J. Transcriptome sequencing of purple petal spot region in tree peony reveals differentially expressed anthocyanin structural genes. Front Plant Sci. 2015, 8, 964. [Google Scholar] [CrossRef] [PubMed]
- Hassani, D.; Liu, H.L.; Chen, Y.N.; Wan, Z.B.; Zhuge, Q.; Li, S.X. Analysis of biochemical compounds and differentially expressed genes of the anthocyanin biosynthetic pathway in variegated peach flowers. Genet. Mol. Res. 2015, 14, 13425–13436. [Google Scholar] [CrossRef] [PubMed]
- Koseki, M.; Goto, K.; Masuta, C.; Kanazawa, A. The star-type color pattern in Petunia hybrida ‘Red Star’ flowers is induced by sequence-specific degradation of Chalcone Synthase RNA. Plant Cell Physiol. 2005, 46, 1879–1883. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Liao, L.; Zhou, H.; Gu, C.; Wang, L.; Han, Y.P. A small indel mutation in an anthocyanin transporter causes variegated colouration of peach flowers. J. Exp. Bot. 2015, 66, 7227–7239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uematsu, C.; Katayama, H.; Makino, I.; Inagaki, A.; Arakawa, O.; Martin, C. Peace, a MYB-like transcription factor, regulates petal pigmentation in flowering peach ‘Genpei’ bearing variegated and fully pigmented flowers. J. Exp. Bot. 2014, 65, 1081–1094. [Google Scholar] [CrossRef] [PubMed]
- Meyer, P.; Heidmann, I.; Niedenhof, I. Differences in DNA-methylation are associated with a paramutation phenomenon in transgenic petunia. Plant J. 1993, 4, 89–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.J.; Chuang, Y.N.; Chiou, C.Y.; Chin, D.C.; Shen, F.Q.; Yeh, K.W. Methylation effect on chalcone synthase gene expression determines anthocyanin pigmentation in floral tissues of two Oncidium orchid cultivars. Planta 2012, 236, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Takai, D. The role of DNA methylation in mammalian epigenetics. Science 2001, 293, 1068–1070. [Google Scholar] [CrossRef] [PubMed]
- Reik, W.; Dean, W.; Walter, J. Epigenetic reprogramming in mammalian development. Science 2001, 293, 1089–1093. [Google Scholar] [CrossRef] [PubMed]
- Reik, W.; Santos, F.; Mitsuya, K.; Morgan, H.P.; Dean, W. Epigenetic asymmetry in the mammalian zygote and early embryo: Relationship to lineage commitment? Philos. Trans. R. Soc. B 2003, 358, 1403–1409. [Google Scholar] [CrossRef] [PubMed]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bocchini, M.; Galla, G.; Pupilli, F. The vesicle traffcking regulator PN_SCD1 is demethylated and overexpressed in florets of apomictic Paspalum notatum genotypes. Sci. Rep. 2018, 8, 3030. [Google Scholar] [CrossRef] [PubMed]
- Marfil, C.F.; Camadro, E.L.; Masuelli, R.W. Phenotypic instability and epigenetic variability in a diploid potato of hybrid origin, Solanum ruiz-lealii. BMC Plant Biol. 2009, 9, 21. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.F.; Song, Y.P.; Jiang, X.B.; Zhang, Z.Y.; Li, B.L.; Zhang, D.Q. Photosynthetic response to genome methylation affects the growth of Chinese white poplar. Tree Genet. Genomes 2012, 8, 1407–1421. [Google Scholar] [CrossRef]
