TGF-β and BMPR2 Signaling in PAH: Two Black Sheep in One Family


Abstract
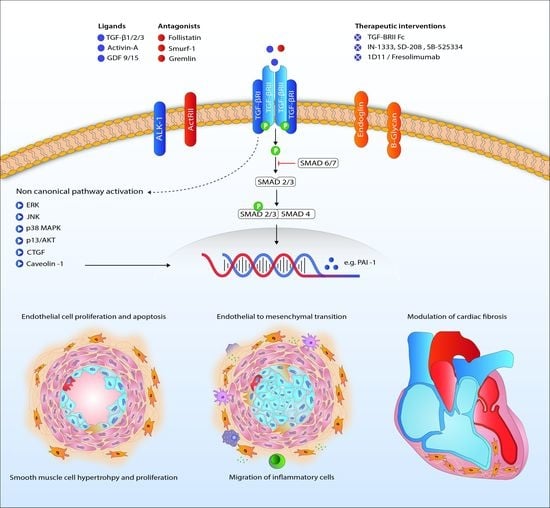
:
1. Introduction
2. TGF-β Signaling
3. Role of TGF-β Ligands in Pulmonary Arterial Hypertension
4. Endothelial-to-Mesenchymal Transition in Pulmonary Arterial Hypertension
5. Receptors in Pulmonary Arterial Hypertension
6. Canonical TGF-β Signaling in Pulmonary Arterial Hypertension
7. Non-Canonical TGF-β Signaling
8. Downstream Targets of TGF-β in the Lung
9. TGF-β Signaling in the Heart in Pulmonary Arterial Hypertension
10. Therapeutic Interventions Relevant in PAH
11. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Galie, N.; Humbert, M.; Vachiery, J.L.; Gibbs, S.; Lang, I.; Torbicki, A.; Simonneau, G.; Peacock, A.; Vonk, N.A.; Beghetti, M.; et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur. Heart J. 2016, 37, 67–119. [Google Scholar] [PubMed]
- Rich, S.; Kaufmann, E.; Levy, P.S. The effect of high doses of calcium-channel blockers on survival in primary pulmonary hypertension. N. Engl. J. Med. 1992, 327, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Tantini, B.; Manes, A.; Fiumana, E.; Pignatti, C.; Guarnieri, C.; Zannoli, R.; Branzi, A.; Galie, N. Antiproliferative effect of sildenafil on human pulmonary artery smooth muscle cells. Basic. Res. Cardiol. 2005, 100, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Ten, D.P.; Arthur, H.M. Extracellular control of TGFβ signalling in vascular development and disease. Nat. Rev. Mol. Cell. Biol. 2007, 8, 857–869. [Google Scholar]
- Ten, D.P.; Goumans, M.J.; Pardali, E. Endoglin in angiogenesis and vascular diseases. Angiogenesis 2008, 11, 79–89. [Google Scholar]
- Liang, H.; Zhang, C.; Ban, T.; Liu, Y.; Mei, L.; Piao, X.; Zhao, D.; Lu, Y.; Chu, W.; Yang, B. A novel reciprocal loop between microRNA-21 and TGFβRIII is involved in cardiac fibrosis. Int. J. Biochem. Cell. Biol. 2012, 44, 2152–2160. [Google Scholar] [CrossRef] [PubMed]
- Goumans, M.J.; Ten Dijke, P. TGF-β signaling in control of cardiovascular function. Cold Spring Harb Perspect. Biol. 2018, 10, a022210. [Google Scholar] [CrossRef] [PubMed]
- Goumans, M.J.; Zwijsen, A.; Ten Dijke, P.; Bailly, S. Bone morphogenetic proteins in vascular homeostasis and disease. Cold Spring Harb. Perspect. Biol. 2018, 10, a031989. [Google Scholar] [CrossRef] [PubMed]
- Lane, K.B.; Machado, R.D.; Pauciulo, M.W.; Thomson, J.R.; Phillips, J.A., III; Loyd, J.E.; Nichols, W.C.; Trembath, R.C. Heterozygous germline mutations in BMPR2, encoding a TGF-β receptor, cause familial primary pulmonary hypertension. Nat. Genet. 2000, 26, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Morse, J.H.; Slager, S.L.; Cuervo, N.; Moore, K.J.; Venetos, G.; Kalachikov, S.; Cayanis, E.; Fischer, S.G.; Barst, R.J.; et al. Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am. J. Hum. Genet. 2000, 67, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Long, L.; Ormiston, M.L.; Yang, X.; Southwood, M.; Graf, S.; Machado, R.D.; Mueller, M.; Kinzel, B.; Yung, L.M.; Wilkinson, J.M.; et al. Selective enhancement of endothelial BMPR-II with BMP9 reverses pulmonary arterial hypertension. Nat. Med. 2015, 21, 777–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yung, L.M.; Nikolic, I.; Paskin-Flerlage, S.D.; Pearsall, R.S.; Kumar, R.; Yu, P.B. A Selective TGFβ Ligand Trap Attenuates Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2016, 134, A19307. [Google Scholar]
- Ogo, T.; Chowdhury, H.M.; Yang, J.; Long, L.; Li, X.; Torres Cleuren, Y.N.; Morrell, N.W.; Schermuly, R.T.; Trembath, R.C.; Nasim, M.T. Inhibition of overactive transforming growth factor-β signaling by prostacyclin analogs in pulmonary arterial hypertension. Am. J. Respir. Cell Mol. Biol. 2013, 48, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Zabini, D.; Granton, E.; Hu, Y.; Miranda, M.Z.; Weichelt, U.; Breuils Bonnet, S.; Bonnet, S.; Morrell, N.W.; Connelly, K.A.; Provencher, S.; et al. Loss of SMAD3 Promotes Vascular Remodeling in Pulmonary Arterial Hypertension via MRTF Disinhibition. Am. J. Respir Crit. Care Med. 2018, 197, 244–260. [Google Scholar] [CrossRef] [PubMed]
- Bellaye, P.S.; Yanagihara, T.; Granton, E.; Sato, S.; Shimbori, C.; Upagupta, C.; Imani, J.; Hambly, N.; Ask, K.; Gauldie, J.; et al. Macitentan reduces progression of TGF-β1-induced pulmonary fibrosis and pulmonary hypertension. Eur. Respir. J. 2018, 2018, 1701857. [Google Scholar] [CrossRef] [PubMed]
