Full-Length Transcriptome Survey and Expression Analysis of Cassia obtusifolia to Discover Putative Genes Related to Aurantio-Obtusin Biosynthesis, Seed Formation and Development, and Stress Response


Abstract
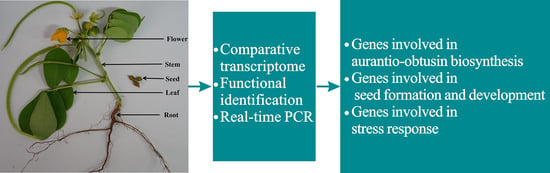
:
1. Introduction
2. Results
2.1. Combined Sequencing Approach to the Various Organs of C. obtusifolia
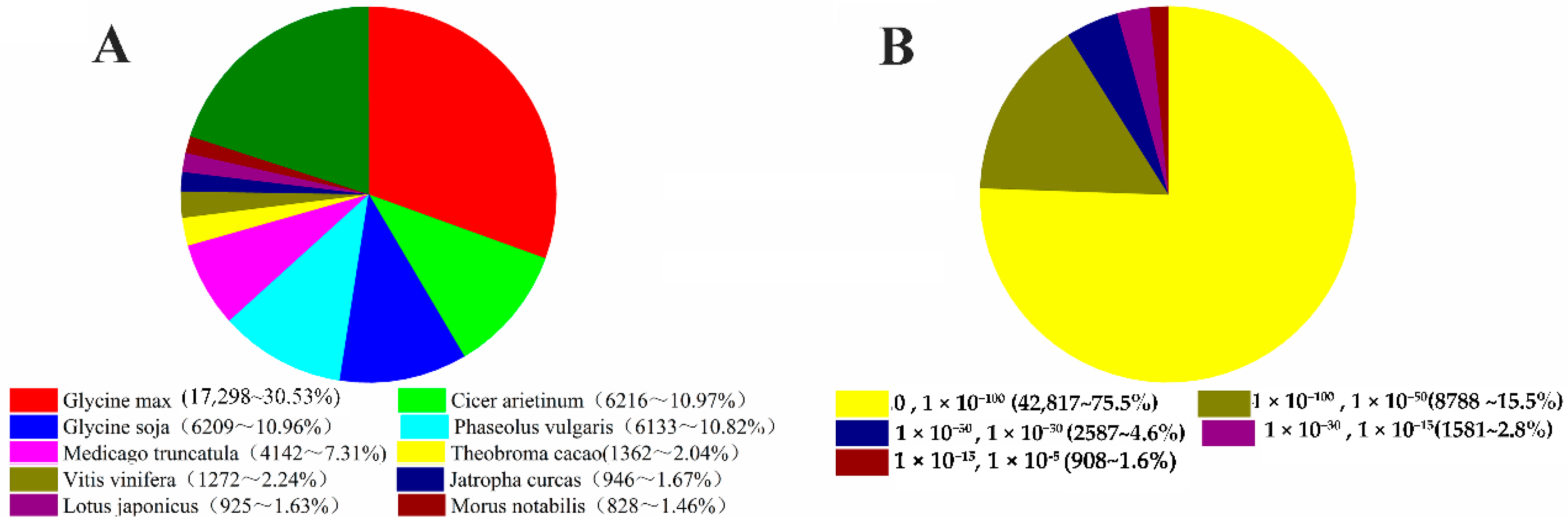
2.2. Functional Annotation Based on Searches against Public Databases
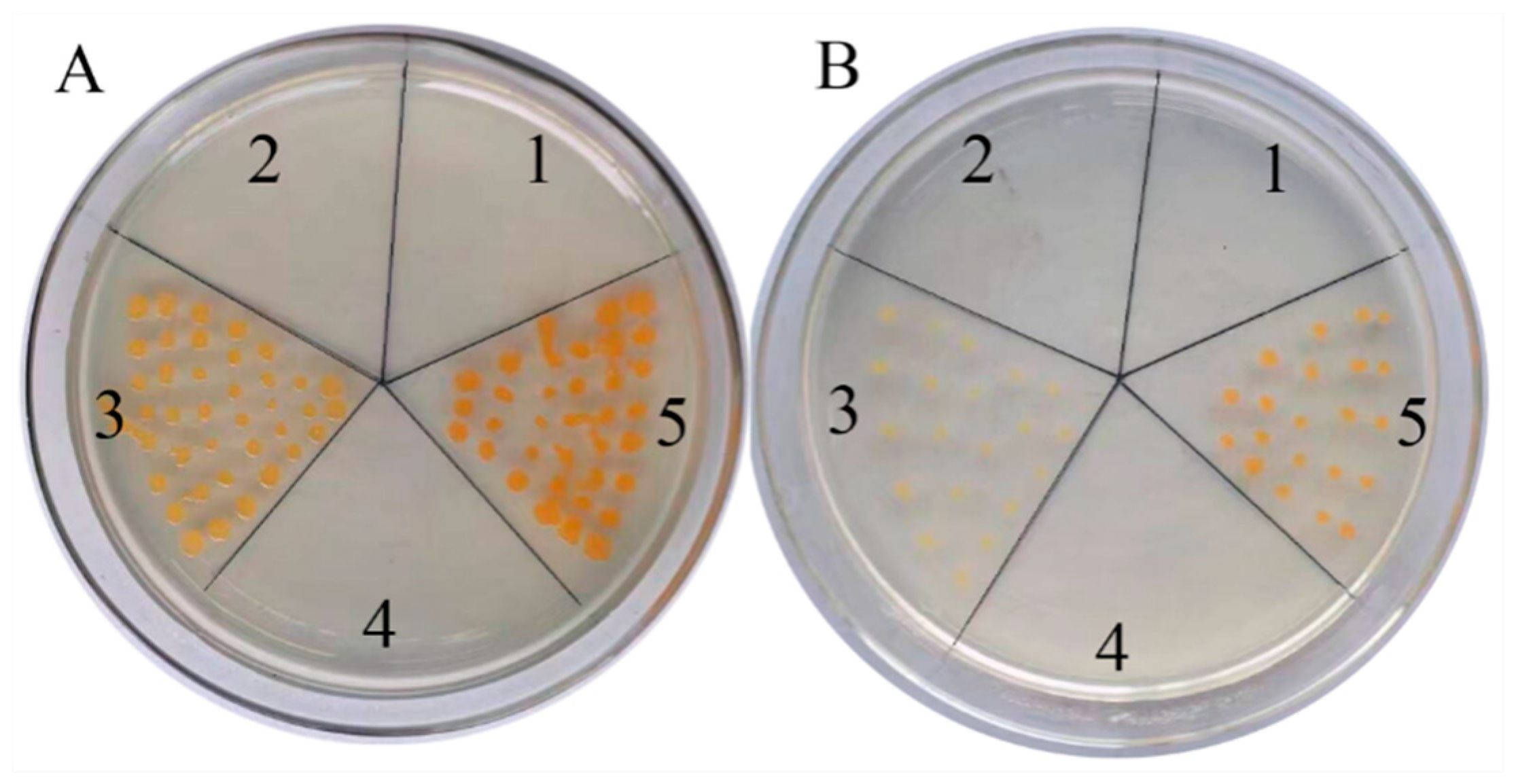
2.3. Functional Analysis of CoDXS and CoDXR Genes
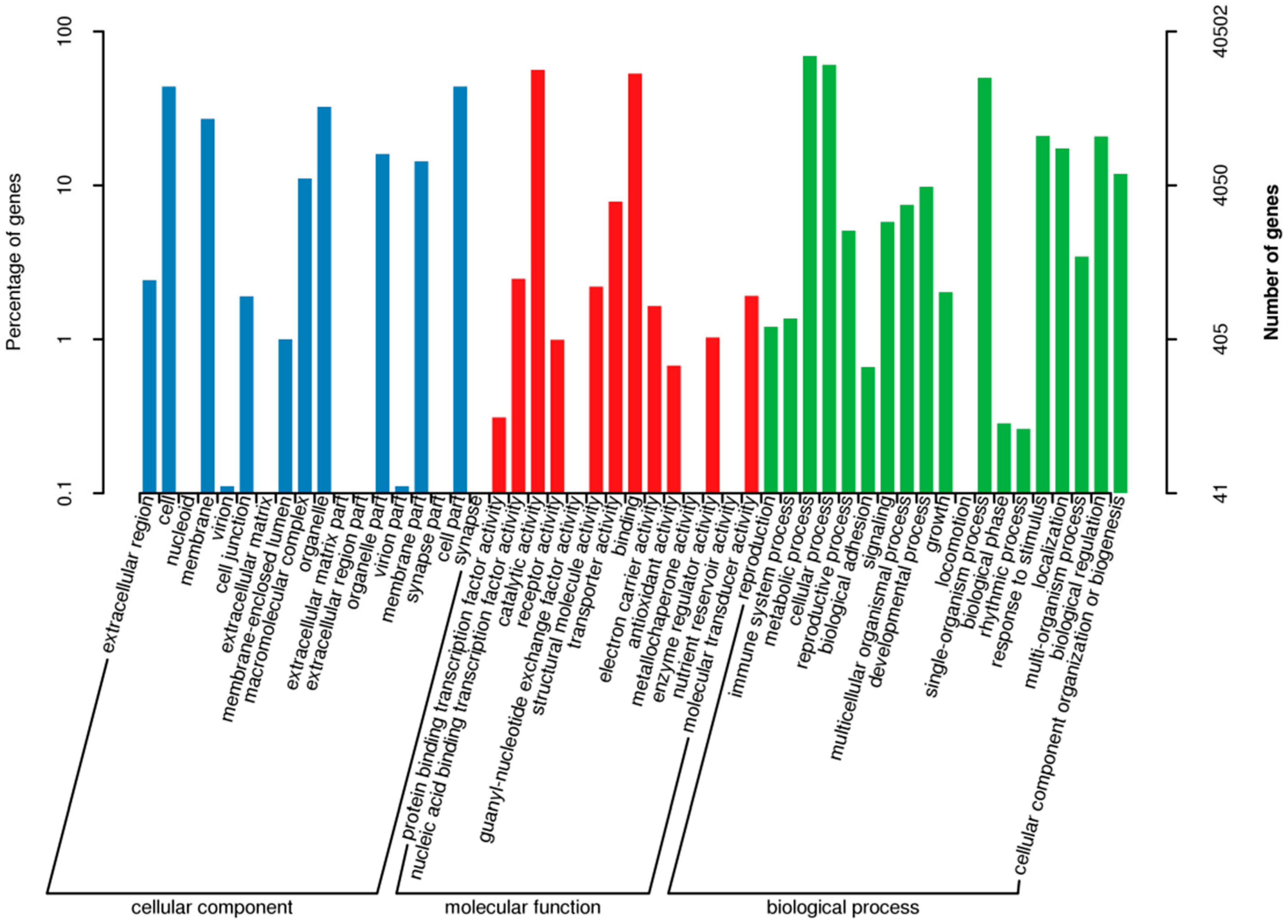
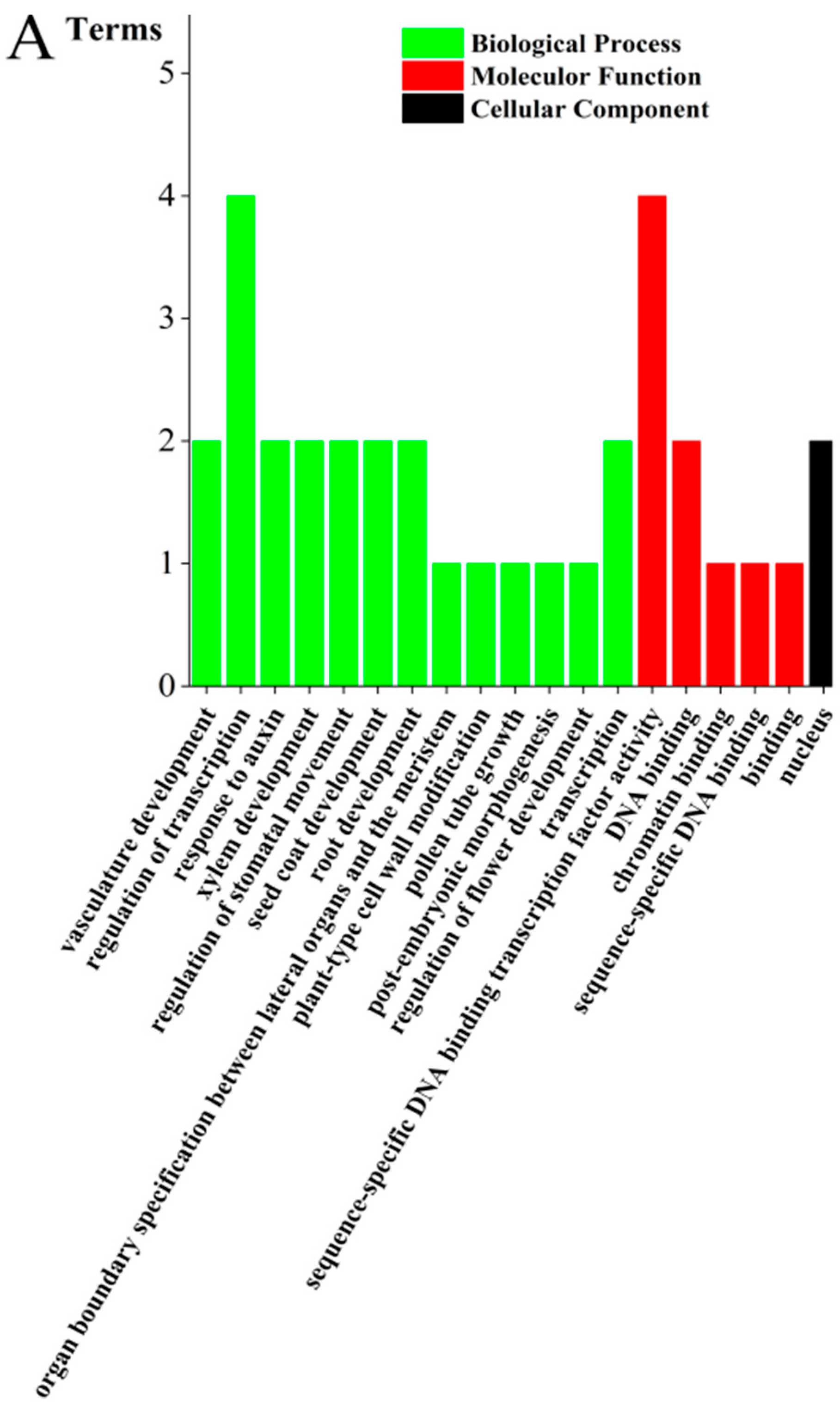
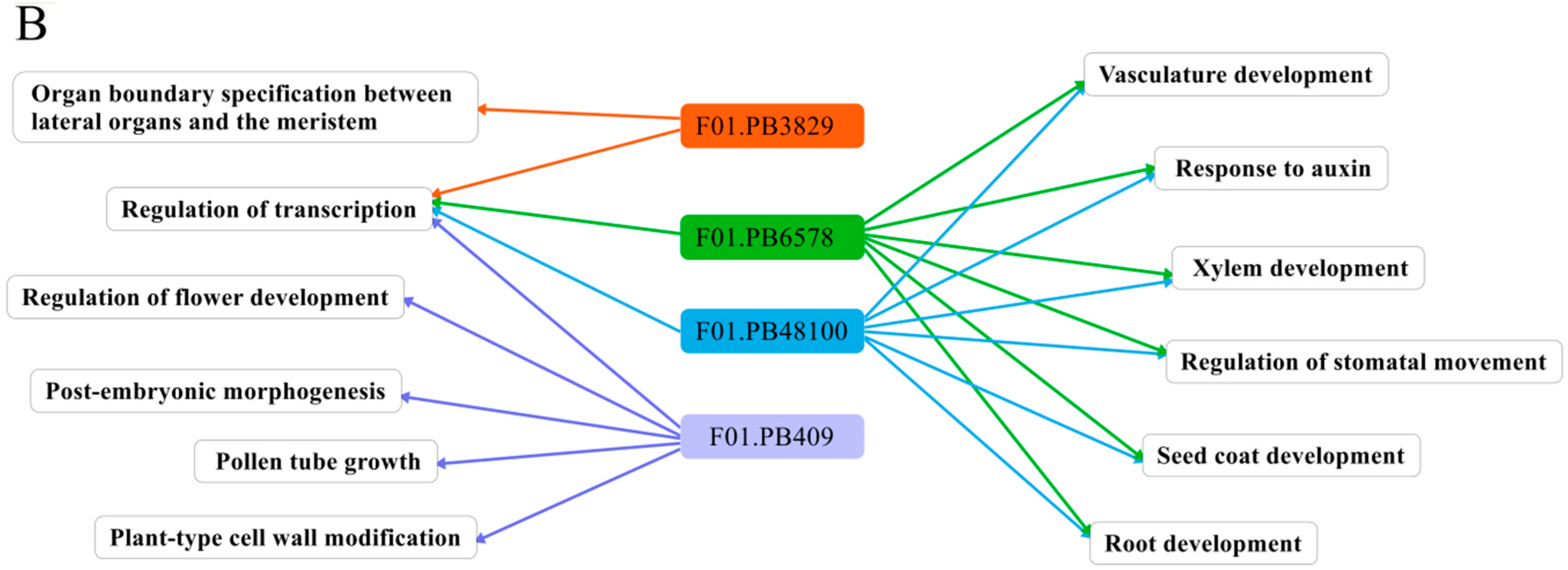
2.4. Gene Ontology (GO) Classification
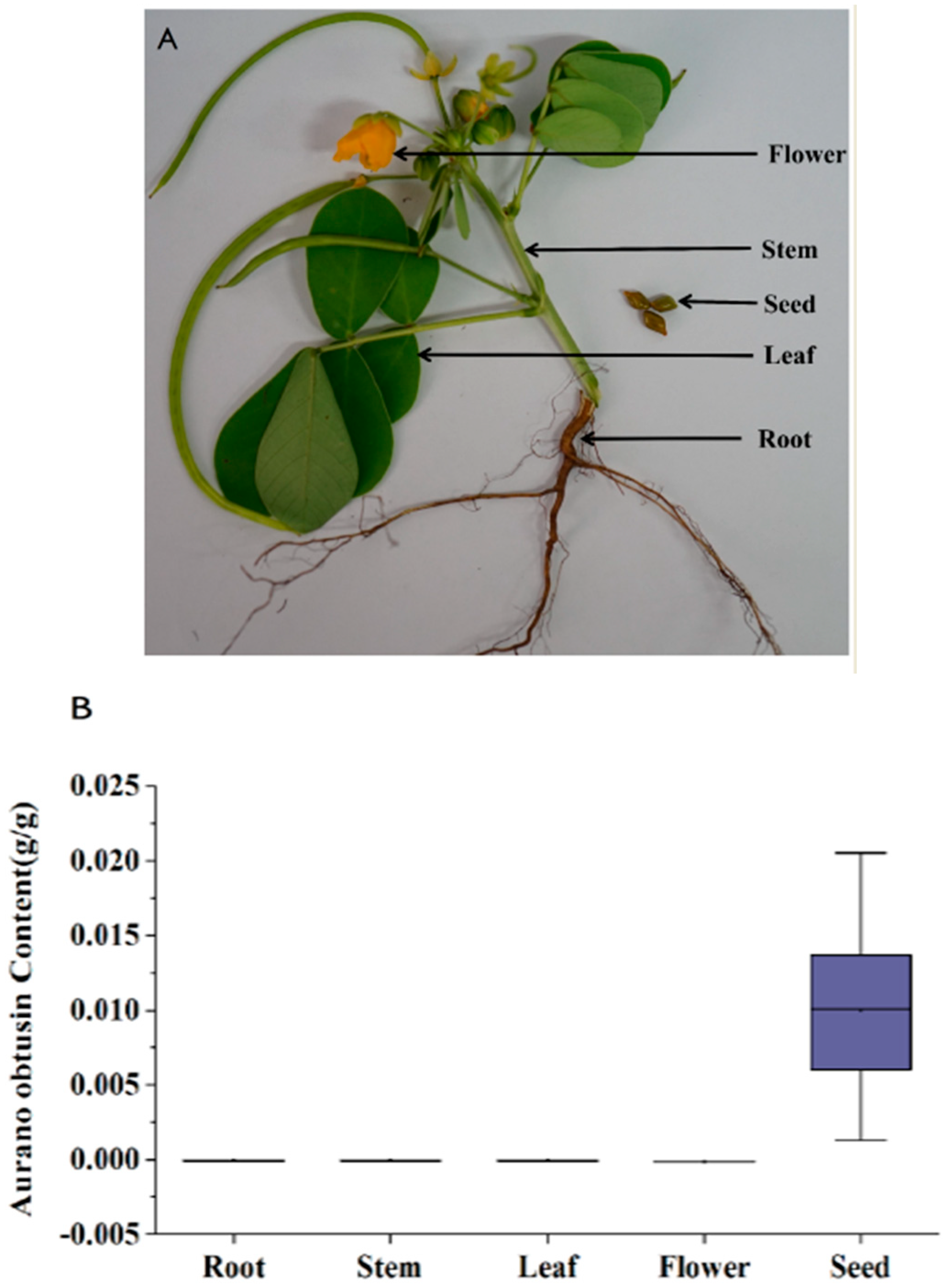
2.5. Localization of Aurantio-Obtusin Accumulation and Gene Profiles Involved in Aurantio-Obtusin Biosynthesis
2.6. Expression Analysis Indicates Colocalization of Aurantio-Obtusin Biosynthesis and Accumulation
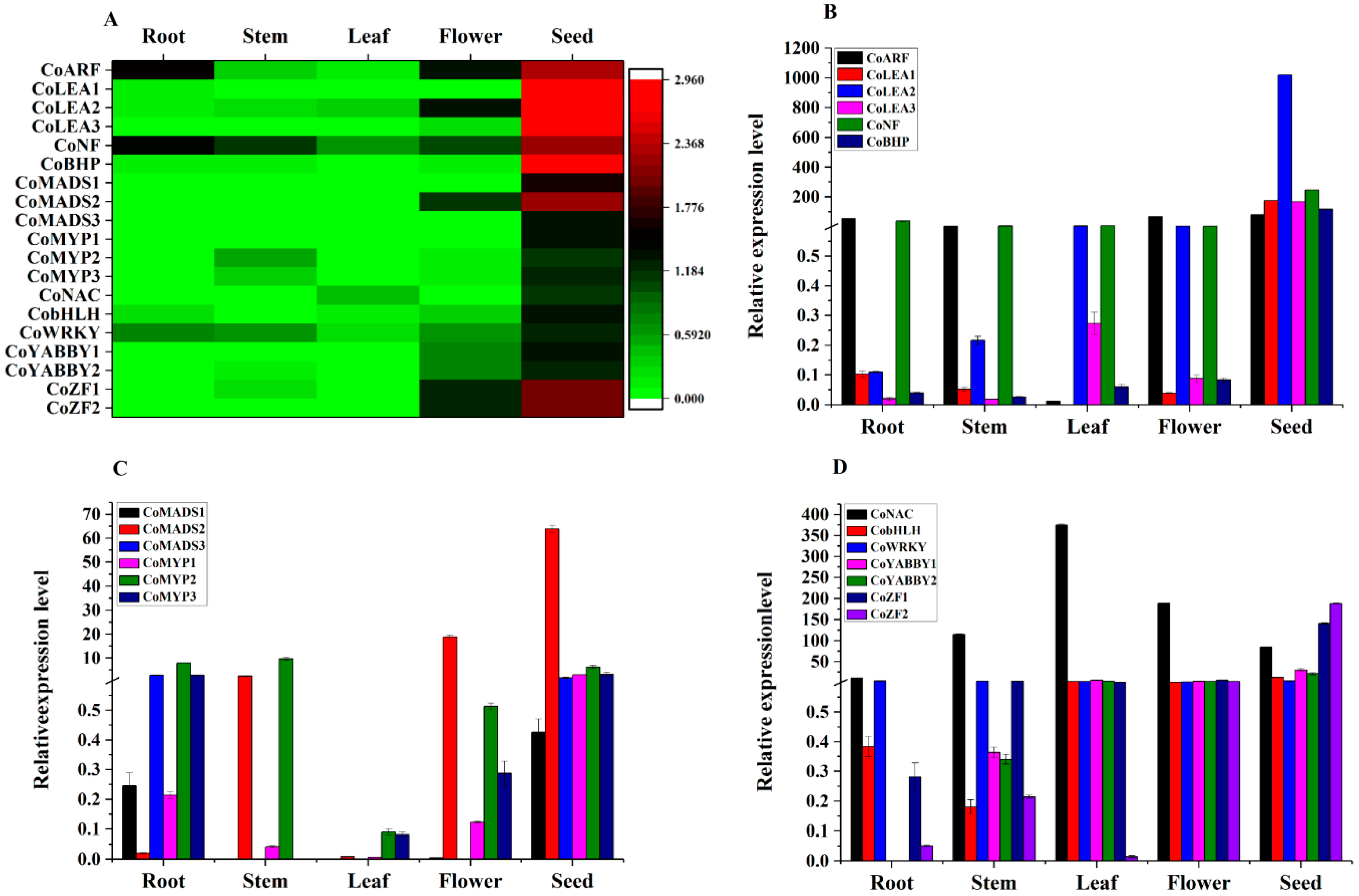
2.7. Expression Pattern of Transcription Factors (TFs) in Seed
2.8. Expression Pattern of CoHsp20 Genes in Various Organs and under Different Stress
3. Discussion
4. Methods and Materials
4.1. Plant Materials
4.2. Determination the Content of Aurantio-Obtusin
4.3. Illumina Library Construction and Sequencing
4.4. Single-Molecule Real-Time Library Construction and Sequencing
4.5. Error Correction of PacBio Reads
4.6. Functional Annotation and Classification
4.7. Determination of Relative Gene Expression Level
4.8. De Novo Detection and Validation of Alternative Splicing Events
4.9. Long Noncoding RNA Identification
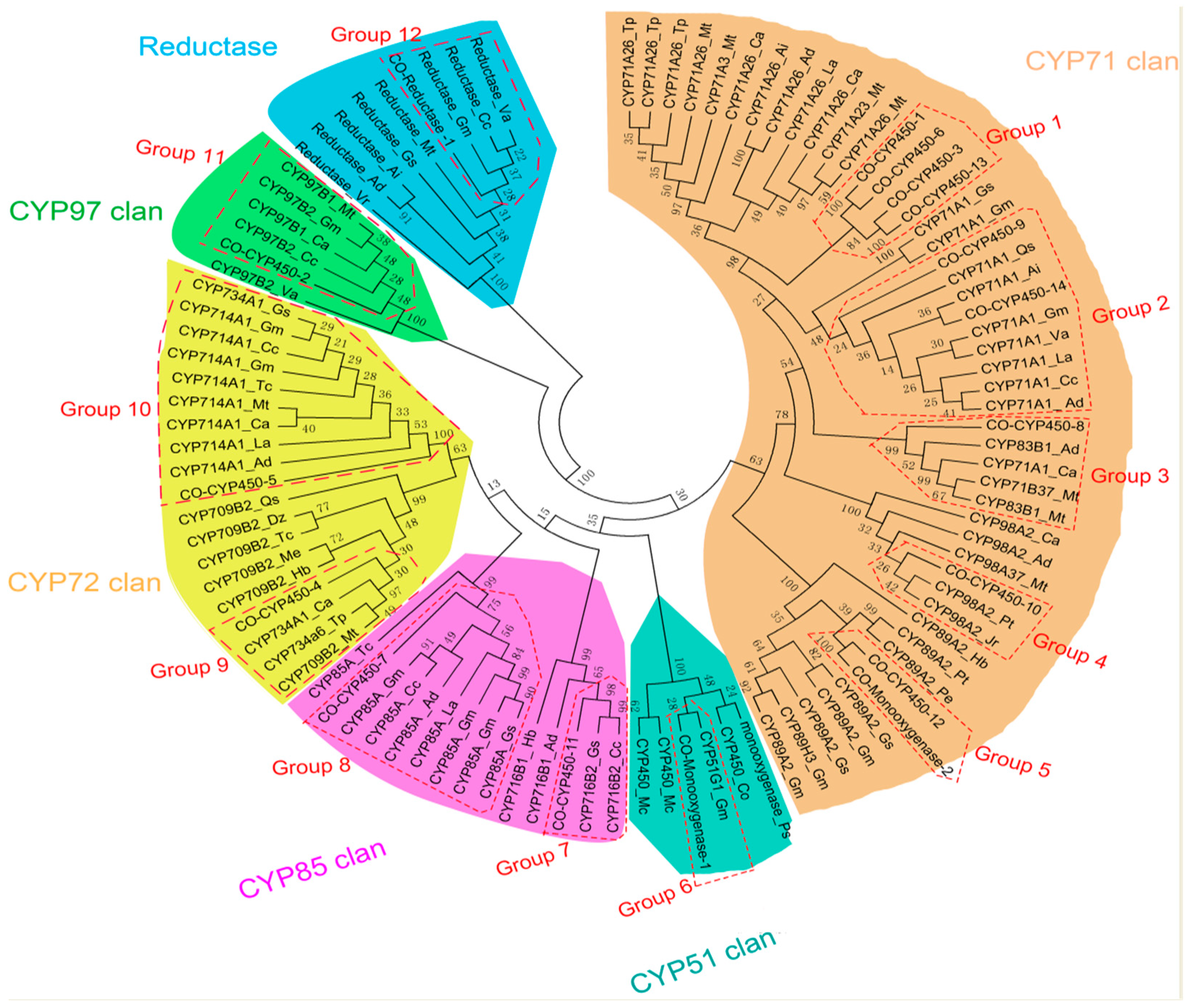
4.10. Phylogenetic Analysis
4.11. Functional Analysis of CoDXS and CoDXR Genes
4.12. Quantitative Real-Time PCR (qRT-PCR) Assay
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| C. obtusifolia | Cassia obtusifolia |
| Co | Cassia obtusifolia |
| TFs | Transcription Factor |
| CYPs | Cytochrome p450/Cytochrome p450 Monooxygenase/NADPH-cytochrome p450 reductase |
| CoCYP450 | Cassia obtusifolia Cytochrome p450 |
| CoHsp20 | Cassia obtusifolia heat shock protein |
| CoReductase | Cassia obtusifolia Cytochrome p450 reductase |
| CoARF | Cassia obtusifolia Auxin-Responsive Transcription Factor |
| CoLEA | Cassia obtusifolia Late Embryogenesis Abundant Transcription Factor |
| CoNF | Cassia obtusifolia Nuclear Transcription Factor |
| CoHBP | Cassia obtusifolia HBP Transcription Factor |
| CoMADS | Cassia obtusifolia MADS-box Transcription Factor |
| CoMYB | Cassia obtusifolia MYB Transcription Factor |
| CoNAC | Cassia obtusifolia Nac Transcription Factor |
| CobHLH | Cassia obtusifolia bHLH Transcription Factor |
| CoWRKY | Cassia obtusifolia WRKY Transcription Factor |
| CoYABBY | Cassia obtusifolia YABBY Transcription Factor |
| CoZF | Cassia obtusifolia Zinc Finger Transcription Factor |
| CoSAMT | Cassia obtusifolia di-O-methyltransferase |
| GATA | GATA transcription factor |
| Trihelix TFs | Trihelix Transcription Factor |
| ACAT | Acetyl-CoA C-acetyltansferase |
| HMGS | Hydroxymethylglutaryl-CoA synthase |
| HMGR | Hydroxymethylglutaryl-CoA reductase |
| MK | Mevalonate kinase |
| PMK | Phosphomevalonate kinase |
| MPD | Diphosphomevalonate decarboxylase |
| DXS | 1-deoxy-d-xylulose-5-phosphate synthase |
| DXR | 1-deoxy-d-xylulose-5-phosphate reductoisomerase |
| ISPD | 1-C-methyl-d-erythritol 4-phosphate cytidylyltransferase |
| CDPMEK | 4-diphosphocytidyl-2-C-methyl-d-erythritol kinase |
| ISPF | 1-C-methyl-D-erythritol 2,4-cyclodiphosphate Synthase |
| HDS | (E)-4-hydroxy-3-methylbut-2-enyl-diphosphase synthase |
| HDR | 4-hydroxy-3-methylbut-2-enyl-diphosphase reductase |
| IPPS | Isopentenyl-diphosphate delta-isomerase |
| DAHPS | 3-deoxy-7-phosphoheptulonate synthase |
| DHQS | 3-dehydroquinate synthase |
| SDH | 3-dehydroquinate dehydratase |
| DHQD | Shikimate dehydrogenase |
| SMK | Shikimate kinase |
| EPSP synthase | 3-phosphoshikimate 1-carboxyvinyltransferase |
| CS | Chorismate synthase |
| ICS | Menaquinone-specific isochorismate synthase |
| menC | 2-succiny-6-hydroxy-2,4-cyclohexadiene-1-carboxylate synthase |
| PHYLLO | O-succinyibenzoate synthase |
| MenH | 2-succinyl-6-hydroxy-2,4-cyclohexadiene-1-carboxylate synthase |
| menD | 2-succinyl-5-enolpyruvyl-6-hydroxy-3-cyclohexene-1-carboxylate synthase |
| menE | Acyl-activating enzyme |
| menB | Naphthoate synthase |
| PSKIII | Polyketide synthaseIII |
| PKC | Polyketide cyclase/dehydratase |
| UDPG | UDP-Glucosyl transferase |
References
