Genome-Wide Association Study and Selection Signatures Detect Genomic Regions Associated with Seed Yield and Oil Quality in Flax

 ,
,  ,
,
Abstract
:1. Introduction
2. Results
2.1. Re-Sequencing and Genome-Wide SNPs
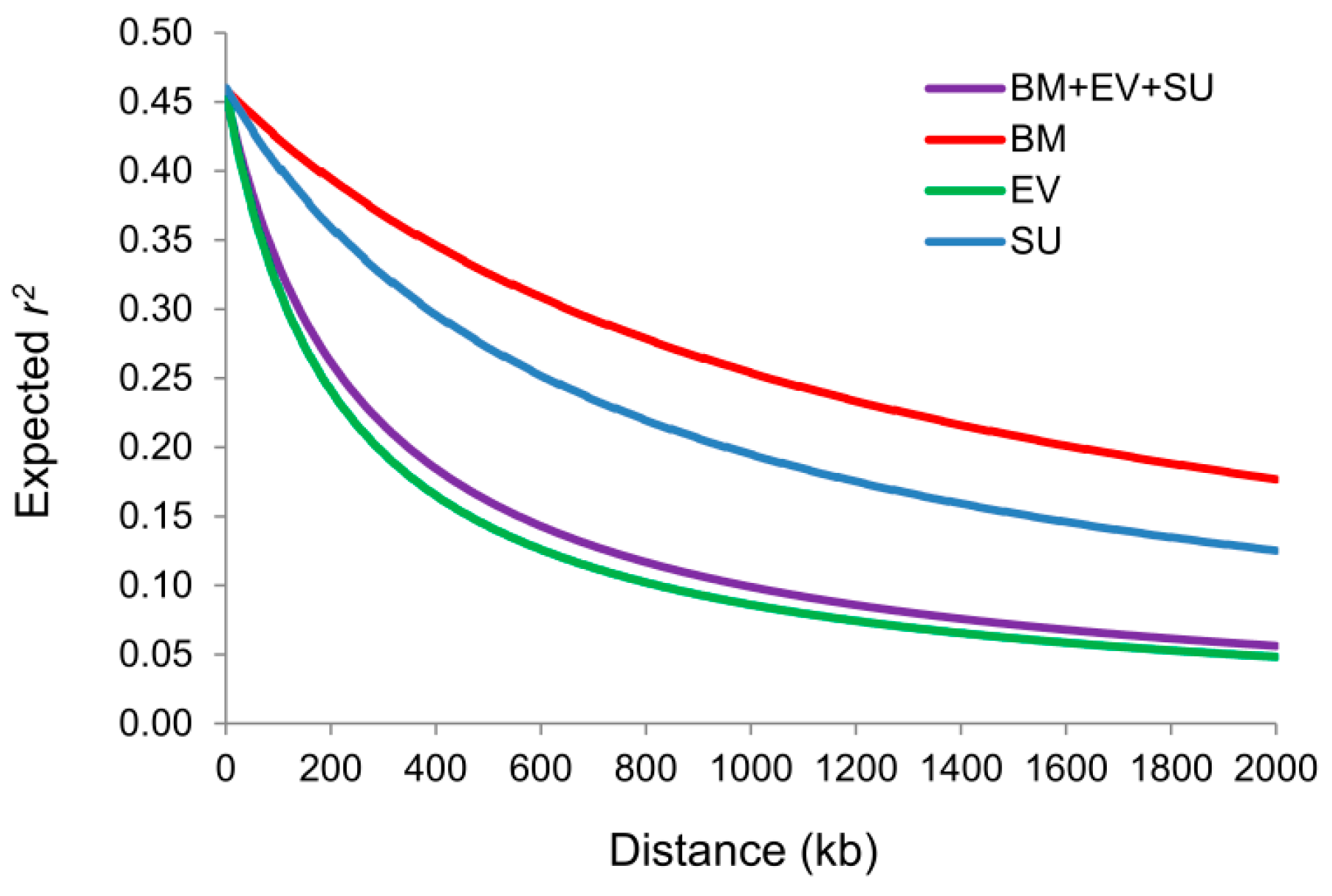
2.2. Whole-Genome Pattern of LD
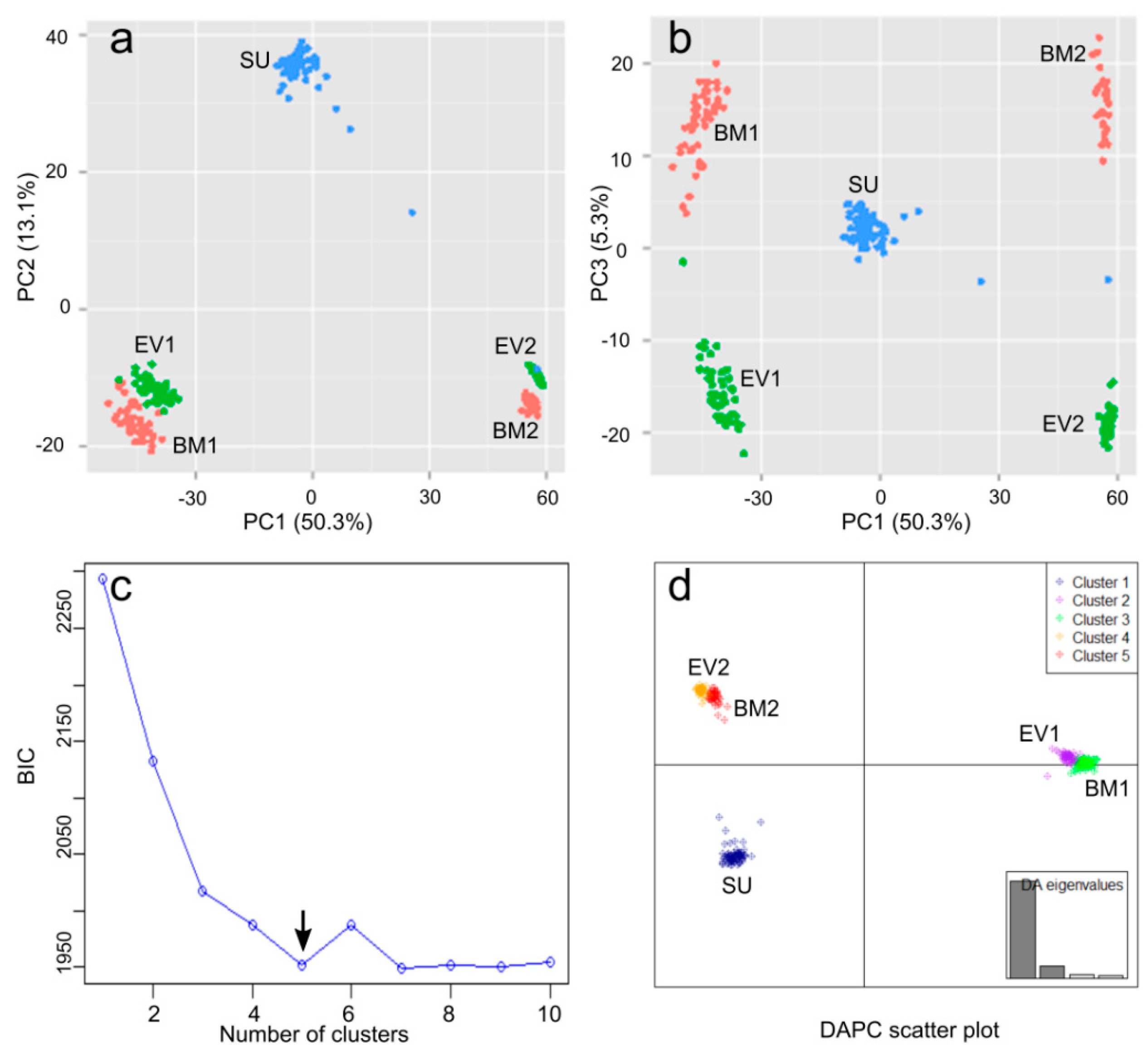
2.3. Genetic Diversity and Population Structure
2.4.