- Cubas, P.; Vincent, C.; Coen, E. An epigenetic mutation responsible for natural variation in floral symmetry. Nature 1999, 401, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Telias, A.; Linwang, K.; Stevenson, D.E.; Cooney, J.M.; Hellens, R.P.; Allan, A.C.; Hoover, E.; Bradeen, J.M. Apple skin patterning is associated with differential expression of MYB10. BMC Plant Biol. 2011, 11, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.G.; Meng, D.; Wang, A.D.; Li, T.L.; Jiang, S.L.; Cong, P.H.; Li, T.Z. The methylation of the PcMYB10 promoter is associated with green-skinned sport in Max red bartlett pear. Plant Physiol. 2013, 162, 885–896. [Google Scholar] [CrossRef] [PubMed]
- Bai, S.L.; Tuan, P.A.; Saito, T.; Honda, C.; Hatsuyama, Y.; Ito, A.; Moriguchi, T. Epigenetic regulation of MdMYB1 is associated with paper bagging-induced red pigmentation of apples. Planta 2016, 244, 573–586. [Google Scholar] [CrossRef] [PubMed]
- Hoekenga, O.A.; Muszynski, M.G.; Cone, K.C. Developmental patterns of chromatin structure and DNA methylation responsible for epigenetic expression of a maize regulatory gene. Genetics 2000, 155, 1889–1902. [Google Scholar] [PubMed]
- Kankel, M.W.; Ramsey, D.E.; Stokes, T.L.; Flowers, S.K.; Haag, J.R.; Jeddeloh, J.A.; Riddle, N.C.; Verbsky, M.L.; Richards, E.J. Arabidopsis MET1 cytosine methyltransferase mutants. Genetics 2003, 163, 1109–1122. [Google Scholar] [PubMed]
- Rudenko, G.N.; Ono, A.; Walbot, V. Initiation of silencing of maize MuDR/Mu transposable elements. Plant J. 2003, 33, 1013–1025. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.W.; Henderson, I.R.; Jacobsen, S.E. Gardening the genome: DNA methylation in Arabidopsis thaliana. Nat. Rev. Genet. 2005, 6, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Zhong, S.L.; Fei, Z.J.; Chen, Y.R.; Zheng, Y.; Huang, M.Y.; Vrebalov, J.; Mcquinn, R.; Gapper, N.E.; Liu, B.; Xiang, J.; et al. Single-base resolution methylomes of tomato fruit development reveal epigenome modifications associated with ripening. Nat. Biotechnol. 2013, 31, 154–159. [Google Scholar] [CrossRef] [PubMed]
- An, Y.C.; Goettel, W.; Han, Q.; Bartels, A.; Liu, Z.R.; Xiao, W.Y. Dynamic changes of genome-wide DNA methylation during soybean seed development. Sci. Rep. 2017, 7, 12263. [Google Scholar] [CrossRef] [PubMed]
- Song, Q.; Zhang, T.; Stelly, D.M.; Chen, Z.J. Epigenomic and functional analyses reveal roles for epialleles in the loss of photoperiod sensitivity during domestication of allotetraploid cottons. Genome Biol. 2017, 18, 99. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.S.; Qin, Q.; Sun, F.; Wang, Y.X.; Xu, D.D.; Li, Z.K.; Fu, B.Y. Genome-wide differences in DNA methylation changes in two contrasting rice genotypes in response to drought conditions. Front. Plant Sci. 2016, 7, 1675. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.K.; Wang, X.G.; Chen, X.G.; Shu, N.; Wang, J.J.; Wang, D.L.; Wang, S.; Fan, W.L.; Guo, L.X.; Guo, X.N.; et al. Single-base resolution methylomes of upland cotton (Gossypium hirsutum L.) reveal epigenome modifications in response to drought stress. BMC Genom. 2017, 18, 297. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.D.; Zhou, S.S.; Gong, X.Q.; Song, Y.; Van Nocker, S.; Ma, F.W.; Guan, Q.M. Single-base methylome analysis reveals dynamic epigenomic differences associated with water deficit in apple. Plant Biotechnol. J. 2018, 16, 672–687. [Google Scholar] [CrossRef] [PubMed]
- Bock, C. Analysing and interpreting DNA methylation data. Nat. Rev. Genet. 2012, 13, 705–719. [Google Scholar] [CrossRef] [PubMed]