- Lee, H.H.; Ahn, J.H.; Kwon, A.R.; Lee, E.S.; Kwak, J.H.; Min, Y.H. Chemical composition and antimicrobial activity of the essential oil of apricot seed. Phytother. Res. 2014, 28, 1867–1872. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.X.; Zhang, Q.Y.; Cui, S.Y.; Cui, X.Y.; Zhang, J.; Zhang, Y.H.; Bai, Y.J.; Zhao, Y.Y. Hypnotic effect of jujubosides from Semen Ziziphi Spinosae. J. Ethnopharmacol. 2010, 130, 163–166. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Li, W.; Qi, D.; Wang, D. Lycium barbarum polysaccharide protects against LPS-induced ARDS by inhibiting apoptosis, oxidative stress and inflammation in pulmonary endothelial cells. Free Radic. Res. 2018, 4, 1–171. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Shi, P.; Qu, L.; Ruan, J.; Yang, S.; Yu, H.; Zhang, Y.; Wang, T. Bioactive constituents obtained from the seeds of Lepidium apetalum Willd. Molecules 2017, 22, 540. [Google Scholar] [CrossRef] [PubMed]
- Luan, G.; Wang, Y.; Wang, Z.; Zhou, W.; Hu, N.; Li, G.; Wang, H. Flavonoid glycosides from Fenugreek seeds regulate glycolipid metabolism by improving mitochondrial function in 3T3-L1 adipocytes in vitro. J. Agric. Food Chem. 2018, 66, 3169–3178. [Google Scholar] [CrossRef] [PubMed]
- Maehara, S.; Agusta, A.; Kitamura, C.; Ohashi, K.; Shibuya, H. Composition of the endophytic filamentous fungi associated with Cinchonaled geriana seeds and production of Cinchona alkaloids. J. Nat. Med. 2016, 70, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Shim, C.K.; Kim, Y.K.; Hong, S.J.; Park, J.H.; Han, E.J.; Kim, S.C. Enhancement of seed dehiscence by seed treatment with Talaromycesflavus GG01 and GG04 in Ginseng (Panaxginseng). Plant Pathol. J. 2017, 33, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Leisner, C.P.; Yendrek, C.R.; Ainsworth, E.A. Physiological and transcriptomic responses in the seed coat of field-grown soybean (Glycine max L. Merr.) to abiotic stress. BMC Plant Biol. 2017, 17, 242. [Google Scholar] [CrossRef] [PubMed]
- Belmonte, M.F.; Kirkbride, R.C.; Stone, S.L.; Pelletier, J.M.; Bui, A.Q.; Yeung, E.C.; Hashimoto, M.; Fei, J.; Harada, C.M.; Munoz, M.D.; et al. Comprehensive developmental profiles of gene activity in regions and subregions of the Arabidopsis seed. Proc. Natl. Acad. Sci. USA 2013, 110, E435–E444. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Kim, S.; Jung, W.Y.; Park, S.J.; Park, D.H.; Kim, J.M.; Cheong, J.H.; Ryu, J.H. The neuroprotective effects of the seeds of Cassia obtusifolia on transient cerebral global ischemia in mice. Food Chem. Toxicol. 2009, 47, 1473–1479. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, S.Y.; Wu, M.L.; Wei, P.J.; Cao, Z.P.; Xiao, P.; Li, C.H. Changes in plasma lipid levels and antioxidant activities in rats after supplementation of obtusifolin. Planta Med. 2016, 82, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.A.; Ali, M.Y.; Choi, J.S. Promising inhibitory effects of anthraquinones, naphthopyrone, and naphthalene glycosides, from Cassia obtusifolia on α-glucosidase and human protein tyrosine phosphatases 1B. Molecules 2016, 22, 28. [Google Scholar] [CrossRef] [PubMed]
- Chai, M.; Zhou, C.; Molina, I.; Fu, C.; Nakashima, J.; Li, G.; Zhang, W.; Park, J.; Tang, Y.; Jiang, Q.; et al. A class II KNOX gene, KNOX4, controls seed physical dormancy. Proc. Natl. Acad. Sci. USA 2016, 113, 6997–7002. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, S.; Mesarich, C.H.; Saccomanno, B.; Vaisberg, A.; Wit, P.J.G.M.; Cox, R.; Collemare, J. Elucidation of cladofulvin biosynthesis reveals a cytochrome P450 monooxygenase required for anthraquinone dimerization. Proc. Natl. Acad. Sci. USA 2016, 113, 6851–6856. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.S.; Roytrakula, S.; Verbernea, M.C.; Heijdenb, R.V.D.; Linthorstc, H.J.M.; Verpoorte, R. Cloning of a cDNA encoding 1-deoxy-d-xylulose 5-phosphate synthase from Morinda citrifolia and analysis of its expression in relation to anthraquinone accumulation. Plant Sci. 2003, 164, 911–917. [Google Scholar] [CrossRef]
- Li, M.Y.; Tan, H.W.; Wang, F.; Jiang, Q.; Xu, Z.S.; Tian, C.; Xiong, A.S. De novo transcriptome sequence assembly and identification of AP2/ERF transcription factor related to abiotic stress in parsley (Petroselinum crispum). PLoS ONE 2014, 9, e108977. [Google Scholar] [CrossRef] [PubMed]