- Lu, A.; Zuo, C.; He, Y.; Chen, G.; Piao, L.; Zhang, J.; Xiao, B.; Shen, Y.; Tang, J.; Kong, D.; et al. EP3 receptor deficiency attenuates pulmonary hypertension through suppression of Rho/TGF-β1 signaling. J. Clin. Investing. 2015, 125, 1228–1242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Rivas, G.; Jerjes-Sanchez, C.; Rodriguez, D.; Garcia-Pelaez, J.; Trevino, V. A systematic review of genetic mutations in pulmonary arterial hypertension. BMC Med. Genet. 2017, 18, 82. [Google Scholar] [CrossRef] [PubMed]
- Selimovic, N.; Bergh, C.H.; Andersson, B.; Sakiniene, E.; Carlsten, H.; Rundqvist, B. Growth factors and interleukin-6 across the lung circulation in pulmonary hypertension. Eur. Respir. J. 2009, 34, 662–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gore, B.; Izikki, M.; Mercier, O.; Dewachter, L.; Fadel, E.; Humbert, M.; Dartevelle, P.; Simonneau, G.; Naeije, R.; Lebrin, F.; et al. Key role of the endothelial TGF-β/ALK1/endoglin signaling pathway in humans and rodents pulmonary hypertension. PLoS ONE 2014, 9, e100310. [Google Scholar] [CrossRef] [PubMed]
- Botney, M.D.; Bahadori, L.; Gold, L.I. Vascular remodeling in primary pulmonary hypertension. Potential role for transforming growth factor-β. Am. J. Pathol. 1994, 144, 286–295. [Google Scholar] [PubMed]
- Ranchoux, B.; Antigny, F.; Rucker-Martin, C.; Hautefort, A.; Pechoux, C.; Bogaard, H.J.; Dorfmuller, P.; Remy, S.; Lecerf, F.; Plante, S.; et al. Endothelial-to-mesenchymal transition in pulmonary hypertension. Circulation 2015, 131, 1006–1018. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.B.; Wang, W.; Zhu, X.C.; Ye, W.J.; Cai, H.; Wu, P.L.; Huang, X.Y.; Wang, L.X. The potential of asiaticoside for TGF-β1/Smad signaling inhibition in prevention and progression of hypoxia-induced pulmonary hypertension. Life Sci. 2015, 137, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Li, Y.; Hu, H.; Shi, L.; Chen, J.; Wang, B.; Chen, C.; Zhu, H.; Li, Y.; Li, Q.; et al. Potential therapeutic targets for hypoxia-induced pulmonary artery hypertension. J. Transl. Med. 2014, 12, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, B.B.; Chabon, J.; Gebreab, L.; Poole, J.; Debella, E.; Davis, L.; Tanaka, T.; Sanders, L.; Dropcho, N.; Bandeira, A.; et al. Transforming growth factor-β signaling promotes pulmonary hypertension caused by Schistosoma mansoni. Circulation 2013, 128, 1354–1364. [Google Scholar] [CrossRef] [PubMed]
- Long, L.; Crosby, A.; Yang, X.; Southwood, M.; Upton, P.D.; Kim, D.K.; Morrell, N.W. Altered bone morphogenetic protein and transforming growth factor-β signaling in rat models of pulmonary hypertension: Potential for activin receptor-like kinase-5 inhibition in prevention and progression of disease. Circulation 2009, 119, 566–576. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, L.A.; Obaid, A.A.; Zaki, H.F.; Agha, A.M. Role of oxidative stress, inflammation, nitric oxide and transforming growth factor-β in the protective effect of diosgenin in monocrotaline-induced pulmonary hypertension in rats. Eur. J. Pharmacol. 2014, 740, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, R.; Konishi, K.; Richards, T.J.; Ishizawar, D.C.; Wiechert, A.C.; Kaminski, N.; Ahmad, F. Genomewide RNA expression profiling in lung identifies distinct signatures in idiopathic pulmonary arterial hypertension and secondary pulmonary hypertension. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H1235–H1248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geraci, M.W.; Moore, M.; Gesell, T.; Yeager, M.E.; Alger, L.; Golpon, H.; Gao, B.; Loyd, J.E.; Tuder, R.M.; Voelkel, N.F. Gene expression patterns in the lungs of patients with primary pulmonary hypertension: A gene microarray analysis. Circ. Res. 2001, 88, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Hemnes, A.R.; Brittain, E.L.; Trammell, A.W.; Fessel, J.P.; Austin, E.D.; Penner, N.; Maynard, K.B.; Gleaves, L.; Talati, M.; Absi, T.; et al. Evidence for right ventricular lipotoxicity in heritable pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2014, 189, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Vinasco, L.; Gomberg-Maitland, M.; Maitland, M.L.; Desai, A.A.; Singleton, P.A.; Sammani, S.; Sam, L.; Liu, Y.; Husain, A.N.; Lang, R.M.; et al. Genomic assessment of a multikinase inhibitor, sorafenib, in a rodent model of pulmonary hypertension. Physiol. Genom. 2008, 33, 278–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yndestad, A.; Larsen, K.O.; Oie, E.; Ueland, T.; Smith, C.; Halvorsen, B.; Sjaastad, I.; Skjonsberg, O.H.; Pedersen, T.M.; Anfinsen, O.G.; et al. Elevated levels of activin A in clinical and experimental pulmonary hypertension. J. Appl. Physiol. 2009, 106, 1356–1364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nickel, N.; Kempf, T.; Tapken, H.; Tongers, J.; Laenger, F.; Lehmann, U.; Golpon, H.; Olsson, K.; Wilkins, M.R.; Gibbs, J.S.; et al. Growth differentiation factor-15 in idiopathic pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2008, 178, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, C.J.; Wharton, J.; Howard, L.S.; Gibbs, J.S.; Wilkins, M.R. Red cell distribution width outperforms other potential circulating biomarkers in predicting survival in idiopathic pulmonary arterial hypertension. Heart 2011, 97, 1054–1060. [Google Scholar] [CrossRef] [PubMed]
