1. Introduction
Soybean (
Glycine max (L.) Merrill) is one of the world’s most important oil and protein crops, serving as a food source for humans and for animal forage, and it has industrial uses [
1]. Transgenic soybean is a genetically modified (GM) crop that occupies the largest global scale with its food, nutritional, industrial, and pharmaceutical uses. From 1996 to 2015, approximately 1030 million hectares of GM soybean were planted and more than 50 billion US dollars were generated as income to farmers. In 2015, approximately 92.1 million hectares of GM soybean were planted around the world in 11 countries, which accounted for 51% of all GM crop hectares and 83% of the soybean grown all over the world [
2].
Valuable genes and efficient transformation are the key factors for the improvement of GM soybean. As rice varieties are becoming more popular model plants, research on soybeantransgenic lines and the improvement of soybean agronomic characteristics has fallen behind, and that may be possibly due to genome complexity and the lower transformation efficiency [
3].
A new technology for genome editing—the CRISPR (clustered regularly interspaced short palindromic repeat)/Cas (CRISPR-associated) system—has been successfully used for genome engineering in many important crops in recent years [
4,
5,
6,
7,
8,
9,
10,
11,
12]. Since 2015, CRISPR/Cas9-mediated genome editing in soybean has shown an initial success [
13,
14,
15,
16,
17,
18,
19]. This technology provides a powerful tool for accurate genetic modification and gene function identification, but it also relies on transformation efficiency.
Soybean transformation was first reported in 1988 using an
Agrobacterium tumefaciens infection with cotyledonary node plant regeneration [
20] or by particle bombardment of the meristems of immature seeds [
21]. Over more than 20 years, these two methods have been continually improved and developed, and
Agrobacterium-mediated transformation remains the method of choice in soybean.
Many factors are known to affect the efficiency of T-DNA delivery to the plant cell. The factors influencing
Agrobacterium-mediated transformation efficiency include the
Agrobacterium strain,
Agrobacterium inoculation density, explant types, genome, inoculation period, culture medium, and selection system, among others [
22,
23]. According to these factors, much effort has been made to enhance the efficiency of soybean transformation: the utilization of different explants, such as the cotyledonary node, hypocotyls, embryonic tip, half-seed, and immature seed [
24,
25,
26,
27]; different selection agents, such as glufosinate, glyphosate, and hygromycin [
28,
29,
30]; supplementation with amino acids or antioxidants, such as dithiothreitol,
l-cysteine, and sodium thiosulfate [
30,
31], among others [
32,
33].
It has been reported that the addition of
l-glutamine into culture medium alone or in combination with a cold shock pretreatment could enhance
Agrobacterium transformation efficiency [
34].
l-Glutamine was also found to inhibit β-aminobutyric acid (BABA)-induced plant resistance to a bacterial pathogen of
Arabidopsis [
35]. The metabolism of glutamine has been implicated in the plant response to pathogen infection [
36]. The molecular mechanisms by which glutamine affects plant defence responses are still poorly understood, but the results suggest that
l-glutamine could play a role in lessening host defence responses by attenuating the expression of certain pathogenesis-related genes (
PRs) and potentially improve the efficiency of
Agrobacterium-mediated plant transformation [
34,
37].
In the present study, we examined the influence of l-glutamine and/or l-asparagine on Agrobacterium-mediated transformation in soybean and explored the probable role of l-glutamine and/or l-asparagine during the process of Agrobacterium-mediated transformation. We designed experiments to: determine the effects of l-glutamine and/or l-asparagine, which were added to the co-cultivation medium (CCM), recovery medium (SIM0), selection medium (SIM6), shoot elongation medium (SEM), and rooting culture medium (RCM),on shoot regeneration and elongation; to investigate if these modified media could attenuate plant defense response, enhance the transformation frequency, and further contribute to a wider range of soybean genotypes, and also; to investigate the transmission of the transgene to the T1 generation.