- Van Ooijen, G.; Lukasik, E.; Van Den Burg, H.A.; Vossen, J.H.; Cornelissen, B.J.; Takken, F.L. The small heat shock protein 20 RSI2 interacts with and is required for stability and function of tomato resistance protein I-2. Plant J. 2010, 63, 563–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Song, T.; Zhu, Q.; Wang, W.; Zhou, J.; Liao, H. De novo assembly and analysis of Cassia obtusifolia seed transcriptome to identify genes involved in the biosynthesis of active metabolites. Biosci. Biotechnol. Biochem. 2014, 78, 791–799. [Google Scholar] [CrossRef] [PubMed]
- Misawa, N.; Satomi, Y.; Kondo, K.; Yokoyama, A.; Kajiwara, S.; Saito, T.; Ohtani, T.; Miki, W. Structure and functional analysis of a marine bacterial carotenoid biosynthesis gene cluster and astaxanthin biosynthetic pathway proposed at the gene level. J. Bacteriol. 1995, 177, 6575–6584. [Google Scholar] [CrossRef] [PubMed]
- Young, P.R.; Lashbrooke, J.G.; Alexandersson, E.; Jacobson, D.; Moser, C.; Velasco, R.; Melané, A. The genes and enzymes of the carotenoid metabolic pathway in Vitis vinifera L. BMC Genom. 2012, 13, 243. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.R.; Su, P.; Zhao, Y.J.; Zhang, M.; Wang, X.J.; Liu, Y.J.; Zhang, X.N.; Gao, W.; Huang, L.Q. Molecular cloning and characterization of DXS and DXR genes in the terpenoid biosynthetic pathway of Tripterygium wilfordii. Int. J. Mol. Sci. 2015, 16, 25516–25535. [Google Scholar] [CrossRef] [PubMed]
- Baque, M.A.; Elgirban, A.; Lee, E.J.; Paek, K.Y. Sucrose regulated enhanced induction of anthraquinone, phenolics, flavonoids biosynthesis and activities of antioxidant enzymes in adventitious root suspension cultures of Morinda citrifolia (L.). Acta Physiol. Plant. 2012, 34, 405–415. [Google Scholar] [CrossRef]
- Lange, B.M.; Severin, K.; Bechthold, A.; Heide, L. Regulatory role of microsomal 3-hydroxyl-3-methylglutaryl-coenzyme A reductase for shikonin biosynthesis in Lithospermum erythrorhizon cell suspension cultures. Planta 1998, 204, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Perassolo, M.; Quevedo, C.; Busto, V.; Lanone, A.; Giulietti, M. Enhance of anthraquinone production by effect of praline and aminoindan-phosphonic acid in Rubia tinctorum suspension cultures. Enzym. Microb. Technol. 2007, 41, 181–185. [Google Scholar] [CrossRef]
- Han, Y.S.; Heijden, R.; Lefeber, A.W.M.; Erkelens, C.; Verpoorte, R. Biosynthesis of anthraquinones in cell cultures of Cinchona Robusta proceeds via the methylerythritol-phosphate pathway. Phytochemistry 2002, 59, 45–55. [Google Scholar] [CrossRef]
- Van Tegelen Leon, J.; Bongaerts, R.J.M.; Croes, A.F.; Verpoorte, R.; Wullems, G.J. Isochorismate synthase isoforms from elicited cell cultures of Rubia tinctorum. Phytochemistry 1999, 51, 263–269. [Google Scholar] [CrossRef]
- Kuzuyama, T.; Takagi, M.; Takahashi, S.; Seto, H. Cloning and characterization of 1-deoxy-d-xylulose 5-phosphate synthase from Streptomyces sp. Strain CL190, which uses both the mevalonate and nonmevalonate pathways for isopentenyl diphosphate biosynthesis. J. Bacteriol. 2000, 182, 891–897. [Google Scholar] [CrossRef] [PubMed]
- Shkryl, Y.N.; Veremeichik, G.N.; Makhazen, D.S.; Silantieva, S.A.; Mishchenko, N.P.; Vasileva, E.A.; Fedoreyev, S.A.; Bulgakov, V.P. Increase of anthraquinone content in Rubia cordifolia cells transformed by native and constitutively active forms of the AtCPK1 gene. Plant Cell Rep. 2016, 35, 1907–1916. [Google Scholar] [CrossRef] [PubMed]
- Thatcher, L.F.; Powell, J.J.; Aitken, E.A.; Kazan, K.; Manners, J.M. The lateral organ boundaries domain transcription factor LBD20 functions in Fusarium wilt Susceptibility and jasmonate signaling in Arabidopsis. Plant Physiol. 2012, 160, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Valdivia, A.C.; van der Heijden, R.; Verpoorte, R.; Camara, B. Purification and characterization of two isoforms of isopentenyl-diphosphate isomerase from elicitor-treated Cinchona robusta cells. Eur. J. Biochem. 1997, 249, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Pandith, S.A.; Dhar, N.; Rana, S.; Bhat, W.W.; Kushwaha, M.; Gupta, A.P.; Shah, M.A.; Vishwakarma, R.; Lattoo, S.K. Functional promiscuity of two divergent paralogs of type III plant polyketide synthases. Plant Physiol. 2016, 171, 2599–2619. [Google Scholar] [PubMed]