2.5. QTL Identified from 11 Traits
2.6. QTL Effect Significance
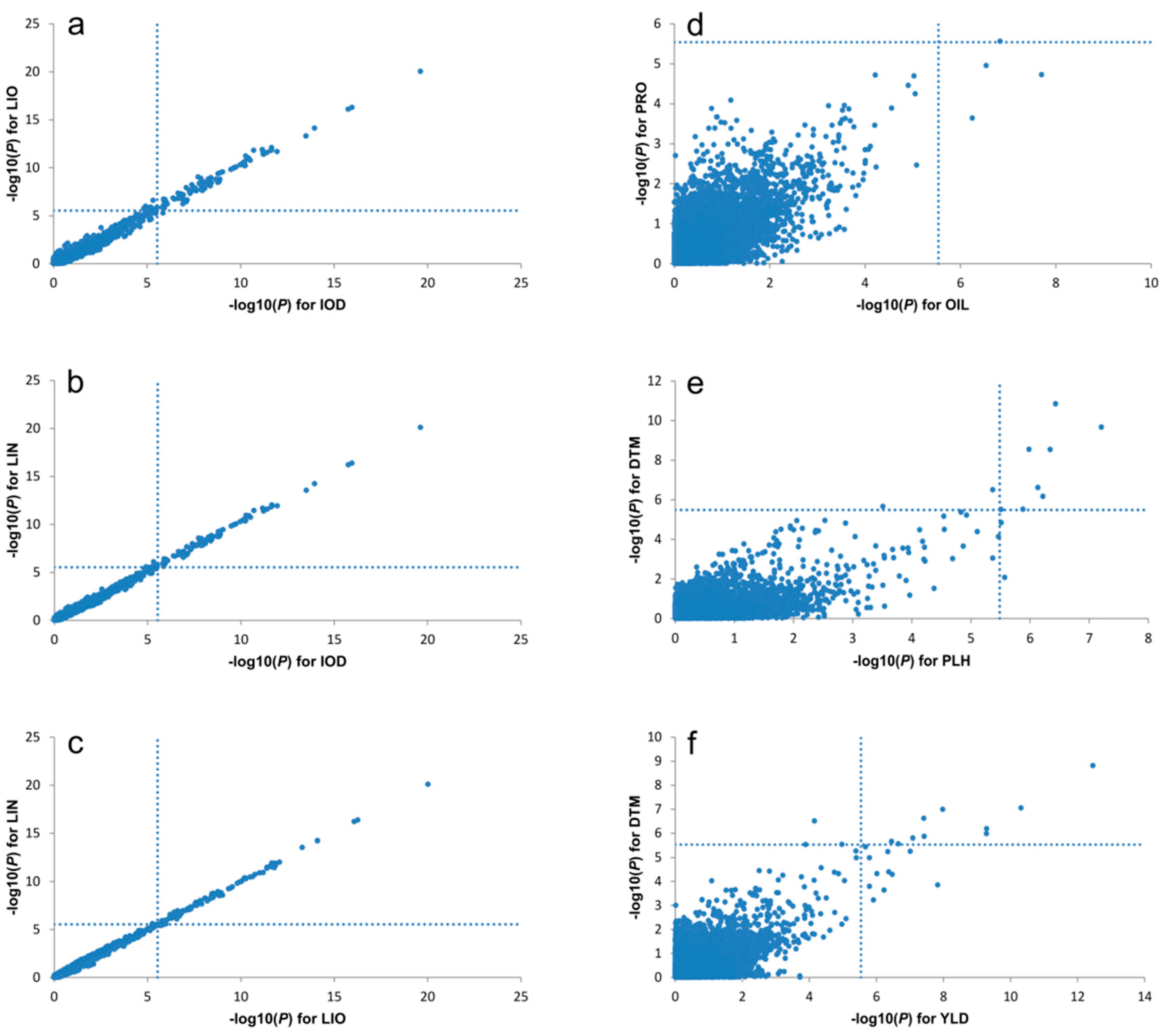
2.7. Pleiotropy of QTL
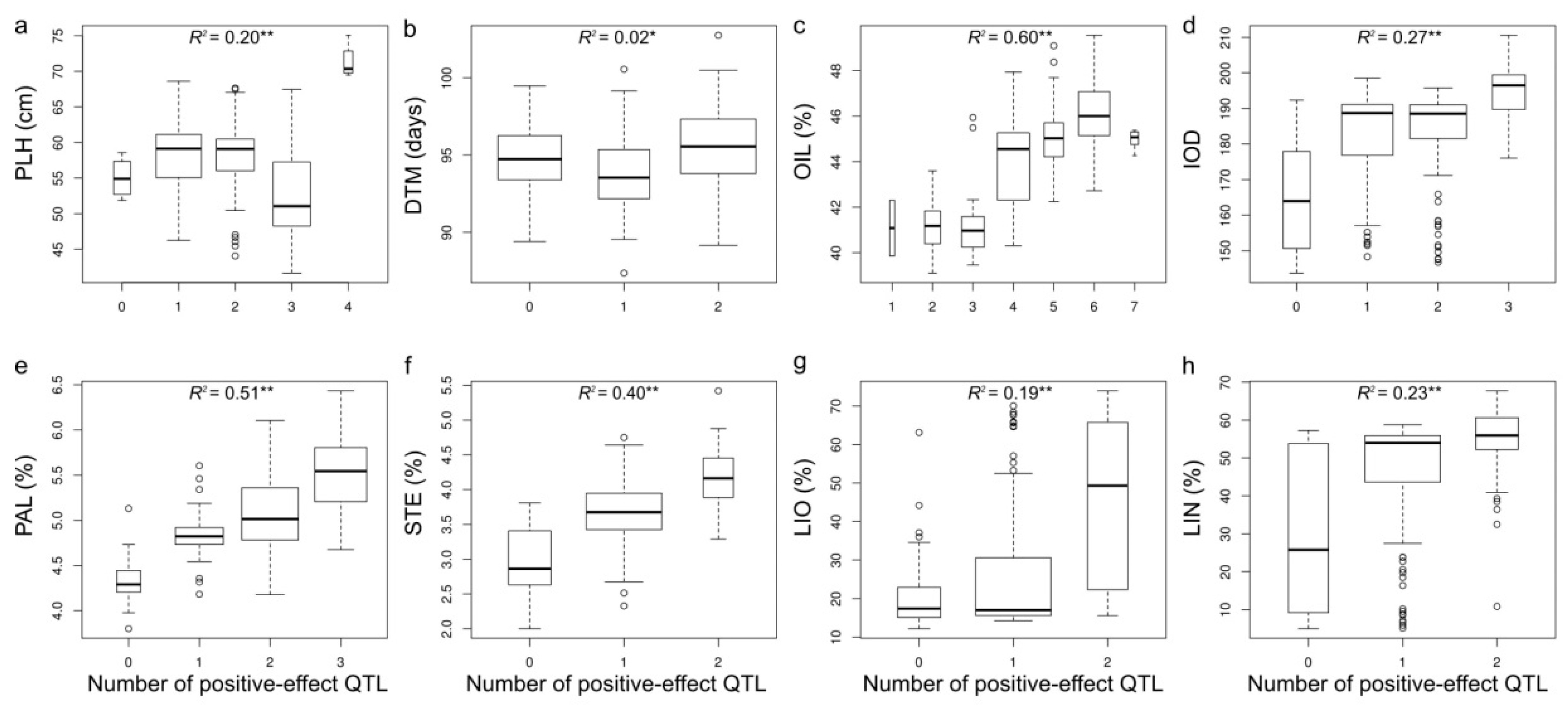
2.8. Phenotypic Variation Explained by QTL
2.9. Candidate Genes Underlying QTL
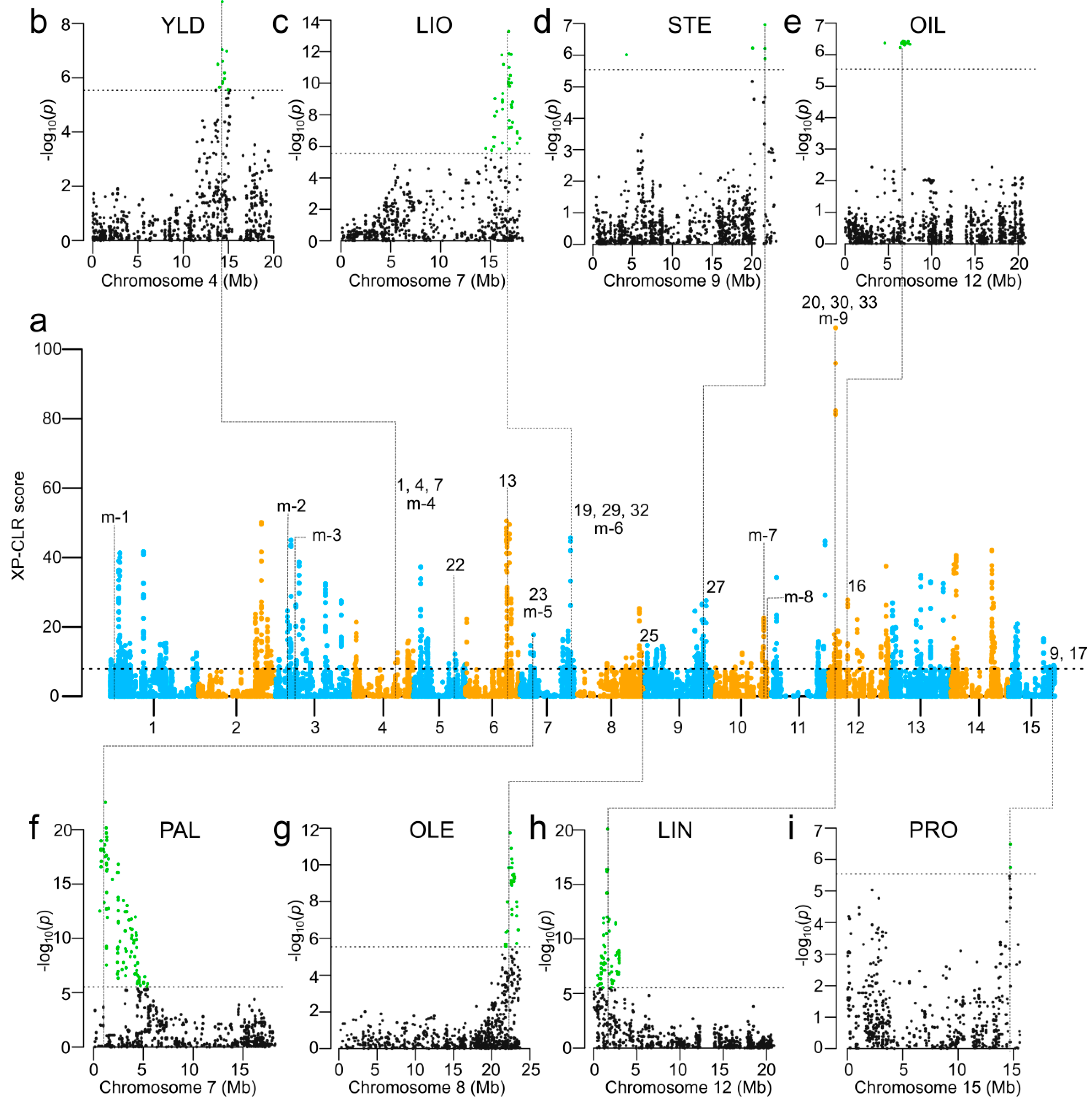
2.10. Selection Signatures in Bi-Parental Populations
3. Discussion
3.1. QTL Associated with Seed Yield and Seed Oil Quality Traits
3.2. Pleotropic QTL Associated with Seed Yield and Quality Traits
3.3. Phenotypic Variation Explained by SNPs and QTL
3.4. Selection Signatures Associated with Seed Yield and Seed Quality Traits
4. Materials and Methods
4.1. Plant Materials
4.2. Whole Genome Resequencing, SNP Calling, SNP Imputation and LD Analysis
4.3. Differentiation and Stratification
4.4. Phenotyping of Bi-Parental Populations
4.5. Phenotypic Variation Explained by All SNPs
4.6. Genome-Wide Association Study
4.7. Candidate Gene Mining
4.8. QTL Validation
4.9. Genome-Wide Selective Sweep Scan
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| DH | doubled haploid |
| GBS | genotyping by sequencing |
| GW3S | genome-wide selective sweep scan |
| GWAS | genome-wide association study |
| IOD | iodine value |
| LD | linkage disequilibrium |
| LIN | linolenic acid |
| LIO | linoleic acid |
| MAF | minor allele frequency |
| MAGIC | multi-parent advanced generation intercross |
| NAM | nested association mapping |
| OIL | oil content |
| OLE | oleic acid |
| PAL | palmitic acid |
| QTL | quantitative trait loci |
| RIL | recombinant inbred line |
| SNP | single nucleotide polymorphism |
| SSR | simple sequence repeat |
| STE | stearic acid |
| YLD | seed yield |
References
- Westcott, N.D.; Muir, A.D. Chemical studies on the constituents of Linum spp. In Flax, the Genus Linum; Muir, A.D., Westcott, N.D., Eds.; Taylor and Francis: New York, NY, USA, 2003; pp. 55–73. [Google Scholar]
- Diederichsen, A.; Kusters, P.M.; Kessler, D.; Bainas, Z.; Gugel, R.K. Assembling a core collection from the flax world collection maintained by Plant Gene resources of Canada. Genet. Resour. Crop Evol. 2013, 60, 1479–1485. [Google Scholar] [CrossRef]
- Green, A.G.; Chen, Y.; Singh, S.P.; Dribnenki, J.C.P. Flax. In Compendium Transgenic Crop Plants: Transgenic Oilseed Crops; Kole, C., Hall, T.C., Eds.; Blackwell Publishing Ltd.: Oxford, UK, 2008; pp. 199–226. [Google Scholar]
- Tolkachev, O.N.; Zhuchenko, A.A. Biologically active substances of flax: Medicinal and nutritional properties. Pharm. Chem. J. 2000, 34, 360–367. [Google Scholar] [CrossRef]
- You, F.M.; Duguid, S.D.; Lam, I.; Cloutier, S.; Rashid, K.Y.; Booker, H. Pedigrees and genetic base of the flax varieties registered in Canada. Can. J. Plant Sci. 2016, 96, 837–852. [Google Scholar] [CrossRef]
- Price, A.H. Believe it or not, QTLs are accurate! Trends Plant Sci. 2006, 11, 213–216. [Google Scholar] [CrossRef] [PubMed]
- Cloutier, S.; Ragupathy, R.; Niu, Z.; Duguid, S.D. SSR-based linkage map of flax (Linum usitatissimum L.) and mapping of QTLs underlying fatty acid composition traits. Mol. Breed. 2011, 28, 437–451. [Google Scholar] [CrossRef]
- Kumar, S.; You, F.M.; Duguid, S.; Booker, H.; Rowland, G.; Cloutier, S. QTL for fatty acid composition and yield in linseed (Linum usitatissimum L.). Theor. Appl. Genet. 2015, 128, 965–984. [Google Scholar] [CrossRef] [PubMed]
- Asgarinia, P.; Cloutier, S.; Duguid, S.; Rashid, K.; Mirlohi, A.; Banik, M.; Saeidi, G. Mapping quantitative trait loci for powdery mildew resistance in flax (Linum usitatissimum L.). Crop Sci. 2013, 53, 2462–2472. [Google Scholar] [CrossRef]
- Fu, Y.-B. Genetic evidence for early flax domestication with capsular dehiscence. Genet. Resour. Crop Evol. 2011, 58, 1119–1128. [Google Scholar] [CrossRef]
- Soto-Cerda, B.J.; Maureira-Butler, I.; Muñoz, G.; Rupayan, A.; Cloutier, S. SSR-based population structure, molecular diversity and linkage disequilibrium analysis of a collection of flax (Linum usitatissimum L.) varying for mucilage seed-coat content. Mol. Breed. 2012, 30, 875–888. [Google Scholar] [CrossRef]
- Wiesnerova, D.; Wiesner, I. ISSR-based clustering of cultivated flax germplasm is statistically correlated to thousand seed mass. Mol. Biotechnol. 2004, 26, 207–213. [Google Scholar] [CrossRef]