- Nickel, N.; Jonigk, D.; Kempf, T.; Bockmeyer, C.L.; Maegel, L.; Rische, J.; Laenger, F.; Lehmann, U.; Sauer, C.; Greer, M.; et al. GDF-15 is abundantly expressed in plexiform lesions in patients with pulmonary arterial hypertension and affects proliferation and apoptosis of pulmonary endothelial cells. Respir. Res. 2011, 12, 62. [Google Scholar] [CrossRef] [PubMed]
- Eyries, M.; Coulet, F.; Girerd, B.; Montani, D.; Humbert, M.; Lacombe, P.; Chinet, T.; Gouya, L.; Roume, J.; Axford, M.M.; et al. ACVRL1 germinal mosaic with two mutant alleles in hereditary hemorrhagic telangiectasia associated with pulmonary arterial hypertension. Clin. Genet. 2012, 82, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Austin, E.D.; Loyd, J.E. The genetics of pulmonary arterial hypertension. Circ. Res. 2014, 115, 189–202. [Google Scholar] [CrossRef] [PubMed]
- Ramos, M.F.; Lame, M.W.; Segall, H.J.; Wilson, D.W. Smad signaling in the rat model of monocrotaline pulmonary hypertension. Toxicol. Pathol. 2008, 36, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Jachec, W.; Foremny, A.; Domal-Kwiatkowska, D.; Smolik, S.; Tomasik, A.; Mazurek, U.; Wodniecki, J. Expression of TGF-β1 and its receptor genes (TβR I, TβR II, and TβR III-βglycan) in peripheral blood leucocytes in patients with idiopathic pulmonary arterial hypertension and Eisenmenger’s syndrome. Int. J. Mol. Med. 2008, 21, 99–107. [Google Scholar] [PubMed]
- Richter, A.; Yeager, M.E.; Zaiman, A.; Cool, C.D.; Voelkel, N.F.; Tuder, R.M. Impaired transforming growth factor-β signaling in idiopathic pulmonary arterial hypertension. Am. J. Respir. Crit Care Med. 2004, 170, 1340–1348. [Google Scholar] [CrossRef] [PubMed]
- Van der Bruggen, C.E.; Happe, C.M.; Dorfmuller, P.; Trip, P.; Spruijt, O.A.; Rol, N.; Hoevenaars, F.P.; Houweling, A.C.; Girerd, B.; Marcus, J.T.; et al. Bone Morphogenetic Protein Receptor Type 2 Mutation in Pulmonary Arterial Hypertension: A View on the Right Ventricle. Circulation 2016, 133, 1747–1760. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Docx, C.; Holmes, A.M.; Beach, S.; Duggan, N.; England, K.; Leblanc, C.; Lebret, C.; Schindler, F.; Raza, F.; et al. Activin-like kinase 5 (ALK5) mediates abnormal proliferation of vascular smooth muscle cells from patients with familial pulmonary arterial hypertension and is involved in the progression of experimental pulmonary arterial hypertension induced by monocrotaline. Am. J. Pathol. 2009, 174, 380–389. [Google Scholar] [PubMed]
- Derrett-Smith, E.C.; Dooley, A.; Gilbane, A.J.; Trinder, S.L.; Khan, K.; Baliga, R.; Holmes, A.M.; Hobbs, A.J.; Abraham, D.; Denton, C.P. Endothelial injury in a transforming growth factor β-dependent mouse model of scleroderma induces pulmonary arterial hypertension. Arthritis Rheum. 2013, 65, 2928–2939. [Google Scholar] [CrossRef] [PubMed]
- Zakrzewicz, A.; Kouri, F.M.; Nejman, B.; Kwapiszewska, G.; Hecker, M.; Sandu, R.; Dony, E.; Seeger, W.; Schermuly, R.T.; Eickelberg, O.; et al. The transforming growth factor-β/Smad2,3 signalling axis is impaired in experimental pulmonary hypertension. Eur. Respir. J. 2007, 29, 1094–1104. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Xu, M.; Dong, Y.; Liu, J.; Li, Y.; Mao, W.; Wang, J.; Wang, L. 1,25(OH)2D3 attenuates pulmonary arterial hypertension via microRNA-204 mediated Tgfbr2/Smad signaling. Exp. Cell Res. 2018, 362, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.A., III; Poling, J.S.; Phillips, C.A.; Stanton, K.C.; Austin, E.D.; Cogan, J.D.; Wheeler, L.; Yu, C.; Newman, J.H.; Dietz, H.C.; et al. Synergistic heterozygosity for TGFβ1 SNPs and BMPR2 mutations modulates the age at diagnosis and penetrance of familial pulmonary arterial hypertension. Genet. Med. 2008, 10, 359–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, W.; Han, W.; Greer, P.A.; Tuder, R.M.; Toque, H.A.; Wang, K.K.; Caldwell, R.W.; Su, Y. Calpain mediates pulmonary vascular remodeling in rodent models of pulmonary hypertension, and its inhibition attenuates pathologic features of disease. J. Clin. Investig. 