- Andersen-Ranberg, J.; Kongstad, K.T.; Nafisi, M.; Staerk, D.; Okkels, F.T.; Mortensen, U.H.; Lindberg Møller, B.; Frandsen, R.J.N.; Kannangara, R. Synthesis of C-glucosylated octaketide anthraquinones in Nicotiana benthamiana by using a multispecies-based biosynthetic pathway. Chembiochem 2017, 18, 1893–1897. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Rahman, I.A.; Beuerle, T.; Ernst, L.; Abdel-Baky, A.M.; Eel-D, D.; Ahmed, A.S.; Beerhues, L. In vitro formation of the anthranoid scaffold by cell-free extracts from yeast-extract-treated Cassia bicapsularis cell cultures. Phytochemistry 2013, 88, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Van Tegelen, L.J.; Moreno, P.R.; Croes, A.F.; Verpoorte, R.; Wullems, G.J. Purification and cDNA cloning of isochorismate synthase from elicited cell cultures of Catharanthus roseus. Plant Physiol. 1999, 119, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Rai, A.; Singh, R.; Shirke, P.A.; Tripathi, R.D.; Trivedi, P.K.; Chakrabarty, D. Expression of rice CYP450-like gene (Os08g01480) in Arabidopsis modulates regulatory network leading to heavy metal and other abiotic stress tolerance. PLoS ONE 2015, 10, e0138574. [Google Scholar] [CrossRef] [PubMed]
- Aharoni, A.; Giri, A.P.; Verstappen, F.W.; Bertea, C.M.; Sevenier, R.; Sun, Z.; Jongsma, M.A.; Schwab, W.; Bouwmeester, H.J. Gain and loss of fruit flavor compounds produced by wild and cultivated strawberry species. Plant Cell 2004, 16, 3110–3131. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Austin, R.S.; Zhou, S.; Brown, D. The root transcriptome for North American ginseng assembled and profiled across seasonal development. BMC Genom. 2013, 14, 564. [Google Scholar] [CrossRef] [PubMed]
- Hallahan, D.L.; Lau, S.M.; Harder, P.A.; Smiley, D.W.; Dawson, G.W.; Pickett, J.A.; Christoffersen, R.E.; O’Keefe, D.P. Cytochrome P-450-catalysed monoterpenoid oxidation in catmint (Nepeta racemosa) and avocado (Persea americana): Evidence for related enzymes with different activities. Biochim. Biophys. Acta 1994, 1201, 94–100. [Google Scholar] [CrossRef]
- Bak, S.; Tax, F.E.; Feldmann, K.A.; Galbraith, D.W.; Feyereisen, R. CYP83B1, a cytochrome P450 at the metabolic branch point in auxin and indole glucosinolate biosynthesis in Arabidopsis. Plant Cell 2001, 13, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Karamat, F.; Olry, A.; Doerper, S.; Vialart, G.; Ullmann, P.; Werck-Reichhart, D.; Bourgaud, F.; Hehn, A. CYP98A22, a phenolic ester 3′-hydroxylase specialized in the synthesis of chlorogenic acid, as a new tool for enhancing the furanocoumarin concentration in Ruta graveolens. BMC Plant Biol. 2012, 12, 152. [Google Scholar] [CrossRef] [PubMed]
- Babineau, M.; Mahmood, K.; Mathiassen, S.K.; Kudsk, P.; Kristensen, M. De novo transcriptome assembly analysis of weed Apera spica-venti from seven tissues and growth stages. BMC Genom. 2017, 18, 128. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Yogendra, K.N.; Karre, S.; Kushalappa, A.C.; Dion, Y.; Choo, T.M. WAX INDUCER1 (HvWIN1) transcription factor regulates free fatty acid biosynthetic genes to reinforce cuticle to resist Fusarium head blight in barley spikelets. J. Exp. Bot. 2016, 67, 4127–4139. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, K.; Kurata, N.; Ohyanagi, H.; Hake, S. Genome-wide study of KNOX regulatory network reveals brassinosteroid catabolic genes important for shoot meristem function in rice. Plant Cell 2014, 26, 3488–3500. [Google Scholar] [CrossRef] [PubMed]
- Nomura, T.; Magome, H.; Hanada, A.; Takeda-Kamiya, N.; Mander, L.N.; Kamiya, Y.; Yamaguchi, S. Functional analysis of Arabidopsis CYP714A1 and CYP714A2 reveals that they are distinct gibberellin modification enzymes. Plant Cell Physiol. 2013, 54, 1837–1851. [Google Scholar] [CrossRef] [PubMed]
- Pérez-España, V.H.; Sánchez-León, N.; Vielle-Calzada, J. CYP85A1 is required for the initiation of female gametogenesis in Arabidopsis thaliana. Plant Signal. Behav. 2011, 6, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Han, Z.; Sun, G.; Hoffman, A.; Wilson, I.W.; Yang, Y.; Gao, Q.; Wu, J.; Xie, D.; Dai, J.; et al. Molecular cloning and characterization of a cytochrome P450 taxoid 9á-hydroxylase in Ginkgo biloba cells. Biochem. Biophys. Res. Commun. 2014, 443, 938–943. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, M.; Chantha, S.C.; Rahier, A.; Matton, D.P. Lipid signaling in plants. Cloning and expression analysis of the obtusifoliol 14alpha-demethylase from Solanum chacoense Bitt., a pollination- and fertilization-induced gene with both obtusifoliol and lanosterol demethylase activity. Plant Physiol. 2005, 139, 734–749. [Google Scholar] [CrossRef] [PubMed]
