1. Introduction
Cisplatin and the clinically accepted platinum drugs have a great importance for the cancer treatment. They have been applied in most anticancer chemotherapeutic regimens [
1,
2,
3,
4]. Intensive studies in this area date back to the mid-sixties of the 20th century, with Rosenberg’s remarkable discovery of the medicinal power of the inorganic coordination compound “nicknamed” cisplatin [
1,
5,
6,
7]. Since then, cisplatin has revolutionized cancer treatment converting the formerly fatal disease, largely curable [
8]. Nowadays, cisplatin is still the most successful anticancer drug in the world, and is widely used in the treatment of a multitude of different cancers. Still, regardless of the achievements of cisplatin and the related platinum-based drugs—carboplatin—a second generation, and oxaliplatin—third generation, and those of regional use in specific countries, such as nedaplatin, lobaplatin, and heptaplatin, their application is limited [
9]. The restrictions are due to their major drawbacks: effectiveness against a limited range of cancers and its intrinsic resistance, development of acquired resistance [
10,
11], severe side-effects [
12,
13], and low solubility. Hence, the efforts in the field of antitumor drugs design target introduction of new formulas possessing both a widened spectrum of chemotherapy and an improved clinical profile [
3,
4,
9,
14]. Intensive research on drug development processes during the past years shows that the invention of universal compounds active against many cancer types is a task difficult to accomplish. The right way is to develop drugs effective against a small group or a subgroup of cancers. Thus, the successful route leading to new efficient drugs inducing a better tumor response in individual patients is to design a pharmacological agent targeting specific abnormalities in particular cancer cells [
4].
There are several approaches to develop new metal-based antitumor agents. Historically, the first one follows the correspondence between anticancer activity and the molecular structure of cisplatin [
3,
4,
15]. Thus, a variety of cisplatin-similar platinum(II) complexes with different ligands have been synthesized and tested. The numerous results achieved have led to a set of rules for constructing a molecular structure that appeared to be required in order to manifest antitumor activity [
7]. According to the rules, the neutral platinum complex with square-planar geometry, containing two cis-am(m)ine carrier ligands (capable of participating in a hydrogen bonding formation) and two cis-coordinated leaving groups (Cl
− ligands) possesses the necessary structural features for intravenous administration into the blood stream. It is considered that the complex should remain largely unchanged during circulation and after entering the cells a substitution of one or two more labile ligands occurs thereby activating the complex. This requirement underlines the necessity to render the complex inactive during the transport and determines that the ligand exchange rates should be compatible to the rates of cell division processes [
1,
16]. The platinum(II) complexes whose structure follows these rules represent the so-called classical platinum chemotherapeutics. Numerous investigations have been conducted in order to determine the mechanism by which these drugs carry out their anticancer action [
17] and most results concerns cisplatin. In general, the main steps cover: cellular uptake by passive diffusion [
18] or active transport via the copper transporters CTR1 and CTR2 [
19,
20]; aquation followed by activation; DNA platination and cellular processing of Pt-DNA lesion leading to apoptosis or to the cell survival. Nuclear DNA is considered the ultimate target of cisplatin and related platinum therapeutics and their capability to form bifunctional DNA crosslinks causing the DNA distortion. Thus, the platinum induced kink in the DNA molecules leads to a chain of events including protein recognition and eventual apoptosis. Unfortunately, some of the platinum compounds can be activated in biological milieu before reaching the tumor cell and they can also interact with nontarget biomolecules. Furthermore, it is clear that DNA is the ultimate pharmacological target of the platinum drugs, but the important issue here is to discern tumor cells from healthy cells. Now it is accepted that the classical platinum compounds can enter in each cell, however the healthy cells and some of the tumor cells can reverse the damage and remove the platinum compounds. Therefore, it is clear that the compounds belonging to this group cannot offer any advantages over cisplatin. Indeed, more than ten other cisplatin related derivatives are currently in clinical trials and the experimental data show [
21,
22] that they have not overcome considerably many of the disadvantages arising from this common structure. However, the achievement analysis in this field demonstrates that metal coordination compounds can play an important part in anticancer treatment regimes.
Another route to create new metal-based anticancer agents is focused on the mechanism of their antitumor action [
23]. In this respect one could distinguish compounds capable of interacting in two different mechanisms: compounds that cause DNA distortion, here including cisplatin- similar and cisplatin dissimilar interactions [
3,
24,
25,
26] and compounds interacting with the key protein targets (including enzymes) that are selective for the specific malignancy and/or that regulate apoptosis, and/or that are responsible for cell invasion and metastasis [
27,
28]. The classical approach based on cell viability assay and characterization of the compounds that bind to DNA and adducts formed has been applied for more than forty years and now this is the first stage for evaluation. Nowadays, the drug design concepts are based on one side on the chemical nature of the compounds and on the other side—on the cancer cells biochemistry. Thus, the new strategies for design and synthesis result in the development of new classes of antitumor agents, the so called “classical nonplatinum metal compounds” and “nonclassical metal compounds” [
3] and thus several compounds (e.g., NKP-1339 [
29], NAMI-A [
30], Satraplatin [
31], etc.) are proposed that are on the “verge” of clinical application. These strategies are consistent with requirements for: a comparatively soluble prodrug’s form; existence of an inactivated form in the blood stream; selective cellular uptake; activation of the drug form in the tumor cells; appropriate tumor cell damage.
Metal coordination compounds with their inherent properties can meet these requirements to a great extent. Their specific three-dimensional structures combined with suitable electronic and ligand exchange properties are attractive for the creation of a successful formula [
3,
4]. The transition series metal complexes having variable oxidation states and coordination numbers, and the ability to bind, of different strength, to a wide variety of donor functional groups are some of the most tested compounds. A special attention is paid to the metals from second and third transition series. They usually exchange their ligands slowly, on the same time scale as the cell division processes. Their comparatively inert behavior keeps them as prodrugs, which have to be activated by ligand substitution reaction in the cells. In general, from a chemical point of view, the successful drug should be a well soluble compound with lipophilicity and acid-base properties determining a selective uptake. The drug should be activated inside the tumor cells and attack the target molecules by ligand substitution reactions. It is necessary to interact predominantly with target molecules and the formed adducts should be stable enough in order to provoke proper cell death. It means that a key element in the design processes is the control on the kinetics of ligand substitution reactions in vivo and the thermodynamic stability of the initial, intermediate, and final complexes produced during the antitumor action. In addition, the redox properties determined in terms of both metal and ligand are of special interest. As a part of antitumor mechanism, the change of the metal oxidation state can trigger ligand release and the participation of the ligands in in vivo redox reactions could produce reactive oxygen species. Also, the possibility to achieve light-triggered activation of an excited-state of the metal complexes in tumor cells instead of their ground-state gives additional advantages in the drugs design. All these considerations give the reason to conclude that a properly selected metal ion with regard to its nature, oxidation state, and coordination polyhedron with right ligands in the inner coordination sphere can create a drug able to overcome some of cisplatin’s disadvantages.