- Meissner, A.; Mikkelsen, T.S.; Gu, H.C.; Wernig, M.; Hanna, J.; Sivachenko, A.; Zhang, X.L.; Bernstein, B.E.; Nusbaum, C.; Jaffe, D.B.; et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 2008, 454, 766–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doi, A.; Park, I.; Wen, B.; Murakami, P.; Aryee, M.J.; Irizarry, R.; Herb, B.R.; Ladd-Acosta, C.; Rho, J.; Loewer, S.; et al. Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nat. Genet. 2009, 41, 1350–1353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Wu, X.; Zhang, Z.; Gao, Z. Comparative proteomic analysis of floral color variegation in peach. Biochem. Biophys. Res. Commun. 2015, 464, 1101–1106. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Suzuki, T.; Nakatsuka, T.; Dohra, H.; Yamagishi, M.; Matsuyama, K.; Matsuura, H. RNA-seq-based evaluation of bicolor tepal pigmentation in Asiatic hybrid lilies (Lilium spp.). BMC Genom. 2016, 17, 611. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Vimolmangkang, S.; Soria-Guerra, R.E.; Korban, S.S. Introduction of apple ANR genes into tobacco inhibits expression of both CHI and DFR genes in flowers, leading to loss of anthocyanin. J. Exp. Bot. 2012, 63, 2437–2447. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Li, H.; Huang, J. Arabidopsis TT19 functions as a carrier to transport anthocyanin from the cytosol to tonoplasts. Mol. Plant 2012, 5, 387–400. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Peng, Q.; Zhao, J.B.; Owiti, A.; Ren, F.; Liao, L.; Wang, L.; Deng, X.B.; Jiang, Q.; Han, Y.P. Multiple R2R3-MYB transcription factors involved in the regulation of anthocyanin accumulation in peach flower. Front. Plant Sci. 2016, 7, 1557. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhou, H.; Linwang, K.; Vimolmangkang, S.; Espley, R.V.; Wang, L.; Allan, A.C.; Han, Y.P. Transcriptome analysis and transient transformation suggest an ancient duplicated MYB transcription factor as a candidate gene for leaf red coloration in peach. BMC Plant Biol. 2014, 14, 388. [Google Scholar] [CrossRef] [PubMed]
- Qi, T.C.; Song, S.S.; Ren, Q.C.; Wu, D.W.; Huang, H.; Chen, Y.; Fan, M.; Peng, W.; Ren, C.M.; Xie, D.X. The Jasmonate-ZIM-Domain proteins interact with the WD-Repeat/bHLH/MYB complexes to regulate jasmonate-mediated anthocyanin accumulation and trichome initiation in Arabidopsis thaliana. Plant Cell 2011, 23, 1795–1814. [Google Scholar] [CrossRef] [PubMed]
- Zilberman, D.; Gehring, M.; Tran, R.K.; Ballinger, T.; Henikoff, S. Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription. Nat. Genet. 2007, 39, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.Y.; Yazaki, J.; Sundaresan, A.; Cokus, S.J.; Chan, S.W.L.; Chen, H.M.; Henderson, I.R.; Shinn, P.; Pellegrini, M.; Jacobsen, S. Genome-wide high-resolution mapping and functional analysis of DNA methylation in Arabidopsis. Cell 2006, 126, 1189–1201. [Google Scholar] [CrossRef] [PubMed]
- Madlung, A.; Comai, L. The effect of stress on genome regulation and structure. Ann. Bot. 2004, 94, 481–495. [Google Scholar] [CrossRef] [PubMed]
- Song, Q.; Decato, B.; Hong, E.E.; Zhou, M.; Fang, F.; Qu, J.H.; Garvin, T.; Kessler, M.; Zhou, J.; Smith, A.D. A reference methylome database and analysis pipeline to facilitate integrative and comparative epigenomics. PLoS ONE 2013, 8, e81148. [Google Scholar] [CrossRef] [PubMed]