2011, 121, 4548–4566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nasim, M.T.; Ogo, T.; Ahmed, M.; Randall, R.; Chowdhury, H.M.; Snape, K.M.; Bradshaw, T.Y.; Southgate, L.; Lee, G.J.; Jackson, I.; et al. Molecular genetic characterization of SMAD signaling molecules in pulmonary arterial hypertension. Hum. Mutat. 2011, 32, 1385–1389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morty, R.E.; Nejman, B.; Kwapiszewska, G.; Hecker, M.; Zakrzewicz, A.; Kouri, F.M.; Peters, D.M.; Dumitrascu, R.; Seeger, W.; Knaus, P.; et al. Dysregulated bone morphogenetic protein signaling in monocrotaline-induced pulmonary arterial hypertension. Arteriosc. Thromb. Vasc. Biol. 2007, 27, 1072–1078. [Google Scholar] [CrossRef] [PubMed]
- Kouri, F.M.; Queisser, M.A.; Konigshoff, M.; Chrobak, I.; Preissner, K.T.; Seeger, W.; Eickelberg, O. Plasminogen activator inhibitor type 1 inhibits smooth muscle cell proliferation in pulmonary arterial hypertension. Int. J. Biochem. Cell Biol. 2008, 40, 1872–1882. [Google Scholar] [CrossRef] [PubMed]
- Welsh, C.H.; Hassell, K.L.; Badesch, D.B.; Kressin, D.C.; Marlar, R.A. Coagulation and fibrinolytic profiles in patients with severe pulmonary hypertension. Chest 1996, 110, 710–717. [Google Scholar] [CrossRef] [PubMed]
- Szulcek, R.; Happe, C.M.; Rol, N.; Fontijn, R.D.; Dickhoff, C.; Hartemink, K.J.; Grunberg, K.; Tu, L.; Timens, W.; Nossent, G.D.; et al. Delayed Microvascular Shear-adaptation in Pulmonary Arterial Hypertension: Role of PECAM-1 Cleavage. Am. J. Respir. Crit. Care Med. 2016, 193, 1410–1420. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Chung, W.K. The role of genetics in pulmonary arterial hypertension. J. Pathol. 2017, 241, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Friedberg, M.K.; Cho, M.Y.; Li, J.; Assad, R.S.; Sun, M.; Rohailla, S.; Honjo, O.; Apitz, C.; Redington, A.N. Adverse biventricular remodeling in isolated right ventricular hypertension is mediated by increased transforming growth factor-β1 signaling and is abrogated by angiotensin receptor blockade. Am. J. Respir. Cell Mol. Biol. 2013, 49, 1019–1028. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Arroyo, J.; Sakagami, M.; Syed, A.A.; Farkas, L.; Van, T.B.; Kraskauskas, D.; Mizuno, S.; Abbate, A.; Bogaard, H.J.; Byron, P.R.; et al. Iloprost reverses established fibrosis in experimental right ventricular failure. Eur. Respir. J. 2015, 45, 449–462. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.H.; Miyazono, K.; Ten, D.P. TGF-β signalling from cell membrane to nucleus through SMAD proteins. Nature 1997, 390, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.H.; Moustakas, A. Role of Smads in TGFβ signaling. Cell. Tissue Res. 2012, 347, 21–36. [Google Scholar] [CrossRef] [PubMed]
- Goumans, M.J.; Liu, Z.; Ten, D.P. TGF-β signaling in vascular biology and dysfunction. Cell Res. 2009, 19, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Kurakula, K.; Goumans, M.J.; Ten, D.P. Regulatory RNAs controlling vascular (dys)function by affecting TGF-β family signalling. EXCLI J. 2015, 14, 832–850. [Google Scholar] [PubMed]
- Schmierer, B.; Hill, C.S. TGFβ-SMAD signal transduction: Molecular specificity and functional flexibility. Nat. Rev. Mol. Cell Biol. 2007, 8, 970–982. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Massague, J. Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef]
- Hata, A.; Chen, Y.G. TGF-β Signaling from Receptors to Smads. Cold Spring Harb. Perspect. Biol. 2016, 8, a022061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heldin, C.H.; Moustakas, A. Signaling Receptors for TGF-β Family Members. Cold Spring Harb. Perspect. Biol. 2016, 8, a022053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, S.H.; Hamel, L.; Chevalier, S.; Philip, A. Endoglin expression on human microvascular endothelial cells association with βglycan and formation of higher order complexes with TGF-β signalling receptors. Eur. J. Biochem. 2000, 267, 5550–5560. [Google Scholar] [CrossRef] [PubMed]
- Goumans, M.J.; Valdimarsdottir, G.; Itoh, S.; Rosendahl, A.; Sideras, P.; Ten, D.P. Balancing the activation state of the endothelium via two distinct TGF-β type I receptors. EMBO J. 2002, 21, 1743–1753. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.P.; Seki, T.; Goss, K.A.; Imamura, T.; Yi, Y.; Donahoe, P.K.; Li, L.; Miyazono, K.; Ten, D.P.; Kim, S.; et al. Activin receptor-like kinase 1 modulates transforming growth factor-β 1 signaling in the regulation of angiogenesis. Proc. Natl. Acad. Sci. USA 2000, 97, 2626–2631. [Google Scholar] [CrossRef] [PubMed]