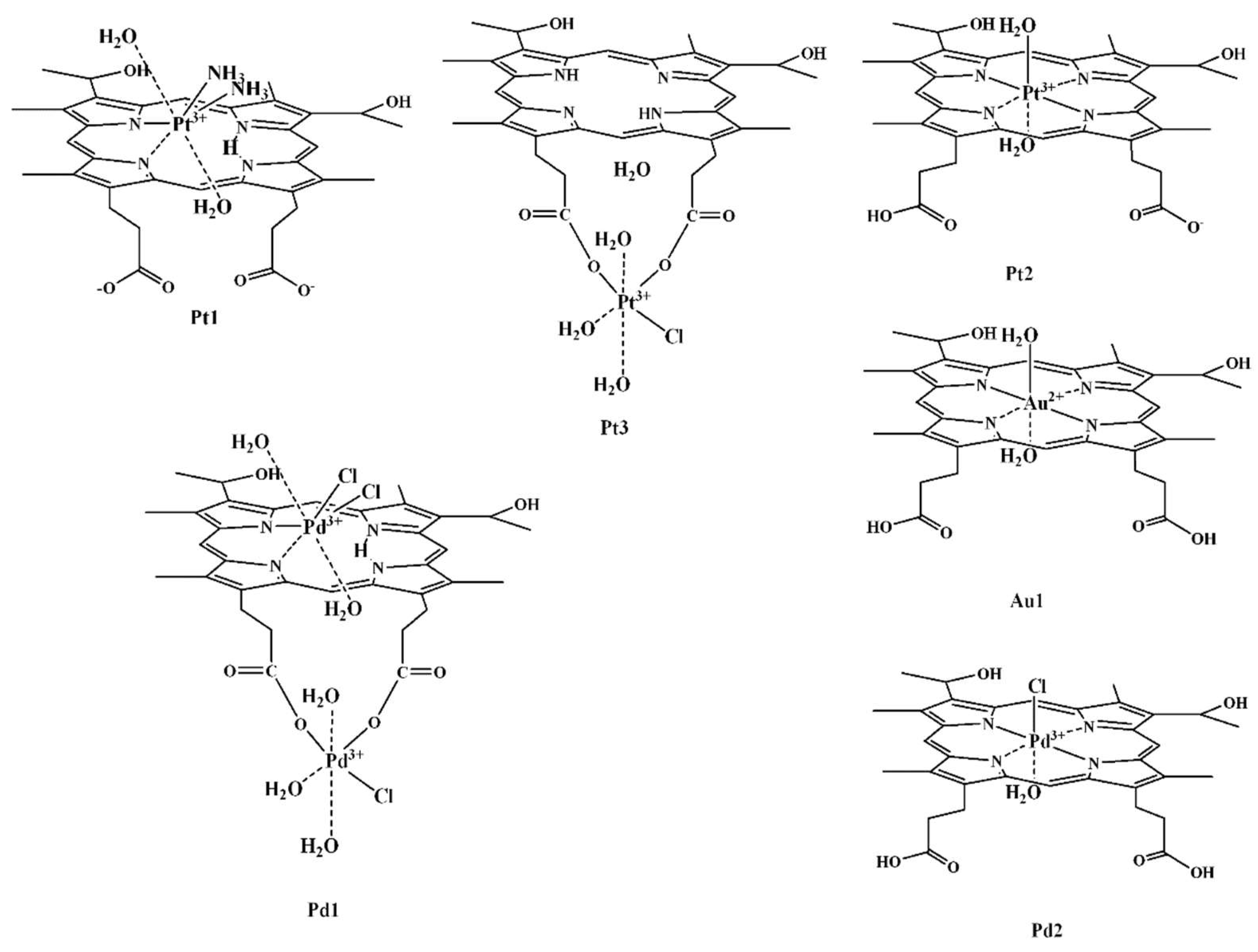
Recently, a series of hematoporphyrin IX (7,12-bis(1-hydroxyethyl)-3,8,13,17-tetramethyl-21H-23H-porphyn-2,18-dipropionic acid, Hp) complexes [
32,
33,
34,
35,
36,
37,
38,
39] of platinum, palladium, and gold (
Scheme 1) have been tested as tumor growth inhibitors. Hematoporphyrin IX and its derivatives are well known for their widespread application in the photodynamic therapy and diagnosis [
40]. It is considered that, due to their acid-base and hydrophobic properties, the porphyrins could preferably accumulate in the neoplastic tissues [
41,
42,
43]. In fact the selective uptake of porphyrins in malignant tissue cells is due to complex mechanisms, the most important of them being the LDL-receptor mediated endocytosis of porphyrin—lipoprotein complexes formed in the systemic circulation [
40].
A new strategy for cytotoxic agents design with improved properties has been proposed in our group, based on the capabilities of this ligand to stabilize unusual oxidation states of the metals. Thus, three Pt
III (d
7) [
32,
33], two Pd
III (d
7) [
34], and one Au
II (d
9) [
35,
36] complexes of Hp were obtained and characterized. The metal ions in the complexes have distorted octahedral coordination. Because of the metals’ electronic configuration, the complexes manifest paramagnetic behavior. Further they show comparatively high cytotoxicity in in vitro tests against a panel of human cell lines [
32,
33,
34,
36]. Great efforts have been made in order to understand the effect of the ligand coordination to the antitumor behavior of the complexes. The ligand Hp has a polydentate nature. Three different modes of coordination have been established [
37] studying its interaction with metal ions representatives of first [
38,
39], second [
34], and third transition series [
32,
33,
35,
36]. Coordination via the four pyrrole N-atoms in the porphyrin framework and forming of metalloporphyrin-type complexes is the most widespread mode of binding, typical of all metal ions under investigation. Coordination via two N-atoms of adjacent pyrrole rings to the metal ions in cis-position leads to the so called “sitting atop” type complexes (SAT). Coordination via the side chains deprotonated propionic COO
− groups, outside the porphyrin macrocycle is a less common mode of coordination. The nitrogen donor atoms of the imino (>N) and aza (=N-) groups of the pyrrole rings, as well as the outside COO
− groups determine the nature and the size of three different coordination modes. The metal ions choose a different mode of coordination as a function of their nature and properties.
Due to its intrinsic inertness, Pt
III forms stable complexes with the three different modes of Hp coordination (
Scheme 1) in the proper reaction conditions [
32]. All these complexes with distinct coordination patterns exert concentration dependent antiproliferative activity against a spectrum of cell lines representing some important types of neoplastic diseases in humans. They have also proven to be far less cytotoxic against the human embryonal kidney cell line HEK-293T compared to cisplatin. It has been discovered that the “sitting atop” complex
Pt1, with PtN
4 coordination plane formed by two adjacent porphyrin pyrrole nitrogens and two NH
3-molecules in cis-position as well as the metalloporphyrin-type complex
Pt2 possess higher potency compared to
Pt3. In the latter, Pt
III is coordinated to the deprotonated propionic carboxylic groups from the side chains of hematoporphyrin IX. The better solubility of
Pt2 compared to
Pt1 makes it preferable in research. Thus, its characteristics, such as superior proapoptotic activity that strongly correlates with its cytotoxicity and significantly higher degree of accumulation in tumor cells compared to the reference drug cisplatin, make it a reliable representative of the group of the “metal-based drugs that break the rules” [
4].
Another promising representative of this group of compounds is the octahedral gold(II) (d
9) complex of hematoporphyrin IX,
Au1 [
35] that is structurally similar to
Pt2. The comparison of its cytotoxicity to that of
Pt2 manifests that the Au
II complex exerts superior activity exactly against the T-cell leukaemia SKW3. These data correlate well with the estimated specific inhibiting effect of gold species upon immune cells and T-cells [
36]. Nevertheless, the compound exerts well-pronounced proapoptotic properties against malignant cells and it is less cytotoxic than cisplatin for the human kidney. Furthermore, this compound demonstrated significant intracellular accumulation presumably mediated by formation of FCS-lipoprotein complexes and subsequent endocytosis.
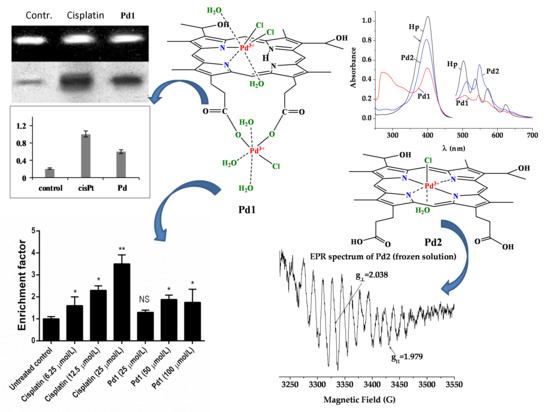
Two relatively new members of this group compounds are the Pd
III (d
7) coordination compounds of hematoporphyrin IX [
34]. The dinuclear [Pd
III2(Hp
-3H)Cl
3(H
2O)
5]·2PdCl
2,
Pd1 and mononuclear [Pd
III(Hp
-2H)Cl(H
2O)]·H
2O,
Pd2 have been obtained during the interaction of the ligand with Pd
IICl
42− in alkaline-aqueous medium. In the dinuclear complex,
Pd1, one of the Pd
III ions is coordinated to the deprotanated COO
− groups from the side chains of the porphyrin ligand and the second Pd
III ion—to two adjacent pyrrole N-atoms on the top of the porphyrin ring. The compound is spontaneously obtained at a large metal excess from the reaction in alkaline-aqueous medium. It is accepted that because of the greater kinetic lability of Pd
III compared to Pt
III both places for coordination are occupied simultaneously and thus a dinuclear Pd
III-Hp-Pd
III system is formed. The Pd
III ion in the mononuclear complex,
Pd2 is incorporated in the porphyrin core. The Pd
III centers in both complexes have a distorted octahedral coordination filled with additional donor species such as Cl
− and H
2O.