- McMullen, M.D.; Kresovich, S.; Villeda, H.S.; Bradbury, P.; Li, H.; Sun, Q.; Flint-Garcia, S.; Thornsberry, J.; Acharya, C.; Bottoms, C. Genetic properties of the maize nested association mapping population. Science 2009, 325, 737–740. [Google Scholar] [CrossRef] [PubMed]
- Bandillo, N.; Raghavan, C.; Muyco, P.A.; Sevilla, M.A.L.; Lobina, I.T.; Dilla-Ermita, C.J.; Tung, C.-W.; McCouch, S.; Thomson, M.; Mauleon, R.; et al. Multi-parent advanced generation inter-cross (MAGIC) populations in rice: Progress and potential for genetics research and breeding. Rice 2013, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Holland, J.B.; McMullen, M.D.; Buckler, E.S. Genetic design and statistical power of nested association mapping in maize. Genetics 2008, 178, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Monir, M.M.; Zhu, J. Dominance and epistasis interactions revealed as important variants for leaf traits of maize NAM population. Front. Plant Sci. 2018, 9, 627. [Google Scholar] [CrossRef] [PubMed]
- Ren, D.; Fang, X.; Jiang, P.; Zhang, G.; Hu, J.; Wang, X.; Meng, Q.; Cui, W.; Lan, S.; Ma, X.; et al. Genetic architecture of nitrogen-deficiency tolerance in wheat seedlings based on a nested association mapping (NAM) population. Front. Plant Sci. 2018, 9, 845. [Google Scholar] [CrossRef] [PubMed]
- Mackay, I.; Powell, W. Methods for linkage disequilibrium mapping in crops. Trends Plant Sci. 2007, 12, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Cavanagh, C.; Morell, M.; Mackay, I.; Powell, W. From mutations to MAGIC: Resources for gene discovery, validation and delivery in crop plants. Curr. Opin. Plant Biol. 2008, 11, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Mathew, B.; Leon, J.; Sannemann, W.; Sillanpaa, M.J. Detection of epistasis for flowering time using Bayesian multilocus estimation in a barley MAGIC population. Genetics 2018, 208, 525–536. [Google Scholar] [CrossRef] [PubMed]
- Camargo, A.V.; Mackay, I.; Mott, R.; Han, J.; Doonan, J.H.; Askew, K.; Corke, F.; Williams, K.; Bentley, A.R. Functional mapping of quantitative trait loci (QTLs) associated with plant performance in a wheat magic mapping population. Front. Plant Sci. 2018, 9, 887. [Google Scholar] [CrossRef] [PubMed]
- Ongom, P.O.; Ejeta, G. Mating design and genetic structure of a multi-parent advanced generation intercross (magic) population of sorghum (Sorghum bicolor (L.) Moench). G3 (Bethesda) 2018, 8, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Huynh, B.L.; Ehlers, J.D.; Huang, B.E.; Munoz-Amatriain, M.; Lonardi, S.; Santos, J.R.P.; Ndeve, A.; Batieno, B.J.; Boukar, O.; Cisse, N.; et al. A multi-parent advanced generation inter-cross (MAGIC) population for genetic analysis and improvement of cowpea (Vigna unguiculata L. Walp.). Plant J. 2018, 93, 1129–1142. [Google Scholar] [CrossRef] [PubMed]
- Ponce, K.S.; Ye, G.; Zhao, X. Qtl identification for cooking and eating quality in indica rice using multi-parent advanced generation intercross (MAGIC) population. Front. Plant Sci. 2018, 9, 868. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Shen, C.; Wen, T.; Gao, B.; Zhu, D.; Li, X.; Ahmed, M.M.; Li, D.; Lin, Z. SSR-based association mapping of fiber quality in upland cotton using an eight-way MAGIC population. Mol. Genet. Genom. 2018, 293, 793–805. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Cardenas, J.A.; Mesa-Valle, C.; Manzano-Agugliaro, F. Trends in plant research using molecular markers. Planta 2018, 247, 543–557. [Google Scholar] [CrossRef] [PubMed]
- Pena, R.N.; Ros-Freixedes, R.; Tor, M.; Estany, J. Genetic marker discovery in complex traits: A field example on fat content and composition in pigs. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.M.; Shao, X.Y.; Pei, Y.H.; Guo, X.M.; Li, J.; Song, X.Y.; Zhao, M.A. Genetic diversity and genome-wide association study of major ear quantitative traits using high-density SNPs in maize. Front. Plant Sci. 2018, 9, 966. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Wan, H.; Qian, J.; Guo, J.; Sun, C.; Wen, J.; Yi, B.; Ma, C.; Tu, J.; Song, L.; et al. Genome-wide association study of cadmium accumulation at the seedling stage in rapeseed (Brassica napus L.). Front. Plant Sci. 2018, 9, 375. [Google Scholar] [CrossRef] [PubMed]