- Cokus, S.J.; Feng, S.H.; Zhang, X.Y.; Chen, Z.G.; Merriman, B.; Haudenschild, C.D.; Pradhan, S.; Nelson, S.F.; Pellegrini, M.; Jacobsen, S.E. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 2008, 452, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.H.; Cokus, S.J.; Zhang, X.Y.; Chen, P.Y.; Bostick, M.; Goll, M.G.; Hetzel, J.; Jain, J.; Strauss, S.H.; Halpern, M.E.; et al. Conservation and divergence of methylation patterning in plants and animals. Proc. Natl. Acad. Sci. USA 2010, 107, 8689–8694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chodavarapu, R.K.; Feng, S.H.; Ding, B.; Simon, S.A.; Lopez, D.; Jia, Y.L.; Wang, G.L.; Meyers, B.C.; Jacobsen, S.E.; Pellegrini, M. Transcriptome and methylome interactions in rice hybrids. Proc. Natl. Acad. Sci. USA 2012, 109, 12040–12045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gent, J.I.; Ellis, N.A.; Guo, L.; Harkess, A.; Yao, Y.Y.; Zhang, X.Y.; Dawe, R.K. CHH islands: De novo DNA methylation in near-gene chromatin regulation in maize. Genome Res. 2013, 23, 628–637. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, R.J.; He, Y.P.; Valdeslopez, O.; Khan, S.M.; Joshi, T.; Urich, M.A.; Nery, J.R.; Diers, B.W.; Xu, D.; Stacey, G.; et al. Epigenome-wide inheritance of cytosine methylation variants in a recombinant inbred population. Genome Res. 2013, 23, 1663–1674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.D.; Baidouri, M.E.; Abernathy, B.; Iwataotsubo, A.; Chavarro, C.; Gonzales, M.; Libault, M.; Grimwood, J.; Jackson, S.A. A comparative epigenomic analysis of polyploidy-derived genes in soybean and common bean. Plant Physiol. 2015, 168, 1433–1447. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Y.; Wang, X.F.; He, K.; Ma, Y.Q.; Su, N.; He, H.; Stolc, V.; Tongprasit, W.; Jin, W.W.; Jiang, J.M.; et al. High-resolution mapping of epigenetic modifications of the rice genome uncovers interplay between DNA methylation, histone methylation, and gene expression. Plant Cell 2008, 20, 259–276. [Google Scholar] [CrossRef] [PubMed]
- Miura, A.; Nakamura, M.; Inagaki, S.; Kobayashi, A.; Saze, H.; Kakutani, T. An ArabidopsisjmjC domain protein protects transcribed genes from DNA methylation at CHG sites. EMBO J. 2009, 28, 1078–1086. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Marowsky, N.C.; Fan, C. Divergence of gene body DNA methylation and evolution of plant duplicate genes. PLoS ONE 2014, 9, e110357. [Google Scholar] [CrossRef] [PubMed]
- Song, Q.X.; Lu, X.; Li, Q.T.; Chen, H.; Hu, X.Y.; Ma, B.; Zhang, W.K.; Chen, S.Y.; Zhang, J. Genome-wide analysis of DNA methylation in soybean. Mol. Plant 2013, 6, 1961–1974. [Google Scholar] [CrossRef] [PubMed]
- Chopra, S.; Brendel, V.; Zhang, J.; Axtell, J.D.; Peterson, T. Molecular characterization of a mutable pigmentation phenotype and isolation of the first active transposable element from Sorghum bicolor. Proc. Natl. Acad. Sci. USA 1999, 96, 15330–15335. [Google Scholar] [CrossRef] [PubMed]
- Feschotte, C.; Jiang, N.; Wessler, S.R. Plant transposable elements: Where genetics meets genomics. Nat. Rev. Genet. 2002, 3, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Chen, L.; Hong, M.Y.; Zhang, Y.; Zu, F.; Wen, J.; Yi, B.; Ma, C.Z.; Shen, J.X.; Tu, J.X.; et al. A large insertion in bHLH transcription factor BrTT8 resulting in yellow seed coat in Brassica rapa. PLoS ONE 2012, 7, e44145. [Google Scholar] [CrossRef] [PubMed]