As a member of the platinum group metals, palladium, with its coordination compounds, is also extensively tested for antitumor activity [
44,
45,
46,
47]. Because of lanthanoid contraction, both metals palladium and platinum in oxidation state +2 have close ionic radii. They adopt a square-planar geometry and behave like soft acids, forming strong bonds with nitrogen and sulfur-containing ligands [
48]. While the equilibrium constants, characterizing the stability of their isostructural complexes differ slightly (only about ten times higher for Pt
II complexes), their kinetic behavior with respect of the ligand substitution is completely dissimilar as a consequence of the lower electron density of Pd
II. The complexes of Pd
II simple analogues of Pt
II-antitumor drugs undergo aquation and ligand exchange reaction 10
4 to 10
5 times more rapidly. The fast hydrolysis of the leaving groups leads to the formation of very reactive Pd
II-aquated-species, unable to reach their pharmacological targets as active compounds. It is accepted that the toxic side effects are a result of inactivation of certain enzymes due to binding to the thiol groups of cysteine residues and obviously the much higher reaction rates typical of Pd
II compounds are favorable for general toxicity.
In order to tune the kinetic behavior and thermodynamic stability of the new compounds proposed as antitumor agents different approaches can be applied. Here, beside the construction of special coordination polyhedra, the intermediate oxidation state +3 of palladium centers has been applied as a factor of great importance. The less common oxidation state of palladium +3 provides many advantages of its complexes, such as a controlled delay of the ligand substitution reactions and reactivity, an octahedral geometry with axial ligands that could alter the redox potential and lipophilicity. The promising antiproliferative activity in micromolar concentration range that has been shown by the Pd
III-Hp complexes [
34] together with the remarkable cytotoxicity against the K-562 cells with more than 4 fold lower IC
50 value compared to cisplatin, characteristic for the dinuclear compound
Pd1, is the basis for further detailed biological investigation on the mechanism of action of these new proposed cisplatin-dissimilar agents.
3. Discussion
In the framework of the design of new antitumor agents, two paramagnetic Pd
III—complexes of hematoporphyrin IX were obtained and biologically tested as representatives of the group of cisplatin-dissimilar metal-based coordination compounds. The reaction conditions for their synthesis were chosen to provide obtaining of Pd
3+ species [
57] and their stabilization in solution and solid state through formation of Hp-complexes [
32,
33,
34,
35]. The complexes were synthesized during the interaction of an initial Pd
II compound as a chloride complex ([PdCl
42−]) with the twofold deprotonated at peripheral propionic acid groups hematoporphyrin IX ligand ([Нр
-2Н]
2−) in alkaline-aqueous medium achieved by adding KOH. All measurements performed during the interaction manifested that a redoxy process takes place together with the coordination reaction. The large spontaneous increase of the acidity of the reaction system together with the appearance of the EPR signals (
Figure 1) proved that the studied process is a very complicated coordination reaction with production of a mixture of paramagnetic metal complexes and stable radicals. The appearance of two-component anisotropic EPR signals with different parameters during the interaction indicates also formation of complex species with different inner coordination sphere of the paramagnetic metal centers. The mode of Hp coordination can be distinguished by using UV/Vis characterization of the reaction system. Coordination via the side deprotonated propionic COO