- MacGregor, S.; Ong, J.S.; An, J.; Han, X.; Zhou, T.; Siggs, O.M.; Law, M.H.; Souzeau, E.; Sharma, S.; Lynn, D.J.; et al. Genome-wide association study of intraocular pressure uncovers new pathways to glaucoma. Nat. Genet. 2018, 50, 1067–1071. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Wei, X.; Sang, T.; Zhao, Q.; Feng, Q.; Zhao, Y.; Li, C.; Zhu, C.; Lu, T.; Zhang, Z.; et al. Genome-wide association studies of 14 agronomic traits in rice landraces. Nat. Genet. 2010, 42, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Zhao, X.; Ponce, K.; Ye, G.; Leung, H. QTL mapping for agronomic traits using multi-parent advanced generation inter-cross (MAGIC) populations derived from diverse elite indica rice lines. Field Crops Res. 2016, 189, 19–42. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Han, B. Natural variations and genome-wide association studies in crop plants. Annu. Rev. Plant Biol. 2014, 65, 531–551. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Patterson, N.; Reich, D. Population differentiation as a test for selective sweeps. Genome Res. 2010, 20, 393–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gore, M.A.; Chia, J.M.; Elshire, R.J.; Sun, Q.; Ersoz, E.S.; Hurwitz, B.L.; Peiffer, J.A.; McMullen, M.D.; Grills, G.S.; Ross-Ibarra, J.; et al. A first-generation haplotype map of maize. Science 2009, 326, 1115–1117. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Wang, G.; Yuan, M.; Yao, W.; Lyu, K.; Zhao, H.; Yang, M.; Li, P.; Zhang, X.; Yuan, J.; et al. Breeding signatures of rice improvement revealed by a genomic variation map from a large germplasm collection. Proc. Natl. Acad. Sci. USA 2015, 112, E5411–5419. [Google Scholar] [CrossRef] [PubMed]
- Wen, Z.; Boyse, J.F.; Song, Q.; Cregan, P.B.; Wang, D. Genomic consequences of selection and genome-wide association mapping in soybean. BMC Genom. 2015, 16, 671. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Jiang, Y.; Wang, Z.; Gou, Z.; Lyu, J.; Li, W.; Yu, Y.; Shu, L.; Zhao, Y.; Ma, Y.; et al. Resequencing 302 wild and cultivated accessions identifies genes related to domestication and improvement in soybean. Nat. Biotechnol. 2015, 33, 408–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, C.; Fu, J.; Zhang, J.; Li, Y.; Zheng, J.; Zhang, H.; Yang, X.; Wang, J.; Wang, G. A gene-oriented haplotype comparison reveals recently selected genomic regions in temperate and tropical maize germplasm. PLoS ONE 2016, 12, e0169806. [Google Scholar] [CrossRef] [PubMed]
- Jordan, K.W.; Wang, S.; Lun, Y.; Gardiner, L.J.; MacLachlan, R.; Hucl, P.; Wiebe, K.; Wong, D.; Forrest, K.L.; Sharpe, A.G.; et al. A haplotype map of allohexaploid wheat reveals distinct patterns of selection on homoeologous genomes. Genome Biol. 2015, 16, 48. [Google Scholar] [CrossRef] [PubMed]
- Cadzow, M.; Boocock, J.; Nguyen, H.T.; Wilcox, P.; Merriman, T.R.; Black, M.A. A bioinformatics workflow for detecting signatures of selection in genomic data. Front. Genet. 2014, 5, 293. [Google Scholar] [CrossRef] [PubMed]
- Korte, A.; Farlow, A. The advantages and limitations of trait analysis with GWAS: A review. Plant Methods 2013, 9, 29. [Google Scholar] [CrossRef] [PubMed]
- Cloutier, S.; Ragupathy, R.; Miranda, E.; Radovanovic, N.; Reimer, E.; Walichnowski, A.; Ward, K.; Rowland, G.; Duguid, S.; Banik, M. Integrated consensus genetic and physical maps of flax (Linum usitatissimum L.). Theor. Appl. Genet. 2012, 125, 1783–1795. [Google Scholar] [CrossRef] [PubMed]
- You, F.M.; Booker, M.H.; Duguid, D.S.; Jia, G.; Cloutier, S. Accuracy of genomic selection in biparental populations of flax (Linum usitatissimum L.). Crop J. 2016, 4, 290–303. [Google Scholar] [CrossRef]
- You, F.M.; Xiao, J.; Li, P.; Yao, Z.; Jia, G.; He, L.; Zhu, T.; Luo, M.C.; Wang, X.; Deyholos, M.K.; et al. Chromosome-scale pseudomolecules refined by optical, physical and genetic maps in flax. Plant J. 2018, 95, 371–384. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Hobson, N.; Galindo, L.; Zhu, S.; Shi, D.; McDill, J.; Yang, L.; Hawkins, S.; Neutelings, G.; Datla, R.; et al. The genome of flax (Linum usitatissimum) assembled de novo from short shotgun sequence reads. Plant J. 2012, 72, 461–473. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; You, F.M.; Cloutier, S. Genome wide SNP discovery in flax through next generation sequencing of reduced representation libraries. BMC Genom. 2012, 13, 684. [Google Scholar] [CrossRef] [PubMed]