3. Discussion
Agrobacterium-mediated soybean transformations have been previously reported, but the transformation frequency has remained low. The effects of the great complexity of soybean genotypes and different explants selections on soybean transformation have been studied by different laboratories [
24,
25,
26,
27,
31,
32]; however, none of the reported protocols appear to be sufficient.
l-Asparagine and
l-glutamine are the two major nitrogen transport compounds in most plants [
40]. Under normal growth conditions, the C/N ratio in the plant is probably the most important determining factor for whether asparagine or glutamine is selected as the major nitrogen. A high C/N ratio can lead to the production of glutamine for nitrogen transport, and a low C/N ratio lead to the production of asparagine for nitrogen transport [
41,
42]. In addition to providing carbon and nitrogen, glutamine is also involved in other cellular processes, including antioxidative stress, mTOR signalling, autophagy, and plant defence reactions [
34]. The
Agrobacterium-mediated transformation efficiency in perennial ryegrass (
Lolium perenne L.) was highly improved by
myo-inositol removal, cold shock pretreatment and
l-glutamine supplementation, which enhanced
Agrobacterium binding to the cell surface and decreased H
2O
2 production [
34]. As the pathogen competes for host nitrogen reserves during infection, the depletion of
l-glutamine in infected tissues could activate plant defence responses. In addition, a direct application of glutamine on leaves can strongly inhibit H
2O
2 production and could suppress cell death during pathogen infection [
43].
In some labs,
l-asparagine and
l-glutamine have been added into the culture medium of at certain stages, like in shoot elongation medium or rooting medium [
24,
44]. However, in our study, we added the
l-asparagine and/or
l-glutamine from co-culture to rooting, the results showed that
l-asparagine or
l-glutamine added alone to culture media could enhance the regeneration rate, but better results were seen with the combination of
l-asparagine and
l-glutamine (
Table 1). When media was supplemented with both
l-asparagine and
l-glutamine, the shoot induction efficiency was enhanced to 90%. The elongation rate was also increased when the medium had both
l-asparagine and
l-glutamine; the elongation rate was above 40%. For soybean, not all multiple shoots could elongate, only the elongated shoots can further grow into transformant plant. A high elongation rate is the basis for obtaining a high transformation frequency.
For the typical Jack cultivar, utilizing
l-asparagine and
l-glutamine in the culture medium increased the soybean transformation efficiency to approximately 11%, an increase from approximately 3.5–5.5% when no asparagine or glutamine was added to the media (
Table 4 and
Table 5). In addition, this increase in overall transformation efficiency was applicable to other soybean cultivars, with the exception of Zigongdongdou (
Table 5) which for reasons unknown is relatively recalcitrant to
Agrobacterium transformation. Based on this improved transformation, the soybean genome editing (CRISPR/Cas9) was successfully achieved in our lab [
45]. Although the improved transformation efficiency was still not high, we can see that it has the potential to enhance the transformation efficiency by improving the positive rate of elongated shoots. At present, through this transformation process, high shoot induction and elongation rates can be achieved, and elongated shoots can be produced, although many were not positive. Further optimization of glufosinate selection may be the key factor to improving the soybean transformation efficiency.
The increased transformation efficiencies resulted in an overall larger number of T0 and T1 plants under new conditions tested. The number of inheritance transformants was 8 for no supplement versus 34 for
l-asaparagine and glutamine (
Table 7). A larger number of genetically stable transformants can be screened by a piece of leaf for the presence of PAT (strip tested) and confirmed through either PCR or activity of the transgene (GUS) during early seedling development. Perhaps even growing T0 seeds on selection media could help screen transformants. So the T1 plants with the desired outcome can be quickly detected from the larger number of T1 plants.