− groups could be supposed because of the hypsochromic shift of the Soret band owing to the porphyrin plane distortion. The spectral changes that follow are most probably connected to a distortion of the porphyrin ring symmetry owing to the coordination through two adjacent pyrrole N-atoms and formation of lower symmetry complex species. The reduction of the Q-bands number to two at the end of the reaction is owing to degeneration of the exited state S
1 orbitals [
58] and proves the formation of a metalloporphyrin type complex (D
4h symmetry).
Two of the complex species formed during the interaction PdCl
42−-Hp
-2H have been isolated at proper reaction conditions. A dinuclear compound
Pd1 was the main product from the interaction at a metal excess (Pd:Hp ≥ 4) and was spontaneously precipitated at pH ~ 8 and a mononuclear metalloporphyrin type complex
Pd2 was the main product of the interaction at an equimolar ratio of the reagents and was precipitated by adding hydrochloric acid (5 × 10
−2 M HCl). The composition of the complexes was derived from the elemental analyses and the content of H
2O and Cl
− in the inner or the outer coordination sphere was proven by studying their thermal behavior [
34]. The molecular structure of the complexes in solid state was deduced based on detailed investigations of their magnetic properties and spectroscopic characterization, published in [
34].
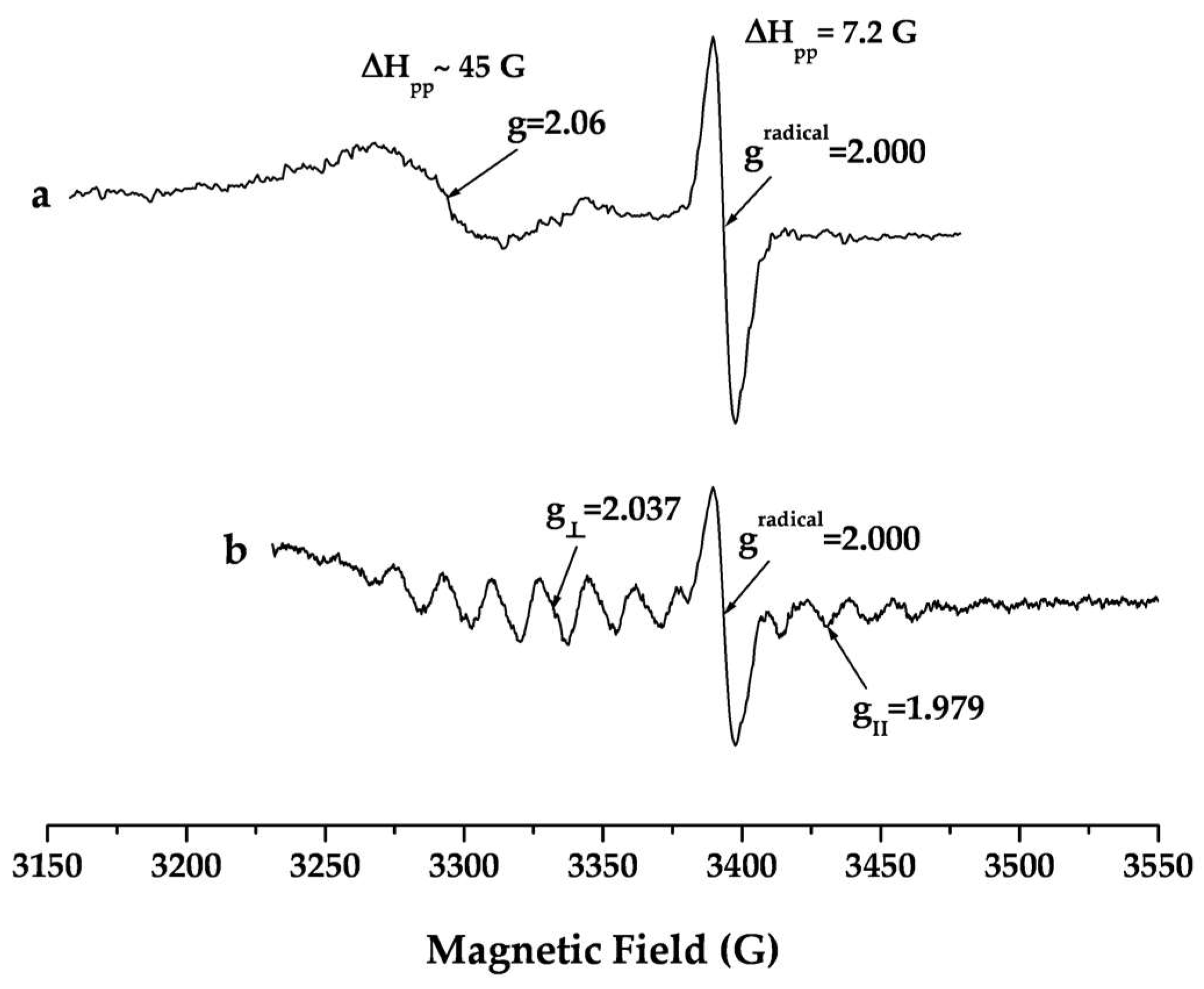
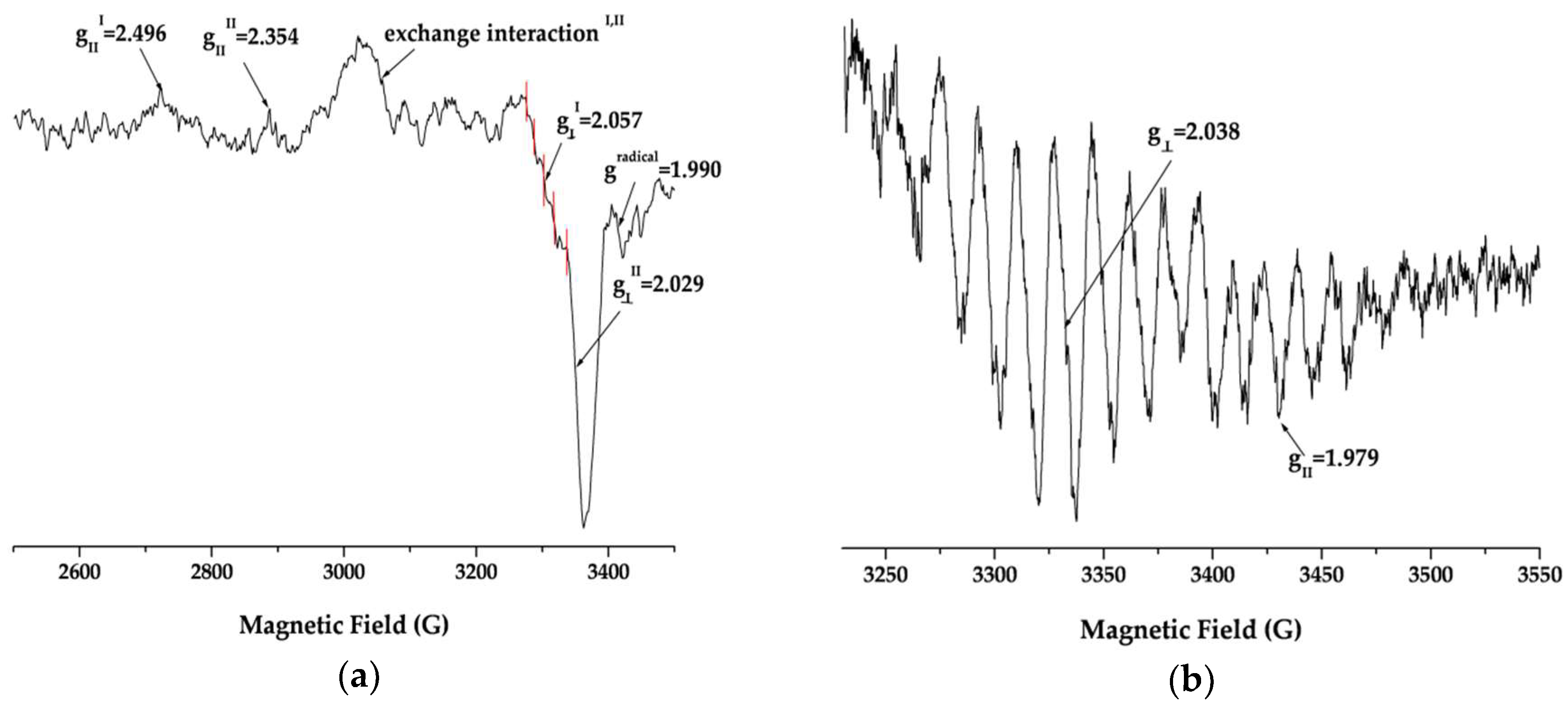
A crucial issue in developing a new drug formulation is to study the structure of the compound in solution and to establish the relationship with the solid state structure. The data presented here relate to the behavior of the complexes in DMSO solutions. The low temperature EPR spectra (
Figure 4a,b) of the complexes dissolved in DMSO show characteristic EPR spectral patterns of paramagnetic Pd-compounds. The EPR spectra recorded several hours (5–8 h) after the dissolving correspond well to the solid state EPR spectra [
34]. The two anisotropic EPR signals observed in the spectrum of
Pd1 possess axial symmetry. The principal values of the g-tensors of the two anisotropic signals g
II > g
⏊ > 2.0023 are consistent with formation of an elongated octahedral coordination with (dz
2)
1-ground state. A five-component superhyperfine structure is observed only in the perpendicular region of the low-field signal. The principal value of the superhyperfine coupling tensor a
⊥(N) = 15.31 × 10
−4 cm
−1 (I(
14N) = 1) and the number of superhyperfine lines typical for interaction of the uncoupled electron with two
14N nuclei proved coordination of one Pd
3+ via two of the pyrrole nitrogen donors of the Hp ligand. The absence of a superhyperfine structure from
14N nuclei on the upfield EPR signal is due to coordination out of the porphyrin ring through the propionic acid groups. The wide signal observed at ~3050 G could be assigned to exchange singlet–triplet interaction between the two unpaired electrons of the differently coordinated Pd
3+-centers in the molecule of
Pd1. Hence the EPR spectra proved the formation of a Pd
3+-L-Pd
3+ system where each of the Pd-ions possesses different coordination. The presence of a signal for a radical could be explained with significant delocalization of the unpaired electron density because of electron-acceptor properties and significant flexibility of the porphyrin moiety. The complex
Pd2 features a completely different EPR spectrum pattern in solution containing exactly one two-component anisotropic signal. The signal displays axial anisotropy with principal values of the g-tensor g
⏊ > 2.0023 > g
II proving formation of a compressed octahedral structure with (dx
2 − y
2)
1 ground state. Superhyperfine lines were observed both in perpendicular and parallel regions. The principal values of the axially symmetrical superhyperfine coupling tensor are typical for the interaction of an uncoupled electron with
14N (I = 1) nuclei. The nine superhyperfine lines are readily distinguished in the perpendicular region and prove coordination of Pd
3+ into the four pyrrole N-donors in the porphyrin ring. Thus the spectrum confirms the existence of a stable metalloporphyrin type complex in solution.