- Kretzschmar, M.; Doody, J.; Massague, J. Opposing BMP and EGF signalling pathways converge on the TGF-β family mediator Smad1. Nature 1997, 389, 618–622. [Google Scholar] [PubMed]
- Labbe, E.; Silvestri, C.; Hoodless, P.A.; Wrana, J.L.; Attisano, L. Smad2 and Smad3 positively and negatively regulate TGF β-dependent transcription through the forkhead DNA-binding protein FAST2. Mol. Cell 1998, 2, 109–120. [Google Scholar] [CrossRef]
- Topper, J.N.; Cai, J.; Qiu, Y.; Anderson, K.R.; Xu, Y.Y.; Deeds, J.D.; Feeley, R.; Gimeno, C.J.; Woolf, E.A.; Tayber, O.; et al. Vascular MADs: Two novel MAD-related genes selectively inducible by flow in human vascular endothelium. Proc. Natl. Acad. Sci. USA 1997, 94, 9314–9319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hata, A.; Lieberman, J. Dysregulation of microRNA biogenesis and gene silencing in cancer. Sci. Signal. 2015, 8, re3. [Google Scholar] [CrossRef] [PubMed]
- Euler, G. Good and bad sides of TGFβ-signaling in myocardial infarction. Front. Physiol. 2015, 6, 66. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Pardali, E.; Sanchez-Duffhues, G.; Ten, D.P. BMP signaling in vascular diseases. FEBS Lett. 2012, 586, 1993–2002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akhurst, R.J.; Hata, A. Targeting the TGFβ signalling pathway in disease. Nat. Rev. Drug Discov. 2012, 11, 790–811. [Google Scholar] [CrossRef] [PubMed]
- Hinck, A.P.; Mueller, T.D.; Springer, T.A. Structural Biology and Evolution of the TGF-β Family. Cold Spring Harb. Perspect. Biol. 2016, 8, a021907. [Google Scholar] [CrossRef] [PubMed]
- Haque, S.; Morris, J.C. Transforming growth factor-β: A therapeutic target for cancer. Hum. Vaccin. Immunother 2017, 13, 1741–1750. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zheng, L.; Yuan, Q.; Zhen, G.; Crane, J.L.; Zhou, X.; Cao, X. Transforming growth factor-β in stem cells and tissue homeostasis. Bone Res. 2018, 6, 2. [Google Scholar] [CrossRef] [PubMed]
- Robertson, I.B.; Rifkin, D.B. Regulation of the Bioavailability of TGF-β and TGF-β-Related Proteins. Cold Spring Harb. Perspect. Biol. 2016, 8, a022103. [Google Scholar] [CrossRef] [PubMed]
- Caja, L.; Dituri, F.; Mancarella, S.; Caballero-Diaz, D.; Moustakas, A.; Giannelli, G.; Fabregat, I. TGF-β and the Tissue Microenvironment: Relevance in Fibrosis and Cancer. Int J. Mol. Sci. 2018, 19, 1294. [Google Scholar] [CrossRef] [PubMed]
- Jonigk, D.; Golpon, H.; Bockmeyer, C.L.; Maegel, L.; Hoeper, M.M.; Gottlieb, J.; Nickel, N.; Hussein, K.; Maus, U.; Lehmann, U.; et al. Plexiform lesions in pulmonary arterial hypertension composition, architecture, and microenvironment. Am. J. Pathol. 2011, 179, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Upton, P.D.; Davies, R.J.; Tajsic, T.; Morrell, N.W. Transforming growth factor-β(1) represses bone morphogenetic protein-mediated Smad signaling in pulmonary artery smooth muscle cells via Smad3. Am. J. Respir. Cell Mol. Biol. 2013, 49, 1135–1145. [Google Scholar] [CrossRef] [PubMed]
- Aykul, S.; Martinez-Hackert, E. Transforming Growth Factor-β Family Ligands Can Function as Antagonists by Competing for Type II Receptor Binding. J. Biol. Chem. 2016, 291, 10792–10804. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Long, L.; Reynolds, P.N.; Morrell, N.W. Expression of mutant BMPR-II in pulmonary endothelial cells promotes apoptosis and a release of factors that stimulate proliferation of pulmonary arterial smooth muscle cells. Pulm. Circ. 2011, 1, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Morrell, N.W.; Yang, X.; Upton, P.D.; Jourdan, K.B.; Morgan, N.; Sheares, K.K.; Trembath, R.C. Altered growth responses of pulmonary artery smooth muscle cells from patients with primary pulmonary hypertension to transforming growth factor-β(1) and bone morphogenetic proteins. Circulation 2001, 104, 790–795. [Google Scholar] [CrossRef] [PubMed]