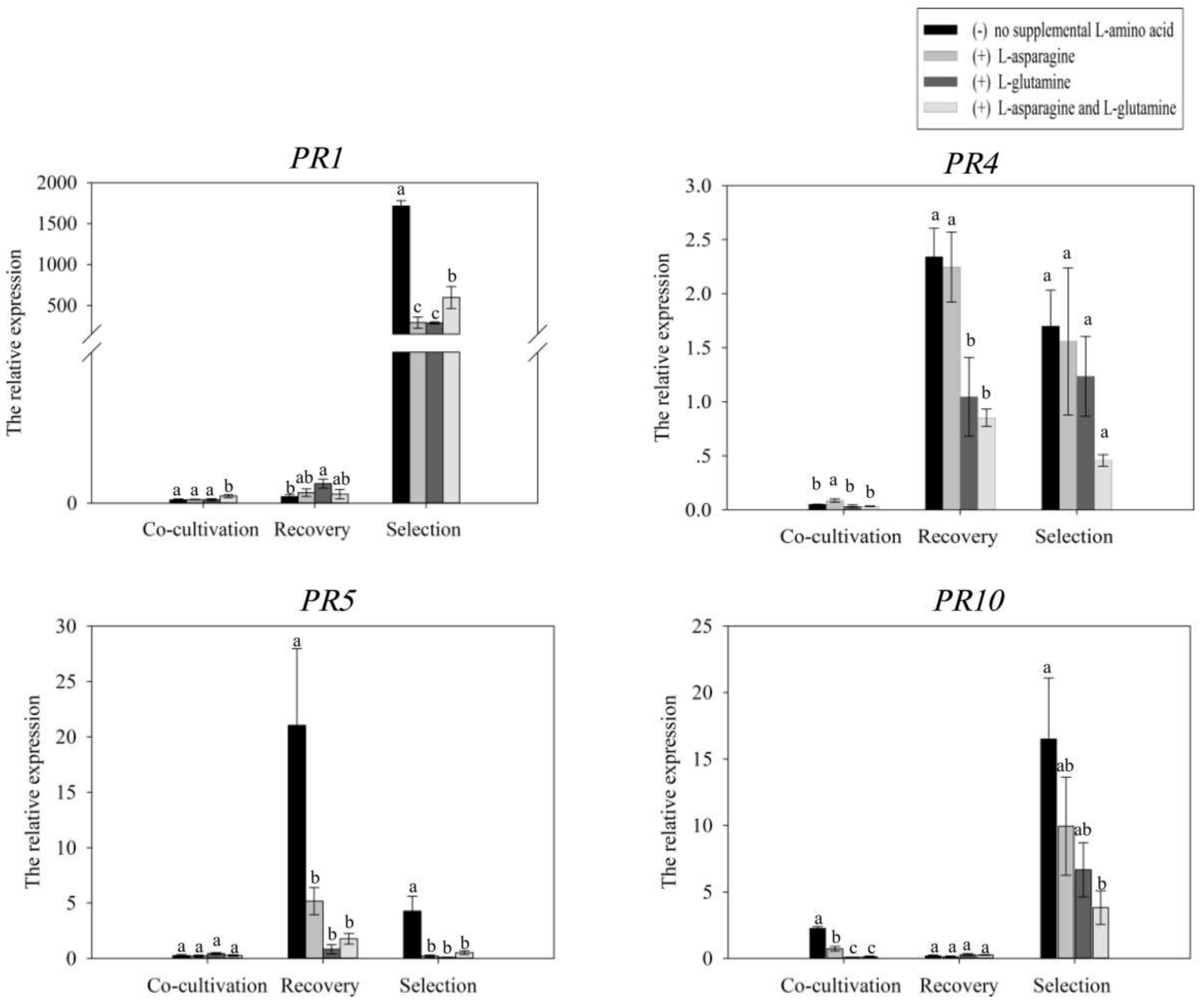
Plant defense responses can lead to cell death at the sites of Agrobacterium infection and lower transformation frequencies. Our results showed that the expression of GmPRs in various treatments was remarkable difference during co-cultivation, recovery and selection culture. The expression level of GmPRs among treatments was relative low during co-cultivation culture. The expression level of GmPR4 and GmPR5 was induced during recovery culture, but it was higher in treatment with no l-asparagine or l-glutamine than in other treatments. During selection culture, the expression level of GmPR1, GmPR4, GmPR5 and GmPR10 was higher in treatment with no l-asparagine or l-glutamine than in other treatments. These results suggested that l-asparagine and l-glutamine could play a role in mitigating plant defense responses by attenuating the expression level of GmPRs. The transformation frequency was enhanced in treatments with l-asparagine or/and l-glutamine. And this enhancement in transformation frequency was likely attributable to attenuation in the expression of GmPRs and suppression of plant defense response by l-asparagine and l-glutamine.