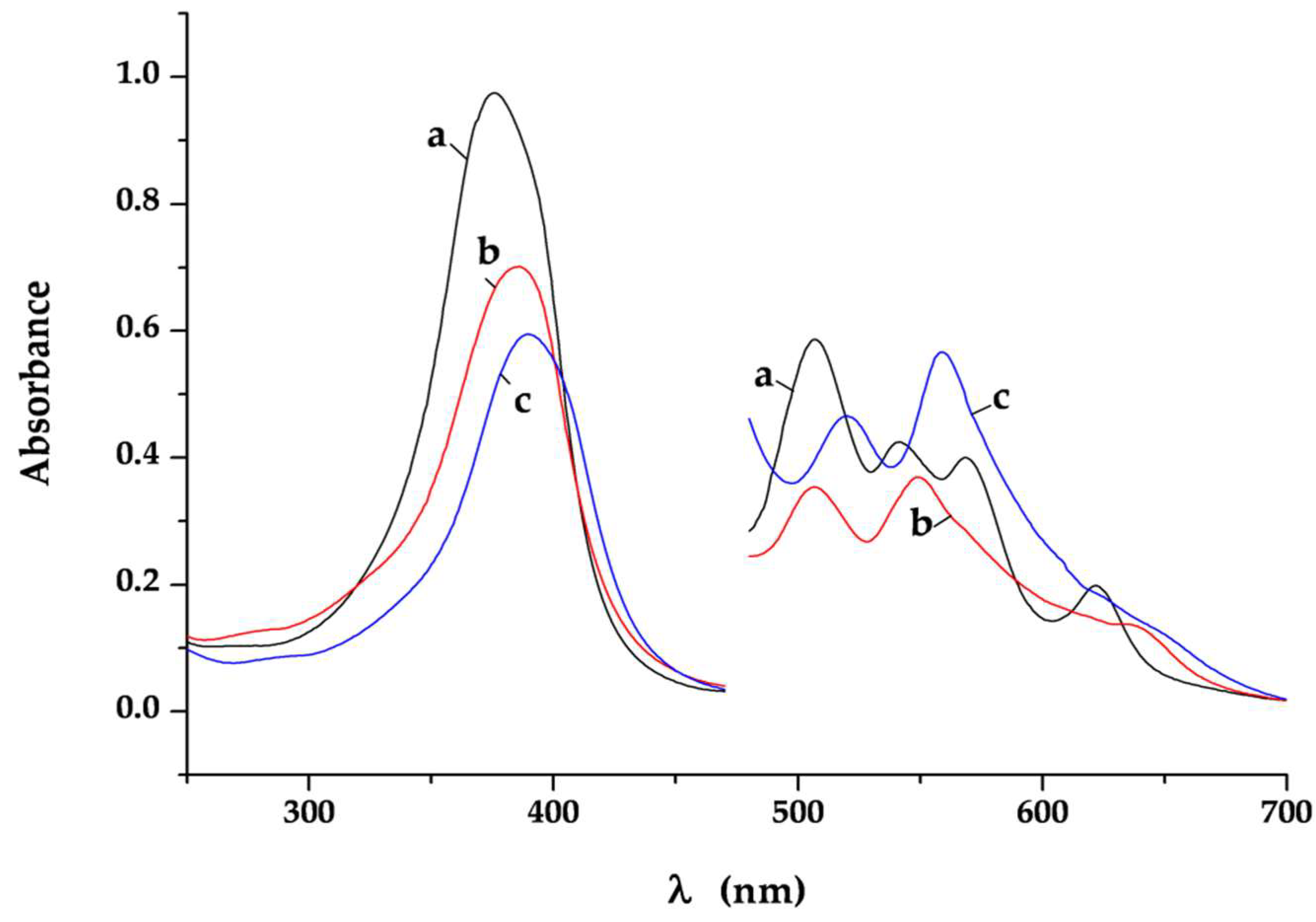
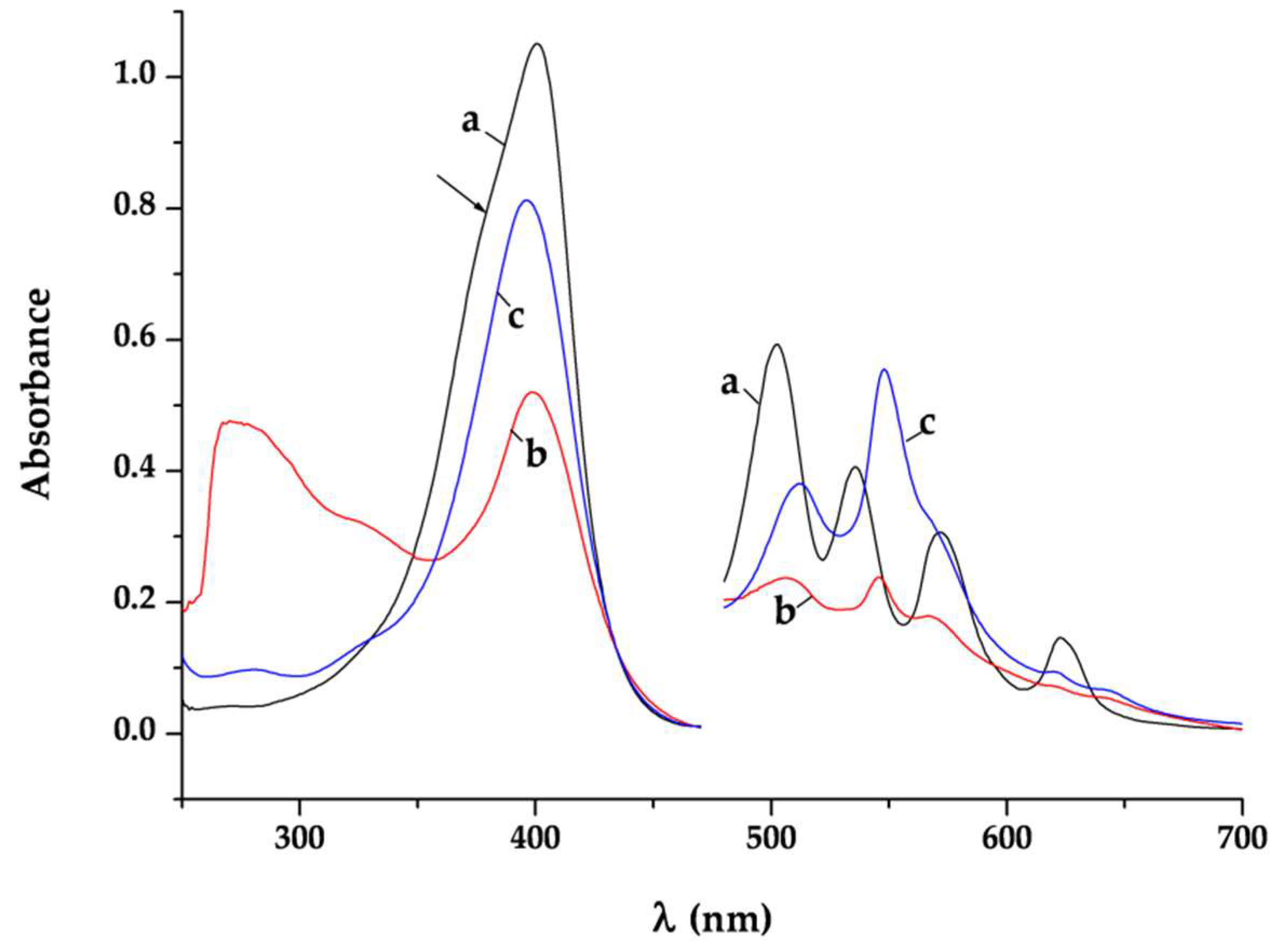
It is well-known that changes in the conjugation and the symmetry of the Hp-ligand can affect the UV/Vis absorption spectra and can be used to obtain data about the mode of Hp coordination at complex species in solution [
32,
33,
34,
35,
36,
37,
38,
39]. While the metal binding through inner nitrogen atoms induces strong changes in the visible region of the spectra, the variations in the peripheral substituents causes minor changes owing to geometrical deformation of the porphyrin ring. The UV/Vis spectra were recorded of the studied compounds dissolved in DMSO. It was found that the position of the Soret band is sensitive to the processes such as acid-base equilibria and aggregation, and also to the modes of metal coordination. It is accepted that the Soret band appears at 376 nm if the propionic acid groups are deprotonated and shifts in accordance with the electronic density distribution caused by protonation and metal-coordination. The major component of the Soret band in the spectrum of the free ligand dissolved in DMSO (400 nm, lgε = 5.02) is batochromically shifted compared to the spectrum registered in alkaline-aqueous medium. The shoulder observed at 376 nm is due to the protolitic equilibrium Hp + 2Solv ⇔ [Hp
-2H]
2− + 2[HSov]
+, which is clearly shifted to the neutral Hp molecules in the weakly proton-acceptor solvent DMSO. In the spectra of the complexes this band is observed at 399 nm (
Pd1) and 396 nm (
Pd2), respectively. Thus it indicates that the propionic acid groups are engaged in the processes of coordination or protonation.