- Davies, R.J.; Holmes, A.M.; Deighton, J.; Long, L.; Yang, X.; Barker, L.; Walker, C.; Budd, D.C.; Upton, P.D.; Morrell, N.W. BMP type II receptor deficiency confers resistance to growth inhibition by TGF-β in pulmonary artery smooth muscle cells: Role of proinflammatory cytokines. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302, L604–L615. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cao, Y.; Sun, S.; Zhu, J.; Gao, S.; Pang, J.; Zhu, D.; Sun, Z. Transforming growth factor-β1 upregulation triggers pulmonary artery smooth muscle cell proliferation and apoptosis imbalance in rats with hypoxic pulmonary hypertension via the PTEN/AKT pathways. Int J. Biochem. Cell Biol. 2016, 77, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Graham, B.B.; Kumar, R. Schistosomiasis and the pulmonary vasculature (2013 Grover Conference series). Pulm. Circ. 2014, 4, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Gilbane, A.J.; Derrett-Smith, E.; Trinder, S.L.; Good, R.B.; Pearce, A.; Denton, C.P.; Holmes, A.M. Impaired bone morphogenetic protein receptor II signaling in a transforming growth factor-β-dependent mouse model of pulmonary hypertension and in systemic sclerosis. Am. J. Respir. Crit. Care Med. 2015, 191, 665–677. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Mickael, C.; Kassa, B.; Gebreab, L.; Robinson, J.C.; Koyanagi, D.E.; Sanders, L.; Barthel, L.; Meadows, C.; Fox, D.; et al. TGF-β activation by bone marrow-derived thrombospondin-1 causes Schistosoma- and hypoxia-induced pulmonary hypertension. Nat. Commun. 2017, 8, 15494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frid, M.G.; Kale, V.A.; Stenmark, K.R. Mature vascular endothelium can give rise to smooth muscle cells via endothelial-mesenchymal transdifferentiation: In vitro analysis. Circ. Res. 2002, 90, 1189–1196. [Google Scholar] [CrossRef] [PubMed]
- Arciniegas, E.; Frid, M.G.; Douglas, I.S.; Stenmark, K.R. Perspectives on endothelial-to-mesenchymal transition: Potential contribution to vascular remodeling in chronic pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, L1–L8. [Google Scholar] [CrossRef] [PubMed]
- Kruithof, B.P.; Duim, S.N.; Moerkamp, A.T.; Goumans, M.J. TGFβ and BMP signaling in cardiac cushion formation: Lessons from mice and chicken. Differentiation 2012, 84, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Good, R.B.; Gilbane, A.J.; Trinder, S.L.; Denton, C.P.; Coghlan, G.; Abraham, D.J.; Holmes, A.M. Endothelial to Mesenchymal Transition Contributes to Endothelial Dysfunction in Pulmonary Arterial Hypertension. Am. J. Pathol. 2015, 185, 1850–1858. [Google Scholar] [CrossRef] [PubMed]
- Diez, M.; Musri, M.M.; Ferrer, E.; Barbera, J.A.; Peinado, V.I. Endothelial progenitor cells undergo an endothelial-to-mesenchymal transition-like process mediated by TGFβRI. Cardiovasc. Res. 2010, 88, 502–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeisberg, E.M.; Tarnavski, O.; Zeisberg, M.; Dorfman, A.L.; McMullen, J.R.; Gustafsson, E.; Chandraker, A.; Yuan, X.; Pu, W.T.; Roberts, A.B.; et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 2007, 13, 952–961. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Xu, Y.; Koya, D.; Kanasaki, K. Role of the endothelial-to-mesenchymal transition in renal fibrosis of chronic kidney disease. Clin. Exp. Nephrol. 2013, 17, 488–497. [Google Scholar] [CrossRef] [PubMed]
- Kitao, A.; Sato, Y.; Sawada-Kitamura, S.; Harada, K.; Sasaki, M.; Morikawa, H.; Shiomi, S.; Honda, M.; Matsui, O.; Nakanuma, Y. Endothelial to mesenchymal transition via transforming growth factor-β1/Smad activation is associated with portal venous stenosis in idiopathic portal hypertension. Am. J. Pathol. 2009, 175, 616–626. [Google Scholar] [CrossRef] [PubMed]
- Kang, Z.; Ji, Y.; Zhang, G.; Qu, Y.; Zhang, L.; Jiang, W. Ponatinib attenuates experimental pulmonary arterial hypertension by modulating Wnt signaling and vasohibin-2/vasohibin-1. Life Sci. 2016, 148, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Vorselaars, V.; Velthuis, S.; van Gent, M.; Westermann, C.; Snijder, R.; Mager, J.; Post, M. Pulmonary Hypertension in a Large Cohort with Hereditary Hemorrhagic Telangiectasia. Respiration 2017, 94, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, M.; Borthwick, G.M.; Hislop, A.A.; Arthur, H.M. Endoglin and activin receptor-like-kinase 1 are co-expressed in the distal vessels of the lung: Implications for two familial vascular dysplasias, HHT and PAH. Lab. Investig. 2009, 89, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Trembath, R.C.; Thomson, J.R.; Machado, R.D.; Morgan, N.V.; Atkinson, C.; Winship, I.; Simonneau, G.; Galie, N.; Loyd, J.E.; Humbert, M.; et al. Clinical and molecular genetic features of pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia. N. Engl. J. Med. 2001, 345, 325–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Upton, P.D.; Morrell, N.W. The transforming growth factor-β-bone morphogenetic protein type signalling pathway in pulmonary vascular homeostasis and disease. Exp. Physiol. 2013, 98, 1262–1266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takagi, K.; Kawaguchi, Y.; Kawamoto, M.; Ota, Y.; Tochimoto, A.; Gono, T.; Katsumata, Y.; Takagi, M.; Hara, M.; Yamanaka, H. Activation of the activin A-ALK-Smad pathway in systemic sclerosis. J. Autoimmun. 