- Terracciano, I.; Cantarella, C.; Fasano, C.; Cardi, T.; Mennella, G.; D’Agostino, N. Liquid-phase sequence capture and targeted re-sequencing revealed novel polymorphisms in tomato genes belonging to the MEP carotenoid pathway. Sci. Rep. 2017, 7, 5616. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.C.; Sung, P.H.; Do, Y.Y.; Huang, P.L. Differential expression and functional characterization of the NADPH cytochrome P450 reductase genes from Nothapodytes foetida. Plant Sci. 2012, 190, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Lepesheva, G.I.; Nes, W.D.; Zhou, W.; Hill, G.C.; Waterman, M.R. CYP51 from Trypanosoma brucei is obtusifoliol-specific. Biochemistry 2004, 43, 10789–10799. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.; Khurana, P. Characterization of a novel zinc finger transcription factor (TaZnF) from wheat conferring heat stress tolerance in Arabidopsis. Cell Stress Chaperones 2018, 23, 253–267. [Google Scholar] [CrossRef] [PubMed]
- Bogamuwa, S.; Jang, J.C. The Arabidopsis tandem CCCH zinc finger proteins AtTZF4, 5 and 6 are involved in light-, abscisic acid- and gibberellic acid-mediated regulation of seed germination. Plant Cell Environ. 2013, 36, 1507–1519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, P.; Yoshida, H.; Yano, K.; Kinoshita, S.; Kawai, K.; Koketsu, E.; Hattori, M.; Takehara, S.; Huang, J.; Hirano, K.; et al. OsIDD2, a zinc finger and INDETERMINATE DOMAIN protein, regulates secondary cell wall formation. J. Integr. Plant Biol. 2018, 60, 130–143. [Google Scholar] [CrossRef] [PubMed]
- Ambawat, S.; Sharma, P.; Yadav, N.R.; Yadav, R.C. MYB transcription factor genes as regulators for plant responses: An overview. Physiol. Mol. Biol. Plants 2013, 19, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Yin, Q.; Liu, J.; Jiang, W.; Di, S.; Pang, Y. GmMYB58 and GmMYB205 are seed-specific activators for isoflavonoid biosynthesis in Glycine max. Plant Cell Rep. 2017, 36, 1889–1902. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Miao, L.; Huo, R.; Song, X.; Johnson, C.; Kong, L.; Sundaresan, V.; Yu, X. ARF2-ARF4 and ARF5 are Essential for Female and Male Gametophyte Development in Arabidopsis. Plant Cell Physiol. 2018, 59, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Orman-Ligeza, B.; Parizot, B.; Gantet, P.P.; Beeckman, T.; Bennett, M.J.; Draye, X. Post-embryonic root organogenesis in cereals: Branching out from model plants. Trends Plant Sci. 2013, 18, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Mathew, I.E.; Das, S.; Mahto, A.; Agarwal, P. Three rice NAC transcription factors heteromerize and are associated with seed size. Front. Plant Sci. 2016, 7, 1638. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Song, C.; Song, L.; Shang, Z.; Yang, S.; Zhang, D.; Sun, W.; Shen, Q.; Zhao, D. RNA sequencing and coexpression analysis reveal key genes involved in α-linolenic acid biosynthesis in Perilla frutescens seed. Int. J. Mol. Sci. 2017, 18, 2433. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Huang, G.Q.; Zou, D.; Yan, J.Q.; Li, Y.; Hu, S.; Li, X.B. The cotton (Gossypium hirsutum) NAC transcription factor (FSN1) as a positive regulator participates in controlling secondary cell wall biosynthesis and modification of fibers. New Phytol. 2018, 217, 625–640. [Google Scholar] [CrossRef] [PubMed]
- Kuijt, S.J.; Greco, R.; Agalou, A.; Shao, J.; ‘t Hoen, C.C.; Overnäs, E.; Osnato, M.; Curiale, S.; Meynard, D.; van Gulik, R.; et al. Interaction between the growth-regulating factor and notted1-like homeobox families of transcription factors. Plant Physiol. 2014, 164, 1952–1966. [Google Scholar] [CrossRef] [PubMed]
- Bakshi, M.; Oelmüller, R. WRKY transcription factors: Jack of many trades in plants. Plant Signal Behav. 2014, 9, e27700. [Google Scholar] [CrossRef] [PubMed]
- Reyero-Saavedra, M.D.R.; Qiao, Z.; Sánchez-Correa, M.D.S.; Díaz-Pineda, M.E.; Reyes, J.L.; Covarrubias, A.A.; Libault, M.; Valdés-López, O. Gene silencing of argonaute5 negatively affects the establishment of the legume-rhizobia symbiosis. Genes 2017, 8, 352. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, W.; Chen, Z.; Han, B.; Haque, M.E.; Liu, A. Gene structure, expression pattern and interaction of Nuclear Factor-Y family in castor bean (Ricinus communis). Planta 2018, 247, 559–572. [Google Scholar] [CrossRef] [PubMed]
- Zhiguo, E.; Li, T.; Zhang, H.; Liu, Z.; Deng, H.; Sharma, S.; Wei, X.; Wang, L.; Niu, B.; Chen, C. A group of nuclear factor Y transcription factors are sub-functionalized during endosperm development in monocots. J. Exp. Bot. 2018. [Google Scholar] [CrossRef]