The spectrum pattern in the area of Q-bands is determined by the symmetry group of the porphyrin macrocycle. The two diagonally located hydrogen atoms at pyrrolic nitrogens in the molecule of the free ligand define a D2h symmetry and hence a four number Q-bands spectrum. The loose of a proton gives a monoprotic ligand form. Many metal ions at proper pH value (6 < pH < 10) interact with the monoprotic ligand anion and bind with its two cis-disposed pyrrole N-donors on the top of the macrocycle forming sitting-atop (SAT) complex. The metal in these complexes is located out of the porphyrin plane and distorts it. The symmetry of the structure is lower (approximately between C4v → C1) than that of both metalloporphyrins (D4h) where the two ammine hydrogens are substituted by the metal located coplanar on the porphyrin ring and the free Hp ligand (D2h). This reflects on the Q-bands number reduction and changes their intensities. Hence the observed spectrum of the complex Pd1 in the area of Q-bands containing mainly three Q-bands (507, 546, and 567 nm) with intensity IV ~ III > II is consistent with coordination through the two cis-disposed pyrrole N-donors on the top of the porphyrin macrocycle substituting one of the pyrrolic nitrogen. Further on, the slight red shift of the Q-bands: IV and III and a blue shift of the Q band II with degeneration of the Qx and Qy orbitals connected with vibronic structure supports simultaneous coordination on the top of the porphyrin ring and out-side the porphyrin macrocycle through the peripheral deprotonated carboxylic groups. The typical two Q-band spectrum of the complex Pd2 indicates unambiguously a D4h symmetry achieved at coordination through the four N-heteronuclei in the porphyrin macrocycle. The intensive bands at 270 (lgε = 4.98) and 323 nm (lgε = 4.81) in the spectrum of Pd1 as well as the one at 280 (lgε = 3.99) in the spectrum of Pd2 were assigned to the ligand-to-metal charge transfer bands of the Pd-Cl bonds. This proves the presence of Cl− ions in the palladium ions inner coordination sphere.
The spectroscopic characteristics observed were unchangeable within more than five days. Hence in this period the compounds, dissolved in DMSO solution are paramagnetic palladium-hematoporphyrin IX complexes as follows: (1) for Pd1—a dinuclear Pd-Hp-Pd system, with two PdIII ions that occupied two coordination places—at the porphyrin ring binding out-of-plan through two cis-disposed N-donors and at the two peripheral propionic acid groups; (2) for Pd2—normal metalloporphyrin of PdIII. Furthermote, this is the reason to accept that during the biological investigation the active species in solution are these palladium complexes of hematoporphyrin IX.
The cytotoxic screening of the two palladium(III) complexes was conducted on a wide spectrum of cell lines—representatives of the main human cancer types. The results of the MTT-dye reduction assay [
50,
51] unambiguously indicate that the two compounds exert concentration-dependent antiproliferative effects against the chosen spectrum of cell lines. Data analysis shows that the tested palladium compounds are generally less active than cisplatin, causing half-maximal inhibition of cell viability at generally higher concentrations (