2011, 36, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, T.; Tada, Y.; Tanabe, N.; Tatsumi, K.; West, J. Rho-kinase inhibition alleviates pulmonary hypertension in transgenic mice expressing a dominant-negative type II bone morphogenetic protein receptor gene. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 301, L667–L674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ha, H.; Oh, E.Y.; Lee, H.B. The role of plasminogen activator inhibitor 1 in renal and cardiovascular diseases. Nat. Rev. Nephrol. 2009, 5, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Hassell, K.L. Altered hemostasis in pulmonary hypertension. Blood Coagul. Fibrinolysis 1998, 9, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Gallione, C.J.; Richards, J.A.; Letteboer, T.G.; Rushlow, D.; Prigoda, N.L.; Leedom, T.P.; Ganguly, A.; Castells, A.; Ploos van Amstel, J.K.; Westermann, C.J.; et al. SMAD4 mutations found in unselected HHT patients. J. Med. Genet. 2006, 43, 793–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Li, X.; Al-Lamki, R.S.; Southwood, M.; Zhao, J.; Lever, A.M.; Grimminger, F.; Schermuly, R.T.; Morrell, N.W. Smad-dependent and smad-independent induction of id1 by prostacyclin analogues inhibits proliferation of pulmonary artery smooth muscle cells in vitro and in vivo. Circ. Res. 2010, 107, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Pardali, E.; Goumans, M.J.; ten Dijke, P. Signaling by members of the TGF-β family in vascular morphogenesis and disease. Trends Cell. Biol. 2010, 20, 556–567. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.E. Non-Smad pathways in TGF-β signaling. Cell Res. 2009, 19, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.E. Non-Smad Signaling Pathways of the TGF-β Family. Cold Spring Harb. Perspect. Biol. 2016, 9, a022129. [Google Scholar] [CrossRef] [PubMed]
- Awad, K.S.; Elinoff, J.M.; Wang, S.; Gairhe, S.; Ferreyra, G.A.; Cai, R.; Sun, J.; Solomon, M.A.; Danner, R.L. Raf/ERK drives the proliferative and invasive phenotype of BMPR2-silenced pulmonary artery endothelial cells. Am. J. Physiol Lung Cell. Mol. Physiol. 2016, 310, L187–L201. [Google Scholar] [CrossRef] [PubMed]
- Lambers, C.; Roth, M.; Zhong, J.; Campregher, C.; Binder, P.; Burian, B.; Petkov, V.; Block, L.H. The interaction of endothelin-1 and TGF-β1 mediates vascular cell remodeling. PLoS ONE 2013, 8, e73399. [Google Scholar] [CrossRef] [PubMed]
- Razani, B.; Zhang, X.L.; Bitzer, M.; von Gersdorff, G.; Bottinger, E.P.; Lisanti, M.P. Caveolin-1 regulates transforming growth factor (TGF)-β/SMAD signaling through an interaction with the TGF-β type I receptor. J. Biol. Chem. 2001, 276, 6727–6738. [Google Scholar] [CrossRef] [PubMed]
- Olsson, K.M.; Delcroix, M.; Ghofrani, H.A.; Tiede, H.; Huscher, D.; Speich, R.; Grunig, E.; Staehler, G.; Rosenkranz, S.; Halank, M.; et al. Anticoagulation and survival in pulmonary arterial hypertension: Results from the Comparative, Prospective Registry of Newly Initiated Therapies for Pulmonary Hypertension (COMPERA). Circulation 2014, 129, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Preston, I.R.; Roberts, K.E.; Miller, D.P.; Sen, G.P.; Selej, M.; Benton, W.W.; Hill, N.S.; Farber, H.W. Effect of Warfarin Treatment on Survival of Patients With Pulmonary Arterial Hypertension (PAH) in the Registry to Evaluate Early and Long-Term PAH Disease Management (REVEAL). Circulation 2015, 132, 2403–2411. [Google Scholar] [CrossRef] [PubMed]
- Hoeper, M.M.; Sosada, M.; Fabel, H. Plasma coagulation profiles in patients with severe primary pulmonary hypertension. Eur. Respir. J. 1998, 12, 1446–1449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, S.R.; Granton, J.T.; Mehta, S. Thrombotic arteriopathy and anticoagulation in pulmonary hypertension. Chest 2006, 130, 545–552. [Google Scholar] [CrossRef]
- Eisenberg, P.R.; Lucore, C.; Kaufman, L.; Sobel, B.E.; Jaffe, A.S.; Rich, S. Fibrinopeptide A levels indicative of pulmonary vascular thrombosis in patients with primary pulmonary hypertension. Circulation 1990, 82, 841–847. [Google Scholar] [CrossRef] [PubMed]
- Huber, K.; Beckmann, R.; Frank, H.; Kneussl, M.; Mlczoch, J.; Binder, B.R. Fibrinogen, t-PA, and PAI-1 plasma levels in patients with pulmonary hypertension. Am. J. Respir. Crit. Care Med. 1994, 150, 929–933. [Google Scholar] [CrossRef] [PubMed]
- Olman, M.A.; Marsh, J.J.; Lang, I.M.; Moser, K.M.; Binder, B.R.; Schleef, R.R. Endogenous fibrinolytic system in chronic large-vessel thromboembolic pulmonary hypertension. Circulation 1992, 86, 1241–1248. [Google Scholar] [CrossRef] [PubMed]