- You, F.M.; Deal, K.R.; Wang, J.; Britton, M.T.; Fass, J.N.; Lin, D.; Dandekar, A.M.; Leslie, C.A.; Aradhya, M.; Luo, M.C.; et al. Genome-wide SNP discovery in walnut with an AGSNP pipeline updated for SNP discovery in allogamous organisms. BMC Genom. 2012, 13, 354. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Wang, B.; Yan, L.; Hu, K.; Liu, S.; Zhou, Y.; Guan, C.; Zhang, Z.; Li, J.; Zhang, J.; et al. Genome-wide association study provides insight into the genetic control of plant height in rapeseed (Brassica napus L.). Front. Plant Sci. 2016, 7, 1102. [Google Scholar] [CrossRef] [PubMed]
- Lamesch, P.; Berardini, T.Z.; Li, D.; Swarbreck, D.; Wilks, C.; Sasidharan, R.; Muller, R.; Dreher, K.; Alexander, D.L.; Garcia-Hernandez, M.; et al. The Arabidopsis Information Resource (TAIR): Improved gene annotation and new tools. Nucleic Acids Res. 2012, 40, D1202–D1210. [Google Scholar] [CrossRef] [PubMed]
- You, F.M.; Li, P.; Kumar, S.; Ragupathy, R.; Li, Z.; Fu, Y.-B.; Cloutier, S. Genome-wide identification and characterization of the gene families controlling fatty acid biosynthesis in flax (Linum usitatissimum L.). J. Proteom. Bioinf. 2014, 7, 310–326. [Google Scholar]
- Vrinten, P.; Hu, Z.; Munchinsky, M.A.; Rowland, G.; Qiu, X. Two FAD3 desaturase genes control the level of linolenic acid in flax seed. Plant Physiol. 2005, 139, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Soto-Cerda, B.J.; Duguid, S.; Booker, H.; Rowland, G.; Diederichsen, A.; Cloutier, S. Association mapping of seed quality traits using the Canadian flax (Linum usitatissimum L.) core collection. Theor. Appl. Genet. 2014, 127, 881–896. [Google Scholar] [CrossRef] [PubMed]
- You, F.M.; Li, P.; Ragupathy, R.; Kumar, S.; Zhu, T.; Luo, M.-C.; Duguid, S.D.; Rashid, K.Y.; Booker, H.M.; Deyholos, M.K.; et al. The Draft Flax Genome Pseudomolecules. In Proceedings of the 66th Flax Institute of the United States, Fargo, ND, USA, 31 March–1 April 2016; pp. 17–24. [Google Scholar]
- Banik, M.; Duguid, S.; Cloutier, S. Transcript profiling and gene characterization of three fatty acid desaturase genes in high, moderate, and low linolenic acid genotypes of flax (Linum usitatissimum L.) and their role in linolenic acid accumulation. Genome 2011, 54, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Song, Q.; Cregan, P.B.; Nelson, R.L.; Wang, X.; Wu, J.; Jiang, G.L. Genome-wide association study for flowering time, maturity dates and plant height in early maturing soybean (Glycine max) germplasm. BMC Genom. 2015, 16, 217. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.K.; Wang, Y.J.; Luo, G.Z.; Zhang, J.S.; He, C.Y.; Wu, X.L.; Gai, J.Y.; Chen, S.Y. QTL mapping of ten agronomic traits on the soybean (Glycine max L. Merr.) genetic map and their association with EST markers. Theor. Appl. Genet. 2004, 108, 1131–1139. [Google Scholar] [CrossRef] [PubMed]
- Eskandari, M.; Cober, E.R.; Rajcan, I. Genetic control of soybean seed oil: Ii. QTL and genes that increase oil concentration without decreasing protein or with increased seed yield. Theor. Appl. Genet. 2013, 126, 1677–1687. [Google Scholar] [CrossRef] [PubMed]
- Hwang, E.Y.; Song, Q.; Jia, G.; Specht, J.E.; Hyten, D.L.; Costa, J.; Cregan, P.B. A genome-wide association study of seed protein and oil content in soybean. BMC Genom. 2014, 15, 1. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Benyamin, B.; McEvoy, B.P.; Gordon, S.; Henders, A.K.; Nyholt, D.R.; Madden, P.A.; Heath, A.C.; Martin, N.G.; Montgomery, G.W.; et al. Common SNPs explain a large proportion of the heritability for human height. Nat. Genet. 2010, 42, 565–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Bakshi, A.; Zhu, Z.; Hemani, G.; Vinkhuyzen, A.A.E.; Lee, S.H.; Robinson, M.R.; Perry, J.R.B.; Nolte, I.M.; van Vliet-Ostaptchouk, J.V.; et al. Genetic variance estimation with imputed variants finds negligible missing heritability for human height and body mass index. Nat. Genet. 2015, 47, 1114–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Manolio, T.A.; Pasquale, L.R.; Boerwinkle, E.; Caporaso, N.; Cunningham, J.M.; de Andrade, M.; Feenstra, B.; Feingold, E.; Hayes, M.G.; et al. Genome partitioning of genetic variation for complex traits using common SNPs. Nat. Genet. 2011, 43, 519–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, A.R.; Esko, T.; Yang, J.; Vedantam, S.; Pers, T.H.; Gustafsson, S.; Chu, A.Y.; Estrada, K.; Luan, J.; Kutalik, Z.; et al. Defining the role of common variation in the genomic and biological architecture of adult human height. Nat. Genet. 2014, 46, 1173–1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duguid, S.D.; Kenaschuk, E.O.; Rashid, K.Y. Macbeth flax. Can. J. Plant Sci. 2003, 83, 803–805. [Google Scholar] [CrossRef] [Green Version]