Table 1). It has also been proven that the dinuclear palladium complex
Pd1 demonstrates superior activity as compared to the metalloporphyrin type complex
Pd2. The latter
Pd2 compound exerts only marginal activity and fails to cause 50% inhibition of malignant cell growth against a half of the cell lines under evaluation. Throughout the panel investigated the anticancer drug cisplatin proved to outclass the novel palladium complexes, with the only exception of K-562 leukemia whereby
Pd1 showed remarkable cytotoxicity [
34].
These findings gave us a reason to conduct more detailed pharmacodynamic evaluation of Pd1 especially regarding its ability to induce apoptosis and to modify DNA. Significant apoptotic fragmentation of genomic DNA was established after treatment of HL-60 cells with both cisplatin and Pd1, whereby the proapoptotic effect of Pd1 required the cell exposure to concentrations significantly higher than its IC50. These data indicate that the cytotoxicity of both compounds is mediated by induction of apoptosis, although its threshold level was significantly higher for the palladium compound.
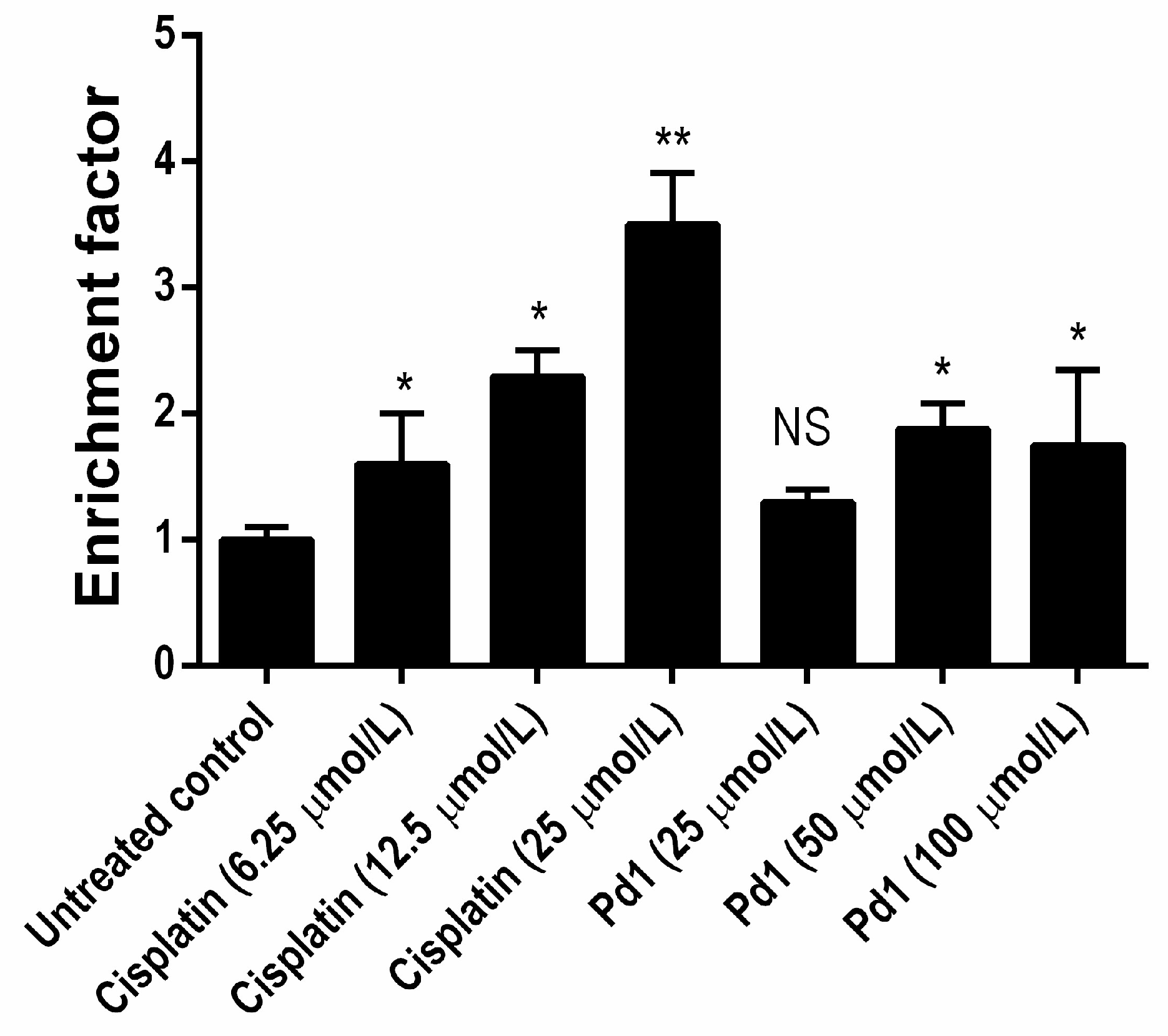
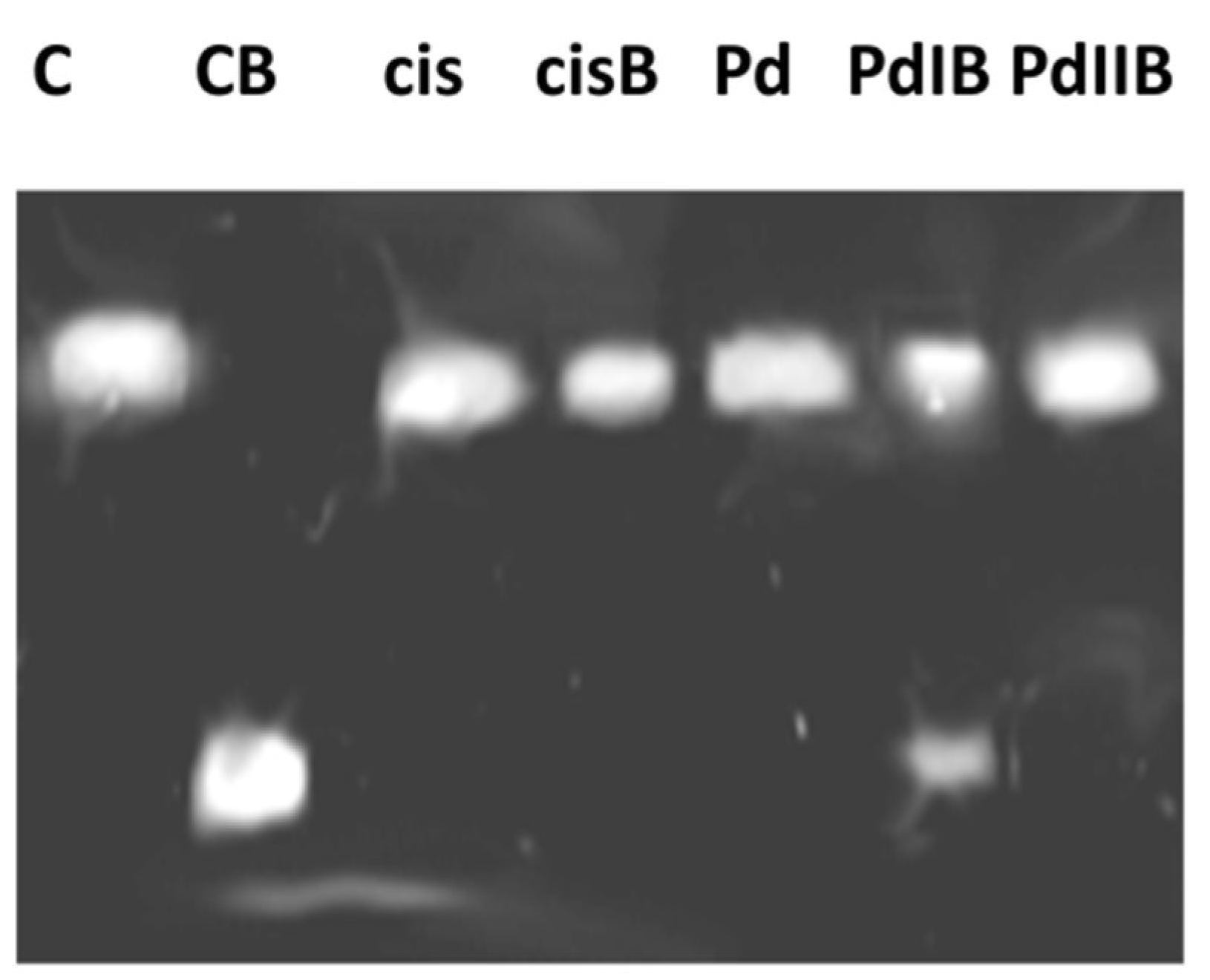
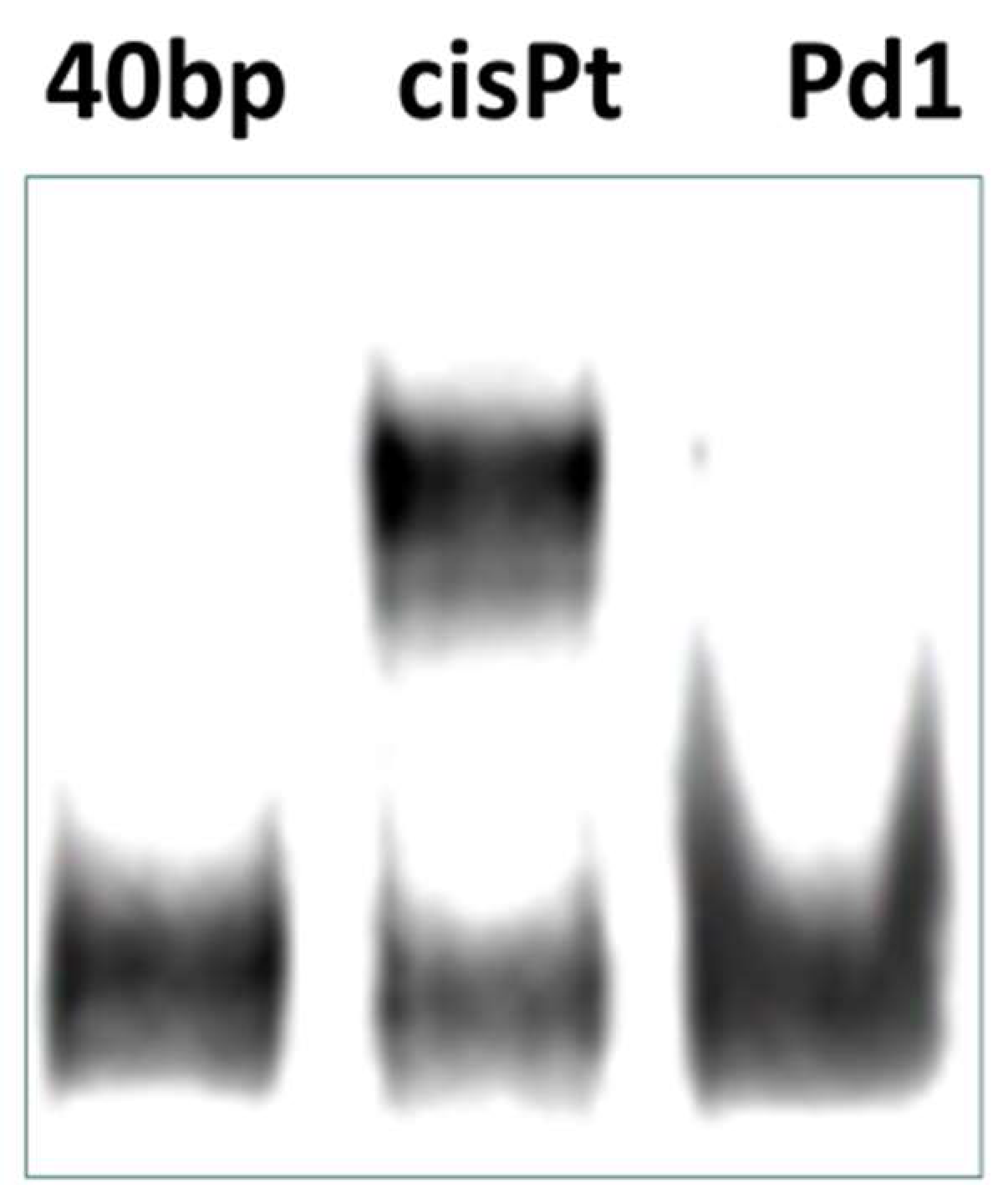
Further experiments were carried out in order to characterize the adduct-forming ability of
Pd1 and the cellular processing of the DNA-lesions. The N7 position of guanine is considered as the ultimate pharmacological target of platinum drugs, leading to formation of intrastrand adducts whose recognition and processing leads to activation of the cell death signaling pathways [
59,
60]. On this ground we studied the metallation of a single strand 40-base DNA fragment in a cell free system. The palladium compound
Pd1 was less capable of forming DNA adducts under the experimental conditions, thus further demonstrating its cisplatin-dissimilar pharmacological properties. These findings indicate that although the DNA-modification plays an important role for the mode of antiproliferative action of
Pd1; its capacity to modify DNA is lower as compared to that of cisplatin.
The structural perturbations induced by the DNA-metallation are recognized by diverse proteins including the DNA-repair enzymatic machinery [
59,
61,
62]. Removal of platinum-DNA adducts by the nucleotide excision repair (NER) is one of the crucial mechanisms of cellular resistance to cisplatin. This prompted us to evaluate the efficiency of NER repair synthesis after modification of pBS plasmids with either cisplatin or
Pd1. The lesions induced by cisplatin were far better recognized and repaired as compared to those of
Pd1, implying that, by virtue of the significant structural differences between the tested complexes, they induce highly dissimilar alterations of DNA conformation. The lower level of NER-mediated removal and repair of
Pd1 modified DNA are an advantageous future of the novel compound as this would condition retained activity against malignant cells overexpressing the NER-enzymatic system.