4. Materials and Methods
4.1. Plant Materials
The soybean cultivars (Jack, Zigongdongdou, Williams 82 and Heihe 27) were utilized for
Agrobacterium-mediated transformation in the present study, and Jack was used for the culture medium experiments. Healthy seeds were surface-sterilized by exposure to chlorine gas for 16 h as described by Di [
46]. Sterilized seeds were placed in germination culture medium (GCM) containing 3.1 g/L Gamborgs Basal Salt Mixture (Phytotech, G768, Lenexa, KS, USA), 20 g/L sugar, 1mL/LGamborgs Vitamin Solution (Phytotech, G219, Lenexa, KS, USA) and 7 g/L agar (Sigma, St. Louis, MO, USA), pH 5.8, and the seeds were germinated at 25 °C for 18–20 h on the light (
Figure 6a).
4.2. Agrobacterium Strain and Vector
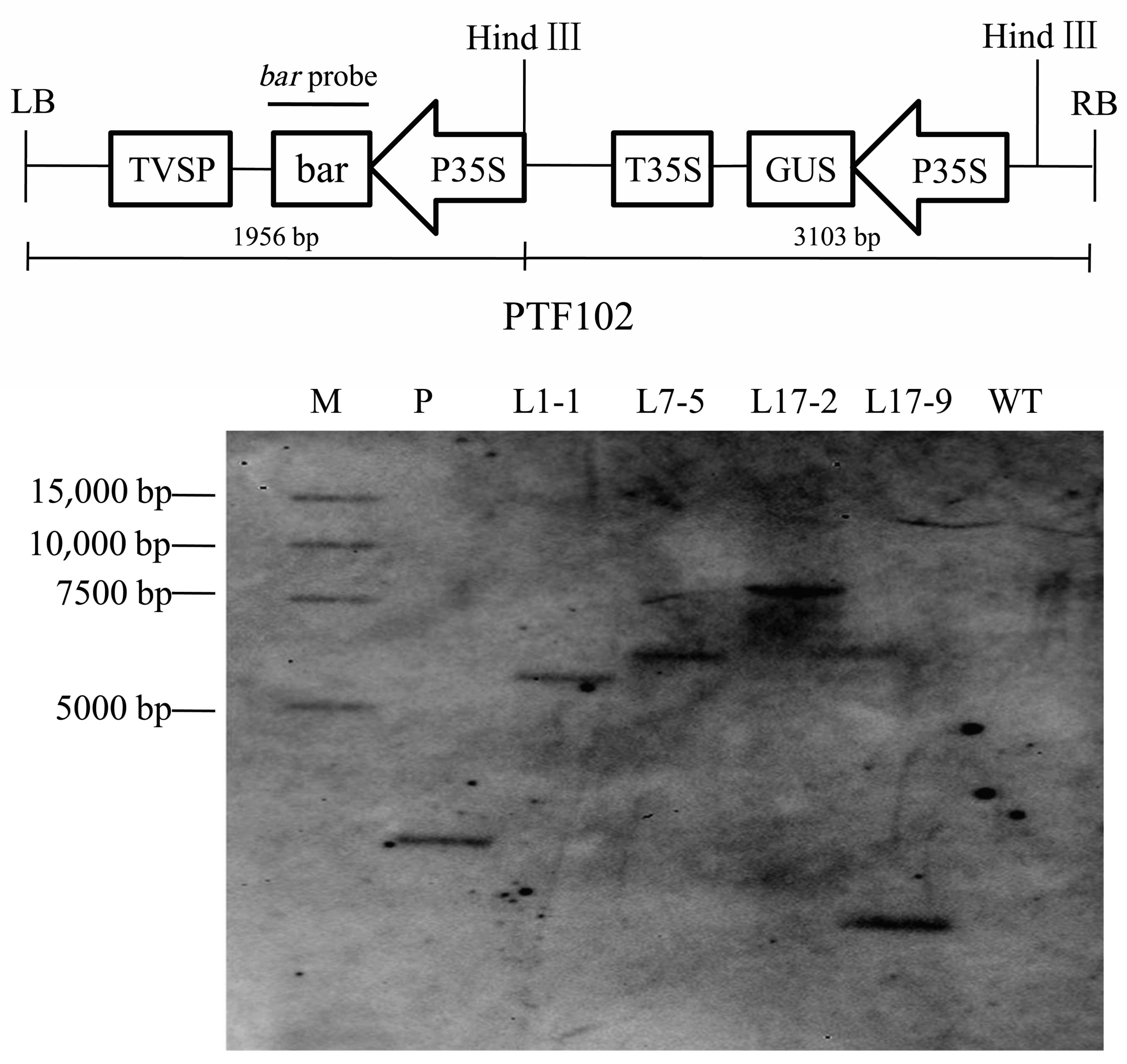
A. tumefaciens strain EHA101 containing the binary vector PTF102 was used in the experiments. The vector carried T-DNA containing the β-glucuronidase gene (
uidA) driven by the 35S CaMV promoter and the
bar gene as an herbicide resistance marker [
44,
47].
4.3. Agrobacterium Preparation
Agrobacterium strain stocks of EHA101/PTF102 stored at −80 °C were streaked on solidified YEP medium plates containing 5 g/L NaCl, 10 g/L tryptone, 5 g/L yeast extract, and 15 g/L agar, with 50 mg/L kanamycin, 75 mg/L chloromycetin, 75 mg/L spectinomycin, and 50 mg/L rifampicin. Plates streaked with
Agrobacterium were incubated at 28 °C for approximately 2 days until colony formation (