- Hong, L.L.; Qian, Q.; Tang, D.; Wang, K.J.; Li, M.; Cheng, Z.K. A mutation in the rice chalcone isomerase gene causes the golden hull and internode 1 phenotype. Planta 2012, 236, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Clegg, M.T.; Durbin, M.L. Tracing floral adaptations from ecology to molecules. Nat. Rev. Genet. 2003, 4, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Zabala, G.; Vodkin, L. The wp mutation of Glycine max carries a gene-fragment-rich transposon of the CACTA superfamily. Plant Cell 2005, 17, 2619–2632. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Hoshino, A.; Park, K.; Park, I.; Iida, S. Spontaneous mutations caused by a Helitron transposon, Hel-It1, in morning glory, Ipomoea tricolor. Plant J. 2007, 49, 924–934. [Google Scholar] [CrossRef] [PubMed]
- Park, K.; Ishikawa, N.; Morita, Y.; Choi, J.; Hoshino, A.; Iida, S. A bHLH regulatory gene in the common morning glory, Ipomoea purpurea, controls anthocyanin biosynthesis in flowers, proanthocyanidin and phytomelanin pigmentation in seeds, and seed trichome formation. Plant J. 2007, 49, 641–654. [Google Scholar] [CrossRef] [PubMed]
- Mirouze, M.; Vitte, C. Transposable elements, a treasure trove to decipher epigenetic variation: Insights from Arabidopsis and crop epigenomes. J. Exp. Bot. 2014, 65, 2801–2812. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.D.; Baidouri, M.E.; Jackson, S.A. Accessing epigenetic variation in the plant methylome. Brief. Funct. Genom. 2014, 13, 318–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujimoto, R.; Kinoshita, Y.; Kawabe, A.; Kinoshita, T.; Takashima, K.; Nordborg, M.; Nasrallah, M.E.; Shimizu, K.K.; Kudoh, H.; Kakutani, T. Evolution and control of imprinted FWA Genes in the genus Arabidopsis. PLoS Genet. 2008, 4, e1000048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsugane, K.; Maekawa, M.; Takagi, K.; Takahara, H.; Qian, Q.; Eun, C.H.; Iida, S. An active DNA transposon nDart causing leaf variegation and mutable dwarfism and its related elements in rice. Plant J. 2006, 45, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, F.; Tanaka, M.; Maeda, H.; Shimizu, K.; Sakata, Y. Characterization of cyanic flower color of Delphinium cultivars. J. Jpn. Soc. Hortic. Sci. 2000, 69, 428–434. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, L.S.; Gao, J.M.; Xu, Y.J.; Li, L.F.; Li, C.H. Rapid separation and identification of anthocyanins from flowers of Viola yedoensis and V. prionantha by high-performance liquid chromatography-photodiode array detection-electrospray ionisationmass spectrometry. Phytochem. Anal. 2012, 23, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Krueger, F.; Andrews, S. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, J.; Duan, J.L.; Gao, X.X.; Zhu, W.; Lu, X.Y.; Yang, L.; Zhang, J.; Li, G.Q.; Ci, W.M.; et al. Programming and inheritance of parental DNA methylomes in mammals. Cell 2014, 157, 979–991. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Mukamel, E.A.; Nery, J.R.; Urich, M.A.; Puddifoot, C.A.; Johnson, N.D.; Lucero, J.; Huang, Y.; Dwork, A.J.; Schultz, M.D.; et al. Global epigenomic reconfiguration during mammalian brain development. Science 2013, 341, 1237905. [Google Scholar] [CrossRef] [PubMed]