- Gramzow, L.; Weilandt, L.; Theißen, G. MADS goes genomic in conifers: Towards determining the ancestral set of MADS-box genes in seed plants. Ann. Bot. 2014, 114, 1407–1429. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Jia, J.; Zhao, X.; Zhang, M.; Huang, X.; E, J.; Ni, L.; Jiang, M. The ascorbate peroxidase APX1 is a direct target of a zinc finger transcription factor ZFP36 and a late embryogenesis abundant protein OsLEA5 interacts with ZFP36 to co-regulate OsAPX1 in seed germination in rice. Biochem. Biophys. Res. Commun. 2018, 495, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Cong, B.; Barrero, L.S.; Tanksley, S.D. Regulatory change in YABBY-like transcription factor led to evolution of extreme fruit size during tomato domestication. Nat. Genet. 2008, 40, 800–804. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, W.; Qin, Y.; Pan, Y.; Wang, X.; Weng, Y.; Chen, P.; Li, Y. The Cytochrome P450 gene CsCYP85A1 is a putative candidate for super compact-1 (scp-1) plant architecture mutation in cucumber (Cucumis sativus L.). Front. Plant Sci. 2017, 8, 266. [Google Scholar] [CrossRef] [PubMed]
- Hudson, D.; Guevara, D.R.; Hand, A.J.; Xu, Z.; Hao, L.; Chen, X.; Zhu, T.; Bi, Y.M.; Rothstein, S.J. Rice cytokinin GATA transcription factor1 regulates chloroplast development and plant architecture. Plant Physiol. 2013, 162, 132–144. [Google Scholar] [CrossRef] [PubMed]
- Vadivel, V.; Janardhanan, K. Nutritional and antinutritional characteristics of seven south Indian wild legumes. Plant Food Hum. Nutr. 2002, 57, 151–164. [Google Scholar] [CrossRef]
- Kaplan-Levy, R.N.; Brewer, P.B.; Quon, T.; Smyth, D.R. The trihelix family of transcription factors-light, stress and development. Trends Plant Sci. 2012, 17, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Lu, D.; Li, C.; Luo, J.; Zhu, B.F.; Zhu, J.; Shangguan, Y.; Wang, Z.; Sang, T.; Zhou, B.; et al. Genetic control of seed shattering in rice by the APETALA2 transcription factor shattering abortion1. Plant Cell 2012, 24, 1034–1048. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Li, C.; Zhang, F.S.; Wu, W.; Zhang, Q.F.; Zhang, L.; Wang, T.; Wang, Q.; Qiu, P.H.; Liang, M.F.; et al. Expression of structural and non-structural proteins of severe fever with thrombocytopenia syndrome bunyavirus. Chin. J. Virol. 2011, 27, 515–520. [Google Scholar]
- Steinbrecher, T.; Leubner-Metzger, G. Tissue and cellular mechanics of seeds. Curr. Opin. Genet. Dev. 2018, 51, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Zhang, A.; Chen, J.; He, X.; Xu, B.; Xie, G.; Miao, Z.; Zhang, X.; Huang, L. Genome-wide analysis of the PvHsp20 family in Switchgrass: Motif, genomic organization, and identification of stress or developmental-related Hsp20s. Front. Plant Sci. 2017, 8, 1024. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Wang, D.; Wang, R.; Kong, N.; Zhang, C.; Yang, C.; Wu, W.; Ma, H.; Chen, Q. Genome-wide analysis of the potato Hsp20 gene family: Identification, genomic organization and expression profiles in response to heat stress. BMC Genom. 2018, 19, 61. [Google Scholar] [CrossRef] [PubMed]
- Van Montfort, R.; Slingsby, C.; Vierling, E. Structure and function of the smallheat shock protein/alpha-crystallin family of molecular chaperones. Adv. Protein Chem. 2001, 59, 105–156. [Google Scholar] [PubMed]
- Yu, J.; Cheng, Y.; Feng, K.; Ruan, M.; Ye, Q.; Wang, Q.; Wang, R.; Li, Z.; Zhou, G.; Yao, Z.; et al. Genome-wide identification and expression profiling of tomato Hsp20 gene family in response to biotic and abiotic stresses. Front. Plant Sci. 2016, 7, 1215. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.; Cho, L.H.; Kim, S.L.; Choi, H.; Koh, H.J.; An, G. The BEL1-type homeobox gene SH5 induces seed shattering by enhancing abscission-zone development and inhibiting lignin biosynthesis. Plant J. 2014, 79, 717–728. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Fu, S.; Xu, Z.; Yan, J.; Zhou, H.; Chen, X.; Li, Y.; Au, K.; Yao, H. Hybrid sequencing of full-length cDNA transcripts of stems and leaves in Dendrobium officinale. Genes 2017, 8, 257. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Harata-Lee, Y.; Denton, M.D.; Feng, Q.; Rathjen, J.R.; Qu, Z.; Adelson, D.L. Long read reference genome-free reconstruction of a full-length transcriptome from Astragalus membranaceus reveals transcript variants involved in bioactive compound biosynthesis. Cell Discov. 2017, 3, 17031. [Google Scholar] [CrossRef] [PubMed]
- Schimpl, F.C.; Kiyota, E.; Mayer, J.L.; Gonçalves, J.F.; da Silva, J.F.; Mazzafera, P. Molecular and biochemical characterization of caffeine synthase and purine alkaloid concentration in guarana fruit. Phytochemistry 2014, 105, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Mbatchou, V.C.; Tchouassi, D.P.; Dickson, R.A.; Annan, K.; Mensah, A.Y.; Amponsah, I.K.; Jacob, J.W.; Cheseto, X.; Habtemariam, S.; Torto, B. Mosquito larvicidal activity of Cassia tora seed extract and its key anthraquinones aurantio-obtusin and obtusin. Parasit. Vectors 2017, 10, 562. [Google Scholar] [CrossRef] [PubMed]