Figure 6b). The colonies were collected by a spreader, daubed onto new solidified YEP medium plates with the same antibiotics and incubated overnight at 28 °C (
Figure 6c). The fresh
Agrobacterium were resuspended in liquid co-cultivation medium (LCCM) containing 1/2 Murashige&Skoog Basal Salt Mixture (Phytotech, M524, Lenexa, KS, USA), 3.9 g/L2-(
N-Morpholino) ethanesulfonic acid(MES), 30 g/L sugar, 1mL/L Gamborgs Vitamin Solution, 150 mg/L
dl-Dithiothreitol (DTT), 2 mg/L zeatin and 40 mg/L 3′,5′-dimethoxy-4-hydroxyacetophenone(AS), pH 5.4.Next, a final optical density of 0.6 was measured at 600 nm, and the
Agrobacterium cultures were prepared for transformation (
Figure 6d).
4.4. Infection and Co-Cultivation
Explants were prepared from one-day-old seedlings following the method described by Paz [
24]. A longitudinal cut along the hilum was made to separate the cotyledons, and the seed coat was removed. The embryonic axis found at the junctions of the hypocotyls and the cotyledon was excised to obtain the half-seed explants. The explant cuttings were immersed in
Agrobacterium for 2 h at 50 rpm. After inoculation, each of the 9 cotyledons were placed in solid co-culture medium (CCM) containing1/2 Murashige&Skoog Basal Salt Mixture, 3.9 g/L MES, 30 g/L sugar, 1mL/L Gamborgs Vitamin Solution, 150 mg/L DTT, 40 mg/L AS, 2 mg/L zeatinand 7 g/L agar, pH 5.4, with a piece of Whatman filter paper and then incubated at 22 °C in the dark for 5 days (
Figure 6e,f).