- Rowland, G.G.; Hormis, Y.A.; Rashid, K.Y. CDC bethune flax. Can. J. Plant Sci. 2002, 82, 101–102. [Google Scholar] [CrossRef]
- Rowland, G.G.; Bhatty, R.S. Ethyl meththane-sulphonate induced fatty acid mutations in flax. J. Am. Oil Chem. Soc. 1990, 67, 213–214. [Google Scholar] [CrossRef]
- You, F.M.; Huo, N.; Deal, K.R.; Gu, Y.Q.; Luo, M.C.; McGuire, P.E.; Dvorak, J.; Anderson, O.D. Annotation-based genome-wide SNP discovery in the large and complex Aegilops tauschii genome using next-generation sequencing without a reference genome sequence. BMC Genom. 2011, 12, 59. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Browning, S.R.; Browning, B.L. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am. J. Hum. Genet. 2007, 81, 1084–1097. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. Plink: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Hill, W.G.; Weir, B.S. Variances and covariances of squared linkage disequilibria in finite populations. Theor. Popul. Biol. 1988, 33, 54–78. [Google Scholar] [CrossRef]
- Pfeifer, B.; Wittelsburger, U.; Ramos-Onsins, S.E.; Lercher, M.J. Popgenome: An efficient swiss army knife for population genomic analyses in R. Mol. Biol. Evol. 2014, 31, 1929–1936. [Google Scholar] [CrossRef] [PubMed]
- Jombart, T.; Devillard, S.; Balloux, F. Discriminant analysis of principal components: A new method for the analysis of genetically structured populations. BMC Genet. 2010, 11, 94. [Google Scholar] [CrossRef] [PubMed]
- Jombart, T. Adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics 2008, 24, 1403–1405. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.S.; Poushinsky, G. A modified augmented design (type 2) for rectangular plots. Can. J. Plant Sci. 1985, 65, 743–749. [Google Scholar] [CrossRef]
- You, F.M.; Duguid, S.D.; Thambugala, D.; Cloutier, S. Statistical analysis and field evaluation of the type 2 modified augmented design (MAD) in phenotyping of flax (Linum usitatissimum) germplasms in multiple environments. Aust. J. Crop Sci. 2013, 7, 1789–1800. [Google Scholar]
- Association of Official Analytical Chemists. Fat (total, saturated and unsaturated) in foods: Hydrolytic extraction gas chromatographic method. In Official Methods of Analysis of AOAC International, 18th ed.; Horwitz, W., Ed.; AOAC International: Gaithersburg, MD, USA, 2001. [Google Scholar]
- Bates, D.; Maechler, M.; Bolker, B.; Walker, S. Fitting linear mixed-effects models using lme4. J. Stat. Softw. 2015, 67, 1–48. [Google Scholar] [CrossRef]
- Zhang, Z.; Ersoz, E.; Lai, C.Q.; Todhunter, R.J.; Tiwari, H.K.; Gore, M.A.; Bradbury, P.J.; Yu, J.; Arnett, D.K.; Ordovas, J.M.; et al. Mixed linear model approach adapted for genome-wide association studies. Nat. Genet. 2010, 42, 355–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Pressoir, G.; Briggs, W.H.; Vroh Bi, I.; Yamasaki, M.; Doebley, J.F.; McMullen, M.D.; Gaut, B.S.; Nielsen, D.M.; Holland, J.B.; et al. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 2006, 38, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. Tassel: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef] [PubMed]
- Turner, S.D. Qqman: An R package for visualizing GWAS results using Q-Q and manhattan plots. biorXiv 2014. [Google Scholar] [CrossRef]
- Thambugala, D.; Duguid, S.; Loewen, E.; Rowland, G.; Booker, H.; You, F.M.; Cloutier, S. Genetic variation of six desaturase genes in flax and their impact on fatty acid composition. Theor. Appl. Genet. 2013, 126, 2627–2641. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Population | BM | EV | SU |
|---|---|---|---|
| BM | 41.52 | 0.04 | 0.44 |
| EV | 38.26 | 0.48 | |
| SU | 3.95 |
| Trait | Population | No. QTL | ||
|---|---|---|---|---|
| YLD | BM + EV + SU | 0.43 ± 0.12 | 1 | 0.14 ± 0.09 § |
| BM | 0.22 ± 0.25 | |||
| EV | 0.15 ± 0.24 | |||
| SU | 0.45 ± 0.21 | |||
| PLH | BM + EV + SU | 0.53 ± 0.12 | 1 | 0.08 ± 0.11 |