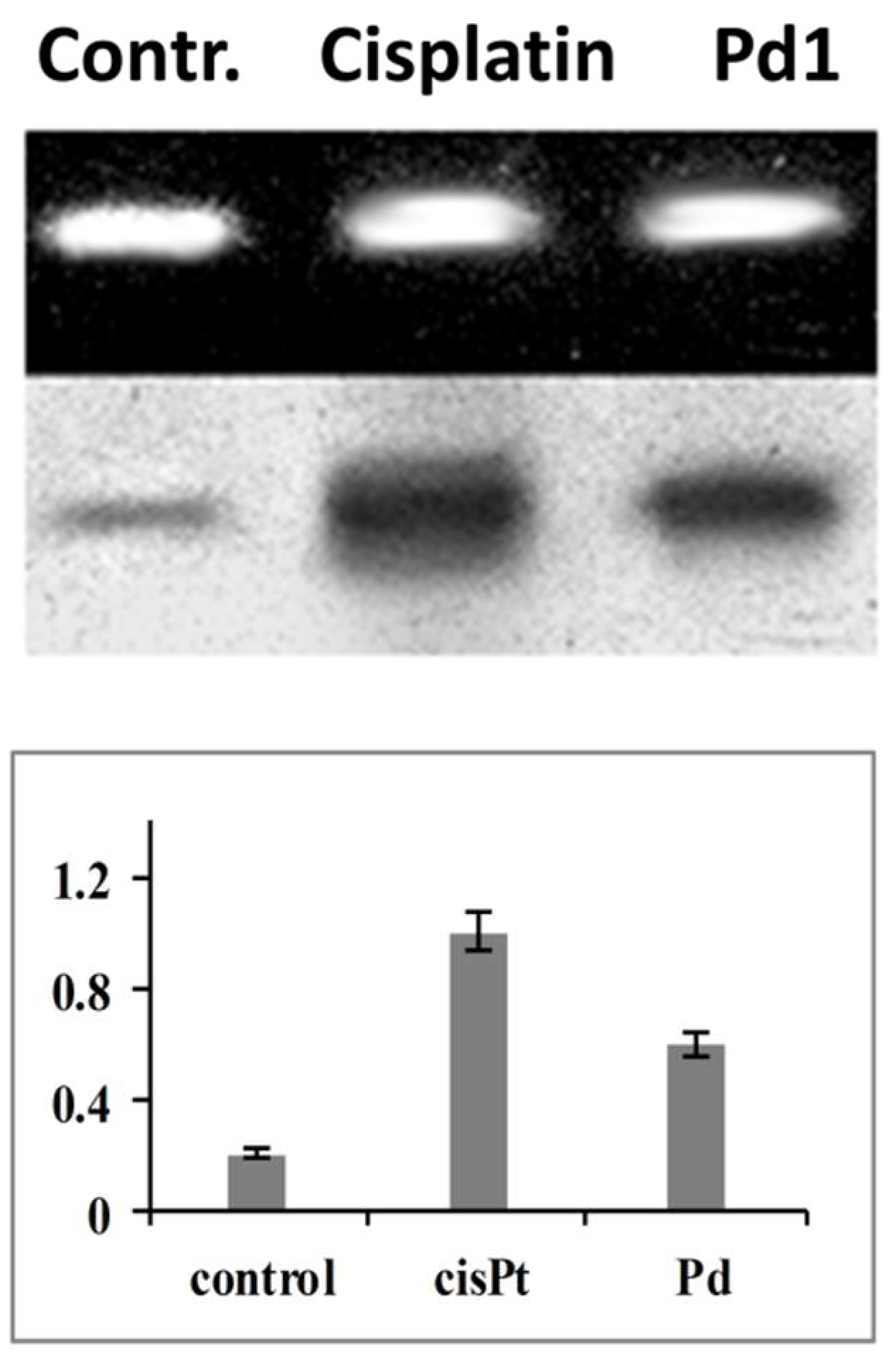
Apart from the DNA-repair enzymes other proteins are also capable of recognizing and binding cisplatin-modified DNA [
59,
61,
63]. Among these special attention has been paid to the high mobility group domain (HMG) proteins [
55,
59,
61,
64]. They are considered to play crucial role for the cytotoxicity of platinum drugs, whereby the proposed mechanisms include: (i) shielding of platinated DNA and steric hindrance against NER-mediated repair of metal-adducts; (ii) “hijacking” i.e., binding of transcription factors or other regulatory proteins to HMG-associated platinum adducts, thus deviating them from their normal targets and compromising their role in signal transduction [
59,
61,
63]. On this ground we evaluated the recognition and binding of HMGB-1 protein to cisplatin or
Pd1 modified DNA probes. As made evident by the results obtained
Pd1-induced modification conditions lower level of HMGB-1 binding, as compared to cisplatin-lesioned DNA. This implies that the high mobility group proteins are most probably less involved in the cytotoxicity of the palladium agent.
5. Conclusions
Two newer members of the group of paramagnetic transition metal complexes of hematoporhyrin IX [
32,
33,
34,
35,
36,
37,
38,
39] have been proposed for extended biological screening: a dinuclear [Pd
III2(Hp
-3H)Cl
3(H
2O)
5]·2PdCl
2 and a mononuclear [Pd
III(Hp
-2H)Cl(H
2O)]·H
2O. The complexes were obtained reproducibly in alkaline-aqueous medium and were isolated as neutral compounds changing the acidity of the reaction system and the M:L molar ratios. Their structure and stability were studied in DMSO solutions in details. It was found that the active species in solution are a dinuclear complex (
Pd1) and a mononuclear (
Pd2) complex. In the dinuclear complex
Pd1, one Pd
III ion is coordinated to the deprotonated COO
− groups from the side chains of the porphyrin ligand and the second Pd
III ion—to two adjacent pyrrole N-atoms on the top of the porphyrin ring and thus a dinuclear Pd
III-Hp-Pd
III system is created. Pd
III in the mononuclear complex,
Pd2, is located in the plane of the porphyrin ring and thus a metalloporphyrin type complex is formed. Pd
III centers in both complexes have a distorted octahedral coordination filled with additional donor species such as Cl
− and solvent molecules.
The compounds tested manifested cell growth inhibitory effects at micromolar concentration against tumor cell lines with distinct cell type and origin. The calculated IC50 values proved that in general, palladium complexes exhibit lower activity compared to that of the referent drug cisplatin and as a rule the metalloporphyrin type complex Pd2 is less active than the dinuclear compound Pd1. Contrarily to the general trend the Pd1 complex exerts remarkable activity against K-562 cells, with 50% cell growth inhibition at more than 4-fold lower concentration compared to cisplatin. It also displays relatively close activity against the HD-MY-Z cell line with regard to the referent drug. The palladium complexes’ ability to induce programmed cell death was evaluated in a comparative experiment of Pd1 and cisplatin in HL-60 cells. The two compounds cause significant increase in the apoptotic histone-associated DNA fragments. However, the novel dinuclear compound causes comparable proapoptotic effects at substantially higher concentrations and that corresponds to the tumor cell line chemosensitivity bioassay. The parallel evaluation of Pd1 and cisplatin’s ability to form intra-strand guanine bis-adducts shows that Pd1 also inhibits the nuclease activity, but failed to totally hamper the fragmentation of the target DNA-molecule. Hence, although the DNA-modification plays an important role for the mode of antiproliferative action of Pd1, its capacity to modify DNA is lower compared to that of cisplatin. The elucidation of DNA-adducts cellular processing by the NER enzymes demonstrated that the lesions induced by cisplatin were far better recognized and repaired as compared to those of Pd1. The lower level of NER-mediated removal and repair of Pd1 modified DNA are an advantageous characteristic of the novel compound and that means that Pd1 would retain the activity against malignant cells, overexpressing the NER-enzymatic system. The ability of HMGB-1 protein to bind metallated 40-base DNA fragment with the reference cytotoxic drug cisplatin resulted in a specific binding. In a dissimilar manner, the Pd1-modified DNA was not recognized and it could be expected that the Pd1-modified DNA would not be shielded by the HMGB-1.
The data analysis of the in-depth biological study unambiguously highlights the differences in molecular pharmacology of the presented “applicants” for antitumor agents in respect to cisplatin. Moreover, the advantages of the new compounds provide grounds for joining them to the “nonclassical metal compounds” group. Their unique structure, based on the octahedral coordination of palladium(III) stabilized with a ligand with favorable properties, is a prerequisite for constructing a new formula with a potential of controlling its kinetic behavior as well as the strength of the M-L bonds of the adducts formed in the biological milieu.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}