4.5. Recovery Culture and Selection Culture
After co-cultivation, explants were then transferred to recovery medium (SIM0) containing 3.1 g/L Gamborgs Basal Salt Mixture, 0.98 g/L MES, 30 g/L sucrose, 1 mL/L Gamborgs Vitamin Solution, 150 mg/L cefotaxime, 450 mg/L timentin, 1 mg/L 6-Benzylaminopurine (6-BA), and 7 g/L agar, pH 5.7, and incubated at 28 °C for 7 days (
Figure 6g,h). Seven days after recovery, the explants were transferred to selection culture medium (SIM6) containing 3.1 g/L Gamborgs Basal Salt Mixture, 0.98 g/L MES, 30 g/L sucrose, 1 mL/L Gamborgs Vitamin Solution, 150 mg/L cefotaxime, 450 mg/L timentin, 1 mg/L 6-BA, 7 g/L agar, and 6 mg/L glufosinate, pH 5.7, and incubated at 28 °C for 21 days (
Figure 6i,j).
4.6. Shoot Elongation and Rooting
After selection culture, the cotyledons and brown leaves were cut from the explants, and the remaining tissues were transferred to shoot elongation medium (SEM) containing 4.0 g/L Murashige&Skoog Basal Salt Mixture, 0.6 g/L MES, 30 g/L sucrose, 1 mL/L Gamborgs Vitamin Solution, 150 mg/L cefotaxime, 450 mg/L timentin, 0.1 mg/L 3-Indoleacetic acid(IAA), 0.5 mg/L Gibberellic acid(GA), 1 mg/L zeatin, 7 g/L agar, and 6 mg/L glufosinate, pH 5.6, and incubated at 28 °C(
Figure 6k). The culture medium was changed every two weeks. Simultaneous with changing the SEM, the elongated shoots (5–8 cm) were cut from the base of the buds, and the stems were dipped in 1 mg/L Indole-3-Butytric acid (IBA) for 1 min, placed ina rooting culture medium (RCM) containing ½ Murashige&Skoog Basal Salt Mixture, 0.6 g/L MES, 20 g/L sucrose, 1 mL/L Gamborgs Vitamin Solution, and 7 g/L agar,3 mg/L glufosinate, pH 5.7, and incubated at 28 °C for 7 days (
Figure 6l). After root production, the plants were transferred topots and grown in the greenhouse.
4.7. Adding l-Asparagine and l-Glutamine
To determine the effects of l-asparagine and l-glutamine on Agrobacterium-mediated transformation efficiency, different experimental treatments were prepared. Starting with the co-cultivation medium, l-asparagine and/or l-glutamine was added to five different medium types (CCM, SIM0, SIM6, SEM, RCM): (1) with no l-asparagine or l-glutamine; (2) with 100 mg/L l-asparagine; (3) with 100 mg/L l-glutamine; (4) with 50 mg/L l-asparagine and 50 mg/L l-glutamine.
4.8. Detection of the Shoot Induction Rate and Elongation Rate
During the selection stage, the shoot induction rate was calculated as follows: shoot induction rate = explants with shoots/total explants for infection × 100%. During the elongation stage, the elongation rate was calculated as follows: elongation rate = total elongated shoots/total explants for infection × 100%.
4.9. Detection of Positive Plants and Transformation Frequency
The transgenic plants were verified by GUS staining, leaf painting, and strip analysis. The leaf was incubated in GUS staining buffer (50 mM sodium phosphate, pH7.0, 0.5 mM potassium ferrocyanide, 0.5 mM potassium ferricyanide, 0.5 mg/mL 5-bromo-4-chloro-3-indolyl-β-
d-glucuronide (X-Gluc), 0.1% Triton X-100 and 20% methanol) for 24 h at 37 °C and then washed in 95% ethanol before being photographed [
48]. Next, 160mg/L of glufosinate was brushed on the leaf for a leaf painting test, and Liberty Link strips were used to detect PAT proteins in the transgenic plants. The plants used to calculate transformation frequency were all positive as determined by these three detection tests. The transformation frequency was calculated as follows: transformation frequency = the number of positive plants/the total explants for infection × 100%.