| BM | 0.76 ± 0.12 | 2 | 0.21 ± 0.15 | |
| EV | 0.76 ± 0.14 | 2 | 0.22 ± 0.18 | |
| SU | 0.06 ± 0.20 | |||
| DTM | BM + EV + SU | 0.43 ± 0.13 | 1 | 0.10 ± 0.07 |
| BM | 0.81 ± 0.11 | 1 | 0.18 ± 0.13 | |
| EV | 0.36 ± 0.24 | 1 | 0.18 ± 0.22 | |
| SU | 0.00 ± 0.20 | |||
| PRO | BM + EV + SU | 0.51 ± 0.11 | 1 | 0.12 ± 0.16 |
| BM | 0.52 ± 0.20 | |||
| EV | 0.34 ± 0.23 | 1 | 0.09 ± 0.12 | |
| SU | 0.58 ± 0.19 | |||
| OIL | BM + EV + SU | 0.66 ± 0.09 | 7 | 0.62 ± 0.14 |
| BM | 0.46 ± 0.22 | |||
| EV | 0.39 ± 0.21 | 1 | 0.08 ± 0.08 | |
| SU | 0.70 ± 0.15 | |||
| IOD | BM + EV + SU | 0.80 ± 0.06 | 3 | 0.57 ± 0.10 |
| BM | 0.49 ± 0.19 | |||
| EV | 0.78 ± 0.12 | 2 | 0.51 ± 0.14 | |
| SU | 0.66 ± 0.17 | 2 | 0.35 ± 0.18 | |
| PAL | BM + EV + SU | 0.79 ± 0.06 | 4 | 0.48 ± 0.11 |
| BM | 0.12 ± 0.26 | |||
| EV | 0.55 ± 0.20 | 1 | 0.09 ± 0.11 | |
| SU | 0.90 ± 0.07 | 1 | 0.56 ± 0.18 | |
| STE | BM + EV + SU | 0.21 ± 0.15 | 2 | 0.41 ± 0.19 |
| BM | 0.85 ± 0.09 | |||
| EV | 0.02 ± 0.14 | |||
| SU | 0.00 ± 0.22 | 1 | ||
| OLE | BM + EV + SU | 0.55 ± 0.10 | 1 | 0.16 ± 0.13 |
| BM | 0.36 ± 0.22 | |||
| EV | 0.09 ± 0.25 | |||
| SU | 0.72 ± 0.16 | 1 | 0.20 ± 0.19 | |
| LIO | BM + EV + SU | 0.80 ± 0.06 | 3 | 0.73 ± 0.07 |
| BM | 0.54 ± 0.20 | |||
| EV | 0.75 ± 0.13 | 2 | 0.54 ± 0.14 | |
| SU | 0.66 ± 0.17 | 2 | 0.36 ± 0.18 | |
| LIN | BM + EV + SU | 0.80 ± 0.06 | 3 | 0.56 ± 0.09 |
| BM | 0.49 ± 0.19 | |||
| EV | 0.76 ± 0.13 | 2 | 0.55 ± 0.14 | |
| SU | 0.66 ± 0.17 | 2 | 0.36 ± 0.18 |
| Trait | QTL No. | QTL Name | Chr. | Start Position (bp) | End Position (bp) | XP-CLR Score | Known QTL or Marker | Candidate Gene IDs | Candidate Gene Location (bp) | Candidate Gene Name | Gene Annotation |
|---|---|---|---|---|---|---|---|---|---|---|---|
| YLD | 1 | QYLD-Lu4.1 | 4 | 13,594,936 | 14,968,389 | 12.54 | QYld.BM.crc-LG4a | ||||
| PLH | 2 | QPLH-Lu1.1 | 1 | 13,887,715 | 13,930,292 | ||||||
| 3 | QPLH-Lu1.2 | 1 | 20,012,490 | 20,012,490 | Lus10020835 | 19,610,837 | BRI1 [49] | Leucine-rich receptor-like protein kinase family protein | |||
| Lus10020865 | 19,790,777 | GA2 [49] | Terpenoid cyclases/Protein prenyltransferases superfamily protein | ||||||||
| 4 | QPLH-Lu4.3 | 4 | 14,305,982 | 15,042,104 | 12.54 | Lus10034358 | 14,006,288 | BIM2 [49] | BES1-interacting Myc-like protein 2 | ||
| Lus10041435 | 14,157,752 | MYB62 [49] | Myb domain protein 62 | ||||||||
| Lus10041481 | 14,398,338 | LMCO4 [49] | Laccase/Diphenol oxidase family protein | ||||||||
| Lus10041794 | 15,920,170 | ROT3 [49] | Cytochrome P450 superfamily protein | ||||||||
| Lus10041801 | 15,948,434 | WAT1 [49] | Walls Are Thin 1 | ||||||||
| 5 | QPLH-Lu13.4 | 13 | 17,243,884 | 17,243,884 | Lus10030567 | 18,680,474 | GA2OX8 [49] | Gibberellin 2-oxidase 8 | |||
| 6 | QPLH-Lu13.5 | 14 | 2,320,469 | 2,320,469 | 40.61 | Lus10021395 | 3,647,029 | HCT [49] | Hydroxycinnamoyl-CoA shikimate/quinate hydroxycinnamoyl transferase | ||
| DTM | 7 | QDTM-Lu4.1 | 4 | 13,171,757 | 15,042,104 | 12.54 | QDm.BM.crc-LG4a | Lus10015766 | 13,094,864 | FLC [50] | K-box region and MADS-box transcription factor family protein |
| Lus10034461 | 13,434,121 | CDF3 [50] | Cycling DOF factor 3 | ||||||||
| Lus10034370 | 13,933,421 | AP1 [50] | K-box region and MADS-box transcription factor family protein | ||||||||
| Lus10041483 | 14,411,103 | PFT1 [50] | Phytochrome and flowering time regulatory protein (PFT1) | ||||||||
| Lus10041500 | 14,512,085 | ATAN11 [50] | Transducin/WD40 repeat-like superfamily protein | ||||||||
| Lus10041540 | 14,716,950 | RGL1 [50] | RGA-like 1 | ||||||||
| Lus10041595 | 14,966,739 | AP2 [50] | Integrase-type DNA-binding superfamily protein | ||||||||
| 8 | QDTM-Lu11.2 | 11 | 14,768,686 | 14,768,686 | |||||||
| PRO | 9 | QPRO-Lu15.1 | 15 | 14,746,288 | 14,746,310 | 8.50 | Lus10030671 | 22,732,660 | WRI [50] | Integrase-type DNA-binding superfamily protein | |
| OIL | 10 | QOIL-Lu2.1 | 2 | 21,913,720 | 21,913,720 | ||||||
| 11 | QOIL-Lu5.2 | 5 | 15,704,607 | 15,705,039 | |||||||
| 12 | QOIL-Lu6.3 | 6 | 4,879,632 | 4,879,632 | |||||||
| 13 | QOIL-Lu6.4 | 6 | 13,799,180 | 13,970,951 | 50.58 | ||||||
| 14 | QOIL-Lu7.4 | 7 | 14,209,179 | 14,209,179 | |||||||
| 15 | QOIL-Lu10.5 | 10 | 6,517,448 | 6,517,448 | |||||||
| 16 | QOIL-Lu12.6 | 12 | 4,591,214 | 7,491,405 | 27.77 | ||||||