- Mehta, R.H.; Ponnuchamy, M.; Kumar, J.; Reddy, N.R. Exploring drought stress-regulated genes in senna (Cassia angustifolia Vahl.): A transcriptomic approach. Funct. Integr. Genom. 2017, 17, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Rama Reddy, N.R.; Mehta, R.H.; Soni, P.H.; Makasana, J.; Gajbhiye, N.A.; Ponnuchamy, M.; Kumar, J. Next generation sequencing and transcriptome analysis predicts biosynthetic pathway of sennosides from Senna (Cassia angustifolia Vahl.), a non-model plant with potent laxative properties. PLoS ONE 2015, 10, e0129422. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Hu, J.; Zhao, Z.; Zhang, D. Transcriptomic analysis of heteromorphic stamens in Cassia biscapsularis L. Sci. Rep. 2016, 6, 31600. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.S.; Van der Heijden, R.; Verpoorte, R. Biosynthesis of anthraquinones in cell cultures of the Rubiaceae. Plant Cell Tissue Organ Cult. 2001, 67, 201–220. [Google Scholar] [CrossRef]
- Rodriguez, T.J.; Verberne, M.C.; Muljono, R.A.B.; Léon, J.P.; Blanca, G.B.; Huub, J.M.; George, J.W.; John, F.B.; Roert, V. Isochorismate synthase transgenic expression in Catharanthus roseus cell suspension. Plant Physiol. Biochem. 2001, 39, 595–602. [Google Scholar] [CrossRef]
- Valdivia, A.C.R.; Heijden, R.; Verpoorte, R. Elicitor-mediated induction of anthraquinone biosynthesis and regulation of isopentenyl diphosphate isomerase and farnesyl diphosphate synthase activities in cell suspension cultures of Cinchona robusta How. Planta 1997, 203, 155–161. [Google Scholar] [CrossRef]
- Hu, Z.; Gallo, S.M. Identification of interacting transcription factors regulating tissue gene expression in human. BMC Genom. 2010, 11, 49. [Google Scholar] [CrossRef] [PubMed]
- Lovisetto, A.; Masiero, S.; Rahim, M.A.; Mendes, M.A.; Casadoro, G. Fleshy seeds form in the basal Angiosperm Magnolia grandiflora and several MADS-box genes are expressed as fleshy seed tissues develop. Evol. Dev. 2015, 17, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liang, W.; Shi, J.; Xu, J.; Zhang, D. MYB56 encoding a R2R3 MYB transcription factor regulates seed size in Arabidopsis thaliana. J. Integr. Plant Biol. 2013, 55, 1166–1178. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhang, X.; Wang, S.; Tan, C.; Zhou, G.; Li, C. Involvement of Alternative Splicing in Barley Seed Germination. PLoS ONE 2016, 11, e0152824. [Google Scholar] [CrossRef] [PubMed]
- Lopes-Caitar, V.S.; de Carvalho, M.C.; Darben, L.M.; Kuwahara, M.K.; Nepomuceno, A.L.; Dias, W.P.; Abdelnoor, R.V.; Marcelino-Guimarães, F.C. Genome-wide analysis of the Hsp20 gene family in soybean: Comprehensive sequence, genomic organization and expression profile analysis under abiotic and biotic stresses. BMC Genom. 2013, 14, 577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, A. Heat shock factor gene family in rice: Genomic organization and transcript expression profiling in response to high temperature, low temperature and oxidative stresses. Plant Physiol. Biochem. 2009, 47, 785–795. [Google Scholar]
- Liu, Z.; Zhu, Q.; Li, J.; Zhang, G.; Jiamahate, A.; Zhou, J.; Liao, H. Isolation, structure modeling and function characterization of a trypsin inhibitor from Cassia obtusifolia. Biotechnol. Lett. 2015, 37, 863–869. [Google Scholar] [CrossRef] [PubMed]
- Scharf, K.D.; Siddique, M.; Vierling, E. The expanding family of Arabidopsis thaliana small heat stress proteins and a new family of proteins containing alpha-crystallin domains (Acd proteins). Cell Stress Chaperones 2001, 6, 225–237. [Google Scholar] [CrossRef]
- Sarkar, N.K.; Kim, Y.K.; Grover, A. Rice sHsp genes: Genomic organization and expression profiling under stress and development. BMC Genom. 2009, 10, 393. [Google Scholar] [CrossRef] [PubMed]
- Lang, S.; Liu, X.; Xue, H.; Wang, X. Functional characterization of BnHSFA4a as a heat shock transcription factor in controlling the re-establishment of desiccation tolerance in seeds. J. Exp. Bot. 2017, 68, 2361–2375. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Mei, W.; Soltis, P.S.; Soltis, D.E.; Barbazuk, W.B. Detecting alternatively spliced transcript isoforms from single-molecule long-read sequences without a reference genome. Mol. Ecol. Resour. 2017, 17, 1243–1256. [Google Scholar] [CrossRef] [PubMed]
- Song, T.; Liu, Z.B.; Li, J.J.; Zhu, Q.K.; Tan, R.; Chen, J.S.; Zhou, J.Y.; Liao, H. Comparative transcriptome of rhizome and leaf in Ligusticum chuanxiong. Plant Syst. Evol. 2015, 301, 2073–2085. [Google Scholar] [CrossRef]