4.10. Detection of GUS Activity
0.1 g tissue samples were homogenized with 1 mL Phosphate buffer saline (PBS) buffer (pH 7.4) at 8000 rpm and 4 °C for 30 min, and the 50 μL supernatant was used for the reaction. The GUS activity was measured using an enzyme-linked immunosorbent assay (ELISA) kit (SU-B91122) according to the kit instructions. The GUS ELISA Kit includes a set of calibration standards. The calibration standards are assayed at the same time as the samples and produce a standard curve of Optical Density versus GUS activity. The GUS activity (U/g) is determined by comparing the Optical Density of the samples to the standard curve generated from standard with kit.
4.11. Plant Defence Analysis by qRT-PCR
The expression of GmPRs was used for analysis of plant defense by qRT-PCR. The 5 days co-cultivated explants, 7 days recovery explants and 21 days selected explants were harvested for RNA extraction. Total RNA was isolated from frozen tissue using TransZolUp Plus RNA Kit (TransGen Biotech, Beijing, China). For reverse transcription, 1 μg of total RNA was used to synthesize single-stranded cDNA using TransScript One-Step gDNA Removal and cDNA Synthesis SuperMix (TransGen Biotech). The GmActin gene was used as internal control. The GmPR1, GmPR4, GmPR5 and GmPR10 were used for analysis of plant defense. The primers used for GmActin amplification were 5′-GAGCTATGAATTGCCTGATGG-3′ (forward) and 5′-CGTTTCATGAATTCCAGTAGC-3′ (reverse). The primers used for GmPR1 amplification were 5′-ACACAGGTCGTTTGGGCTAA-3′ (forward) and 5′-CAACAAAGTTGCCAGGGGGA-3′ (reverse). The primers used for GmPR4 amplification were 5′-TGAACGCCGTGAGTGCTTAT-3′ (forward) and 5′-CTGTATTCGTCACCCGGAGG-3′ (reverse). The primers used for GmPR5 amplification were 5′-GTGCTTGGCGTTGAATACGG-3′ (forward) and 5′-TGTGGGACACGCATTCTTGA-3′ (reverse). The primers used for GmPR10 amplification were 5′-ACAACGTCATCCCAAAGGCT-3′ (forward) and 5′-TCCATTCATTAACATCAGCCTCA-3′ (reverse). qRT-PCR reactions were performed according to the three-step method using SYBR Green I dye and the ABI7500 instrument for qRT-PCR. For qRT-PCR, a total volume of 20 μL was used that contained 10 μL of SYBR Premix Ex Taq (2×), 0.4 μL of dye, 0.4 μL of 10 μM of the upstream or downstream primers, 2 μL of cDNA template, and 6.8 μL of ddH2O. qRT-PCR amplification using the standard three-step procedure for denaturation was performed as follows: 95 °C for 30 s, followed by 40 cycles of 95 °C for 5 s, and 60 °C for 30 s. The relative expression level was calculated using the comparative 2−ΔΔCt method.