| 17 | QOIL-Lu15.7 | 15 | 14,665,900 | 15,429,055 | 8.89 | Lus10039906 | 19,833,852 | KCS14-2 [51] | 3-ketoacyl-CoA synthase | ||
| IOD | 18 | QIOD-Lu4.1 | 4 | 19,909,467 | 19,909,467 | Lus10039906 | 19,833,852 | KCS14-2 [51] | 3-ketoacyl-CoA synthase | ||
| 19 | QIOD-Lu7.2 | 7 | 15,346,458 | 17,977,459 | 45.70 | QIOD.crc-LG7b | Lus10038321 | 16,089,922 | FAD3a [52] | Fatty acid desaturase | |
| 20 | QIOD-Lu12.3 | 12 | 489,561 | 2,981,642 | 106.22 | QIOD.crc-LG16b | Lus10036184 | 1,035,336 | FAD3b [52] | Fatty acid desaturase | |
| Lus10023359 | 1,729,292 | FAH1 [50] | Fatty acid hydroxylase 1 | ||||||||
| PAL | 21 | QPAL-Lu5.1 | 5 | 12,062,376 | 12,182,441 | Lus10029880 | 12,062,376 | KCS12-3 [51] | 3-ketoacyl-CoA synthase | ||
| 22 | QPAL-Lu5.2 | 5 | 13,797,851 | 15,668,995 | 12.14 | ||||||
| 23 | QPAL-Lu7.3 | 7 | 624,461 | 5,423,691 | 17.74 | QPal.BM.crc-LG7a QPAL.crc-LG9b c79-s540_Lu2534c | Lus10001814 | 79,471 | KAS Ic-1 [51] | 3-ketoacyl-acyl carrier protein synthase I | |
| Lus10028925 | 1,085,389 | KAS IIIb-2 [51] | 3-ketoacyl-acyl carrier protein synthase III | ||||||||
| Lus10028885 | 1,262,079 | SUN1 [50] | SAD1/UNC-84 domain protein 1 | ||||||||
| 24 | QPAL-Lu11.4 | 11 | 4,417,685 | 4,429,424 | Lus10026345 | 4,333,672 | KCS7-1 [51] | 3-ketoacyl-CoA synthase | |||
| OLE | 25 | QOLE-Lu8.1 | 8 | 21,782,841 | 23,527,563 | 12.64 | Lus10006636 | 22,165,534 | KCS9-1 [51] | 3-ketoacyl-CoA synthase | |
| Lus10006637 | 22,174,324 | KCS1-1 [51] | 3-ketoacyl-CoA synthase | ||||||||
| Lus10018485 | 23,111,453 | DES-1-LIKE [50] | Fatty acid desaturase family protein | ||||||||
| STE | 26 | QSTE-Lu9.1 | 9 | 4,229,230 | 4,229,230 | Lus10040333 | 4,275,842 | KCS18-2 [51] | 3-ketoacyl-CoA synthase | ||
| 27 | QSTE-Lu9.2 | 9 | 20,080,531 | 21,636,823 | 27.55 | Lus10011877 | 20,059,127 | SAD1 [51] | Stearoyl acyl carrier protein desaturase | ||
| Lus10011839 | 20,227,416 | FatA2-2 [51] | FatA acyl-ACP thioesterase | ||||||||
| LIO | 28 | QLIO-Lu4.1 | 4 | 19,909,467 | 19,909,467 | Lus10039906 | 19,833,852 | KCS14-2 [51] | 3-ketoacyl-CoA synthase | ||
| 29 | QLIO-Lu7.2 | 7 | 14,540,706 | 17,977,459 | 45.70 | QLIO.crc-LG7b c281-s185_ Lu566c | Lus10038321 | 16,089,922 | FAD3a [52] | Fatty acid desaturase | |
| 30 | QLIO-Lu12.3 | 12 | 489,561 | 2,981,642 | 106.22 | QLIO.crc-LG16b Llio-LG12.3c | Lus10036184 | 1,035,336 | FAD3b [52] | Fatty acid desaturase | |
| LIN | 31 | QLIN-Lu4.1 | 4 | 19,909,467 | 19,909,467 | Lus10039906 | 19,833,852 | KCS14-2 [51] | 3-ketoacyl-CoA synthase | ||
| 32 | QLIN-Lu7.2 | 7 | 14,540,719 | 17,977,459 | 45.70 | QLIN.crc-LG7b c281-s185_ Lu566c | Lus10038321 | 16,089,922 | FAD3a [52] | Fatty acid desaturase | |
| 33 | QLIN-Lu12.3 | 12 | 489,561 | 2,981,642 | 106.22 | QLIN.crc-LG16b Llin-LG12.3c | Lus10036184 | 1,035,336 | FAD3b [52] | Fatty acid desaturase | |
| Lus10023359 | 1,729,292 | FAH1 [50] | Fatty acid hydroxylase 1 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
You, F.M.; Xiao, J.; Li, P.; Yao, Z.; Jia, G.; He, L.; Kumar, S.; Soto-Cerda, B.; Duguid, S.D.; Booker, H.M.; et al. Genome-Wide Association Study and Selection Signatures Detect Genomic Regions Associated with Seed Yield and Oil Quality in Flax. Int. J. Mol. Sci. 2018, 19, 2303. https://doi.org/10.3390/ijms19082303
You FM, Xiao J, Li P, Yao Z, Jia G, He L, Kumar S, Soto-Cerda B, Duguid SD, Booker HM, et al. Genome-Wide Association Study and Selection Signatures Detect Genomic Regions Associated with Seed Yield and Oil Quality in Flax. International Journal of Molecular Sciences. 2018; 19(8):2303. https://doi.org/10.3390/ijms19082303
Chicago/Turabian StyleYou, Frank M., Jin Xiao, Pingchuan Li, Zhen Yao, Gaofeng Jia, Liqiang He, Santosh Kumar, Braulio Soto-Cerda, Scott D. Duguid, Helen M. Booker, and et al. 2018. "Genome-Wide Association Study and Selection Signatures Detect Genomic Regions Associated with Seed Yield and Oil Quality in Flax" International Journal of Molecular Sciences 19, no. 8: 2303. https://doi.org/10.3390/ijms19082303
APA StyleYou, F. M., Xiao, J., Li, P., Yao, Z., Jia, G., He, L., Kumar, S., Soto-Cerda, B., Duguid, S. D., Booker, H. M., Rashid, K. Y., & Cloutier, S. (2018). Genome-Wide Association Study and Selection Signatures Detect Genomic Regions Associated with Seed Yield and Oil Quality in Flax. International Journal of Molecular Sciences, 19(8), 2303. https://doi.org/10.3390/ijms19082303