- Krzywinski, M.; Schein, J.E.; Birol, I.; Connors, J.M.; Gascoyne, R.D.; Horsman, D.; Jones, S.J.M.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tontifilippini, J.; Nery, J.R.; Lee, L.K.; Ye, Z.; Ngo, Q.; et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smallwood, S.A.; Lee, H.J.; Angermueller, C.; Krueger, F.; Saadeh, H.; Peat, J.R.; Andrews, S.; Stegle, O.; Reik, W.; Kelsey, G. Single-cell genome-wide bisulfite sequencing for assessing epigenetic heterogeneity. Nat. Methods 2014, 11, 817–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, H.; Conneely, K.N.; Wu, H.A. Bayesian hierarchical model to detect differentially methylated loci from single nucleotide resolution sequencing data. Nucleic Acids Res. 2014, 42, e69. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Xu, T.L.; Feng, H.; Chen, L.; Li, B.; Yao, B.; Qin, Z.H.; Jin, P.; Conneely, K.N. Detection of differentially methylated regions from whole-genome bisulfite sequencing data without replicates. Nucleic Acids Res. 2015, 43, e141. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.; Wu, H. Differential methylation analysis for BS-seq data under general experimental design. Bioinformatics 2016, 32, 1446–1453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2007, 36, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; Van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential Expression of RNA-Seq Data at the Gene Level-the DESeq Package; European Molecular Biology Laboratory (EMBL): Heidelberg, Germany, 2012. [Google Scholar]
- Pérez-Rodríguez, P.; Rianopachon, D.M.; Correa, L.G.G.; Rensing, S.A.; Kersten, B.; Muellerroeber, B. PlnTFDB: Updated content and new features of the plant transcription factor database. Nucleic Acids Res. 2010, 38, 822–827. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Zhang, H.; Kong, L.; Gao, G.; Luo, J. PlantTFDB 3.0: A portal for the functional and evolutionary study of plant transcription factors. Nucleic Acids Res. 2014, 42, 1182–1187. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Jiao, C.; Sun, H.; Rosli, H.G.; Pombo, M.A.; Zhang, P.; Banf, M.; Dai, X.; Martin, G.B.; Giovannoni, J.J.; et al. iTAK: A program for genome-wide prediction and classification of plant transcription factors, transcriptional regulators, and protein kinases. Mol. Plant 2016, 9, 1667–1670. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.J.; Yuan, D.J.; Tu, L.L.; Gao, W.H.; He, Y.H.; Hu, H.Y.; Wang, P.C.; Liu, N.; Lindsey, K.; Zhang, X.L. Long noncoding RNAs and their proposed functions in fibre development of cotton (Gossypium spp.). New Phytol. 2015, 207, 1181–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, C.W.; Yildirim, F.; Yap, Y.S.; Dalin, S.; Matthews, B.J.; Velez, P.J.; Labadorf, A.; Housman, D.E.; Fraenkel, E. Extensive changes in DNA methylation are associated with expression of mutant huntingtin. Proc. Natl. Acad. Sci. USA 2013, 110, 2354–2359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.R.; Zhang, F.F.; Ma, Z.Y.; Huang, H.W.; Jiang, L.; Cai, T.; Zhu, J.K.; Zhang, C.Y.; He, X.J. Folate polyglutamylation is involved in chromatin silencing by maintaining global DNA methylation and histone H3K9 dimethylation in Arabidopsis. Plant Cell 2013, 25, 2545–2559. [Google Scholar] [CrossRef] [PubMed]