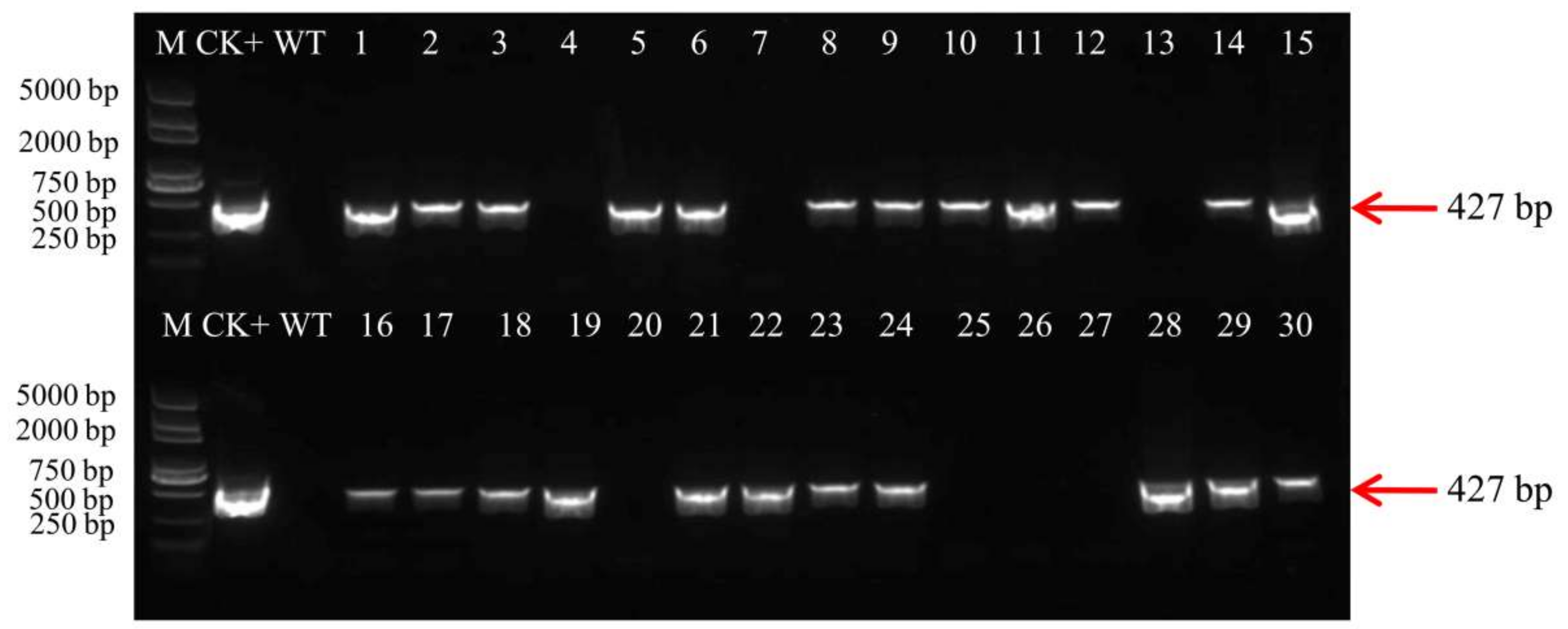
4.12. Molecular Analysis
PCR, strip and southern blot were used to analysis T1 generations of transgenic lines. Total genomic DNA was extracted from the leaves of the T1 transgenic plants using CTAB method. The bar gene detection in transgenic plants was demonstrated by PCR amplification of a 427 bp fragment using the primer pair 5′-GCACCATCGTCAACCACTACATC-3′ and 5′-CAGAAACCCACGTCATGCCAGTT-3′. For southern blot analysis, the 10 μg genomic DNA was digested with HindIII, the restriction products were separated on 0.8% agarose gel and transferred onto a nylon Hybond-N+ membrane (Roche, Mannheim, Germany) with a membrane transfer instrument (Model 785, Bio-Rad, Hercules, CA, USA). The bar (427 bp) PCR products were labelled with Digoxigenin and used as probes to hybridize with the digested DNA on the membrane. The hybridization and detection steps were performed according to the instructions for the DIG High Prime DNA Labeling and Detection Starter KitII (Roche, Mannheim, Germany).
4.13. Statistical Analysis
For all experiments, each treatment contained three replicates. The regeneration frequency, elongation rate and transformation frequency were expressed as the mean ± standard deviation, and the data shown represent the mean of three independent experiments. The data were analysed by ANOVA. The different small letters represent significant differences at p<0.05.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}