- Kong, L.; Zhang, Y.; Ye, Z.; Liu, X.; Zhao, S.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Luo, H.; Bu, D.; Guo, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.G.; Park, H.J.; Dasari, S.; Wang, S.Q.; Kocher, J.P.; Li, W. CPAT: Coding-potential assessment tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013, 41, e74. [Google Scholar] [CrossRef] [PubMed]
- Flowerika, A.; Kumar, J.; Thakur, N.; Pandey, A.; Pandey, A.K.; Upadhyay, S.K.; Tiwari, S. Characterization and expression analysis of phytoene synthase from bread wheat (Triticum aestivum L.). PLoS ONE 2016, 11, e0162443. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhu, Q.; Li, J.; Yu, J.; Li, Y.; Huang, X.; Wang, W.; Tan, R.; Zhou, J.; Liao, H. Selection and evaluation of reference genes for expression analysis of Cassi. Biosci. Biotechnol. Biochem. 2015, 79, 1818–1826. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Peters, R.J.; Weirather, J.; Luo, H.; Liao, B.; Zhang, X.; Zhu, Y.; Ji, A.; Zhang, B.; Hu, S.; et al. Full-length transcriptome sequences and splice variants obtained by a combination of sequencing platforms applied to different root tissues of Salvia miltiorrhiza and tanshinone biosynthesis. Plant J. 2015, 82, 951–961. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| cDNA Size | Number of Consensus Isoforms | Average Consensus Isoforms Read Length | Number of Polished High-Quality Isoforms | Number of Polished Low-Quality Isoforms | Percent of Polished High-Quality Isoforms (%) |
|---|---|---|---|---|---|
| 0–1 kb | 6206 | 897 | 5729 | 477 | 92.31% |
| 1–2 kb | 25,179 | 1371 | 21,937 | 3242 | 87.12% |
| 2–3 kb | 17,457 | 2457 | 14,323 | 3134 | 82.05% |
| 3–6 kb | 17,616 | 3485 | 12,537 | 5079 | 71.17% |
| >6 kb | 764 | 9064 | 7 | 757 | 0.92% |
| All | 67,222 | 2250 | 54,533 | 12,689 | 81.12% |
| Anno Database | Annotated Number | 300 ≤ Length < 1000 | Length ≥ 1000 |
|---|---|---|---|
| COG_Annotation | 24,630 | 1706 | 22,924 |
| GO_Annotation | 40,502 | 3344 | 37,158 |
| KEGG_Annotation | 25,573 | 2147 | 23,426 |
| KOG_Annotation | 36,919 | 2704 | 34,215 |
| Pfam_Annotation | 47,734 | 3576 | 44,158 |
| Swissprot_Annotation | 43,158 | 3383 | 39,775 |
| eggNOG_Annotation | 56,156 | 4598 | 51,558 |
| Nr_Annotation | 56,681 | 4767 | 51,914 |
| All_Annotated | 57,092 | 4800 | 52,292 |
| Biosynthesis of Secondary Metabolites | Transcript Numbers |
|---|---|
| Brassinosteroid biosynthesis | 8 |
| Caffeine metabolism | 14 |
| Carotenoid biosynthesis | 130 |
| Diterpenoid biosynthesis | 37 |
| Flavone and flavonol biosynthesis | 6 |
| Flavonoid biosynthesis | 177 |
| Isoquinoline alkaloid biosynthesis | 51 |
| Limonene and pinene degradation | 28 |
| Monoterpenoid biosynthesis | 5 |
| Phenylpropanoid biosynthesis | 363 |
| Stilbenoid, diarylheptanoid, and gingerol biosynthesis | 52 |
| Terpenoid backbone biosynthesis | 165 |
| Tropane, piperidine, and pyridine alkaloid biosynthesis | 85 |
| Steroid biosynthesis | 79 |
| Zeatin biosynthesis | 14 |
| Total | 1214 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deng, Y.; Zheng, H.; Yan, Z.; Liao, D.; Li, C.; Zhou, J.; Liao, H. Full-Length Transcriptome Survey and Expression Analysis of Cassia obtusifolia to Discover Putative Genes Related to Aurantio-Obtusin Biosynthesis, Seed Formation and Development, and Stress Response. Int. J. Mol. Sci. 2018, 19, 2476. https://doi.org/10.3390/ijms19092476
Deng Y, Zheng H, Yan Z, Liao D, Li C, Zhou J, Liao H. Full-Length Transcriptome Survey and Expression Analysis of Cassia obtusifolia to Discover Putative Genes Related to Aurantio-Obtusin Biosynthesis, Seed Formation and Development, and Stress Response. International Journal of Molecular Sciences. 2018; 19(9):2476. https://doi.org/10.3390/ijms19092476
Chicago/Turabian StyleDeng, Yin, Hui Zheng, Zicheng Yan, Dongying Liao, Chaolin Li, Jiayu Zhou, and Hai Liao. 2018. "Full-Length Transcriptome Survey and Expression Analysis of Cassia obtusifolia to Discover Putative Genes Related to Aurantio-Obtusin Biosynthesis, Seed Formation and Development, and Stress Response" International Journal of Molecular Sciences 19, no. 9: 2476. https://doi.org/10.3390/ijms19092476
APA StyleDeng, Y., Zheng, H., Yan, Z., Liao, D., Li, C., Zhou, J., & Liao, H. (2018). Full-Length Transcriptome Survey and Expression Analysis of Cassia obtusifolia to Discover Putative Genes Related to Aurantio-Obtusin Biosynthesis, Seed Formation and Development, and Stress Response. International Journal of Molecular Sciences, 19(9), 2476. https://doi.org/10.3390/ijms19092476