- Yang, I.V.; Pedersen, B.S.; Rabinovich, E.I.; Hennessy, C.E.; Davidson, E.J.; Murphy, E.; Guardela, B.J.; Tedrow, J.; Zhang, Y.Z.; Singh, M.K.; et al. Relationship of DNA methylation and gene expression in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2014, 190, 1263–1272. [Google Scholar] [CrossRef] [PubMed]
- Kretzmer, H.; Bernhart, S.H.; Wang, W.; Haake, A.; Weniger, M.A.; Bergmann, A.K.; Betts, M.J.; Carrillodesantapau, E.; Doose, G.; Gutwein, J.; et al. DNA methylome analysis in Burkitt and follicular lymphomas identifies differentially methylated regions linked to somatic mutation and transcriptional control. Nat. Genet. 2015, 47, 1316–1325. [Google Scholar] [CrossRef] [PubMed] [Green Version]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C Site | mC (mC/C) | CG | mCG (mCG/CG) | CHG | mCHG (mCHG/CHG) | CHH | mCHH (mCHH/CHH) | |
|---|---|---|---|---|---|---|---|---|
| WT1 | 82368087 | 9302812 11.29% | 8335974 | 3193016 38.30% | 11664623 | 2275905 19.51% | 62367490 | 3833891 6.14% |
| WT2 | 82368087 | 11706910 14.21% | 8335974 | 3153580 37.83% | 11664623 | 2396865 20.54% | 62367490 | 6156465 9.87% |
| WT3 | 82368087 | 10982193 13.33% | 8335974 | 3015038 36.16% | 11664623 | 2258108 19.35% | 62367490 | 5709047 9.15% |
| WT-average | 82368087 | 10663972 12.94% | 8335974 | 3120545 37.43% | 11664623 | 2310293 19.80% | 62367490 | 5233134 8.39% |
| RT1 | 82368087 | 11609228 14.09% | 8335974 | 3086648 37.02% | 11664623 | 2350640 20.15% | 62367490 | 6171940 9.89% |
| RT2 | 82368087 | 11991003 14.55% | 8335974 | 3170368 38.03% | 11664623 | 2434099 20.86% | 62367490 | 6386536 10.24% |
| RT3 | 82368087 | 12222792 14.83% | 8335974 | 3411185 40.92% | 11664623 | 2580335 22.12% | 62367490 | 6231272 9.99% |
| RT-average | 82368087 | 11941008 14.49% | 8335974 | 3222734 38.66% | 11664623 | 2455025 21.04% | 62367490 | 6263249 10.04% |
| Average | 82368087 | 11302490 13.72% | 8335974 | 3171639 38.04% | 11664623 | 2382659 20.42% | 62367490 | 5748192 9.21% |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, K.-F.; Zhang, Q.-X.; Cheng, T.-R.; Yan, X.-L.; Pan, H.-T.; Wang, J. Substantial Epigenetic Variation Causing Flower Color Chimerism in the Ornamental Tree Prunus mume Revealed by Single Base Resolution Methylome Detection and Transcriptome Sequencing. Int. J. Mol. Sci. 2018, 19, 2315. https://doi.org/10.3390/ijms19082315
Ma K-F, Zhang Q-X, Cheng T-R, Yan X-L, Pan H-T, Wang J. Substantial Epigenetic Variation Causing Flower Color Chimerism in the Ornamental Tree Prunus mume Revealed by Single Base Resolution Methylome Detection and Transcriptome Sequencing. International Journal of Molecular Sciences. 2018; 19(8):2315. https://doi.org/10.3390/ijms19082315
Chicago/Turabian StyleMa, Kai-Feng, Qi-Xiang Zhang, Tang-Ren Cheng, Xiao-Lan Yan, Hui-Tang Pan, and Jia Wang. 2018. "Substantial Epigenetic Variation Causing Flower Color Chimerism in the Ornamental Tree Prunus mume Revealed by Single Base Resolution Methylome Detection and Transcriptome Sequencing" International Journal of Molecular Sciences 19, no. 8: 2315. https://doi.org/10.3390/ijms19082315
APA StyleMa, K. -F., Zhang, Q. -X., Cheng, T. -R., Yan, X. -L., Pan, H. -T., & Wang, J. (2018). Substantial Epigenetic Variation Causing Flower Color Chimerism in the Ornamental Tree Prunus mume Revealed by Single Base Resolution Methylome Detection and Transcriptome Sequencing. International Journal of Molecular Sciences, 19(8), 2315. https://doi.org/10.3390/ijms19082315