- Christ, G.; Graf, S.; Huber-Beckmann, R.; Zorn, G.; Lang, I.; Kneussi, M.; Binder, B.R.; Huber, K. Impairment of the plasmin activation system in primary pulmonary hypertension: Evidence for gender differences. Thromb. Haemost. 2001, 86, 557–562. [Google Scholar] [PubMed]
- Yang, J.; Li, X.; Li, Y.; Southwood, M.; Ye, L.; Long, L.; Al-Lamki, R.S.; Morrell, N.W. Id proteins are critical downstream effectors of BMP signaling in human pulmonary arterial smooth muscle cells. Am. J. Physiol Lung Cell. Mol. Physiol. 2013, 305, L312–L321. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Davies, R.J.; Southwood, M.; Long, L.; Yang, X.; Sobolewski, A.; Upton, P.D.; Trembath, R.C.; Morrell, N.W. Mutations in bone morphogenetic protein type II receptor cause dysregulation of Id gene expression in pulmonary artery smooth muscle cells: Implications for familial pulmonary arterial hypertension. Circ. Res. 2008, 102, 1212–1221. [Google Scholar] [CrossRef] [PubMed]
- Riechmann, V.; van Cruchten, I.; Sablitzky, F. The expression pattern of Id4, a novel dominant negative helix-loop-helix protein, is distinct from Id1, Id2 and Id3. Nucleic Acids Res. 1994, 22, 749–755. [Google Scholar] [CrossRef] [PubMed]
- Upton, P.D.; Davies, R.J.; Trembath, R.C.; Morrell, N.W. Bone morphogenetic protein (BMP) and activin type II receptors balance BMP9 signals mediated by activin receptor-like kinase-1 in human pulmonary artery endothelial cells. J. Biol. Chem. 2009, 284, 15794–15804. [Google Scholar] [CrossRef] [PubMed]
- Van de Veerdonk, M.C.; Kind, T.; Marcus, J.T.; Mauritz, G.J.; Heymans, M.W.; Bogaard, H.J.; Boonstra, A.; Marques, K.M.; Westerhof, N.; Vonk-Noordegraaf, A. Progressive right ventricular dysfunction in patients with pulmonary arterial hypertension responding to therapy. J. Am. Coll. Cardiol. 2011, 58, 2511–2519. [Google Scholar] [CrossRef] [PubMed]
- Naeije, R.; Manes, A. The right ventricle in pulmonary arterial hypertension. Eur. Respir. Rev. 2014, 23, 476–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogaard, H.J.; Abe, K.; Vonk, N.A.; Voelkel, N.F. The right ventricle under pressure: Cellular and molecular mechanisms of right-heart failure in pulmonary hypertension. Chest 2009, 135, 794–804. [Google Scholar] [CrossRef] [PubMed]
- Khan, R.; Sheppard, R. Fibrosis in heart disease: Understanding the role of transforming growth factor-β in cardiomyopathy, valvular disease and arrhythmia. Immunology 2006, 118, 10–24. [Google Scholar] [CrossRef] [PubMed]
- Okumura, K.; Kato, H.; Honjo, O.; Breitling, S.; Kuebler, W.M.; Sun, M.; Friedberg, M.K. Carvedilol improves biventricular fibrosis and function in experimental pulmonary hypertension. J. Mol. Med. 2015, 93, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Wollin, L.; Wex, E.; Pautsch, A.; Schnapp, G.; Hostettler, K.E.; Stowasser, S.; Kolb, M. Mode of action of nintedanib in the treatment of idiopathic pulmonary fibrosis. Eur. Respir. J. 2015, 45, 1434–1445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rol, N.; de Raaf, M.A.; Sun, X.; Kuiper, V.P.; da Silva Goncalves Bos, D.; Happe, C.; Kurakula, K.; Dickhoff, C.; Thuillet, R.; Tu, L.; et al. Nintedanib improves cardiac fibrosis but leaves pulmonary vascular remodeling unaltered in experimental pulmonary hypertension. Cardiovasc. Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Megalou, A.J.; Glava, C.; Oikonomidis, D.L.; Vilaeti, A.; Agelaki, M.G.; Baltogiannis, G.G.; Papalois, A.; Vlahos, A.P.; Kolettis, T.M. Transforming growth factor-β inhibition attenuates pulmonary arterial hypertension in rats. Int. J. Clin. Exp. Med. 2010, 3, 332–340. [Google Scholar] [PubMed]
- De Gramont, A.; Faivre, S.; Raymond, E. Novel TGF-β inhibitors ready for prime time in onco-immunology. Oncoimmunology 2017, 6, e1257453. [Google Scholar] [CrossRef] [PubMed]
- Zaiman, A.L.; Podowski, M.; Medicherla, S.; Gordy, K.; Xu, F.; Zhen, L.; Shimoda, L.A.; Neptune, E.; Higgins, L.; Murphy, A.; et al. Role of the TGF-β/Alk5 signaling pathway in monocrotaline-induced pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2008, 177, 896–905. [Google Scholar] [CrossRef] [PubMed]
- Schermuly, R.T.; Yilmaz, H.; Ghofrani, H.A.; Woyda, K.; Pullamsetti, S.; Schulz, A.; Gessler, T.; Dumitrascu, R.; Weissmann, N.; Grimminger, F.; et al. Inhaled iloprost reverses vascular remodeling in chronic experimental pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2005, 172, 358–363. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
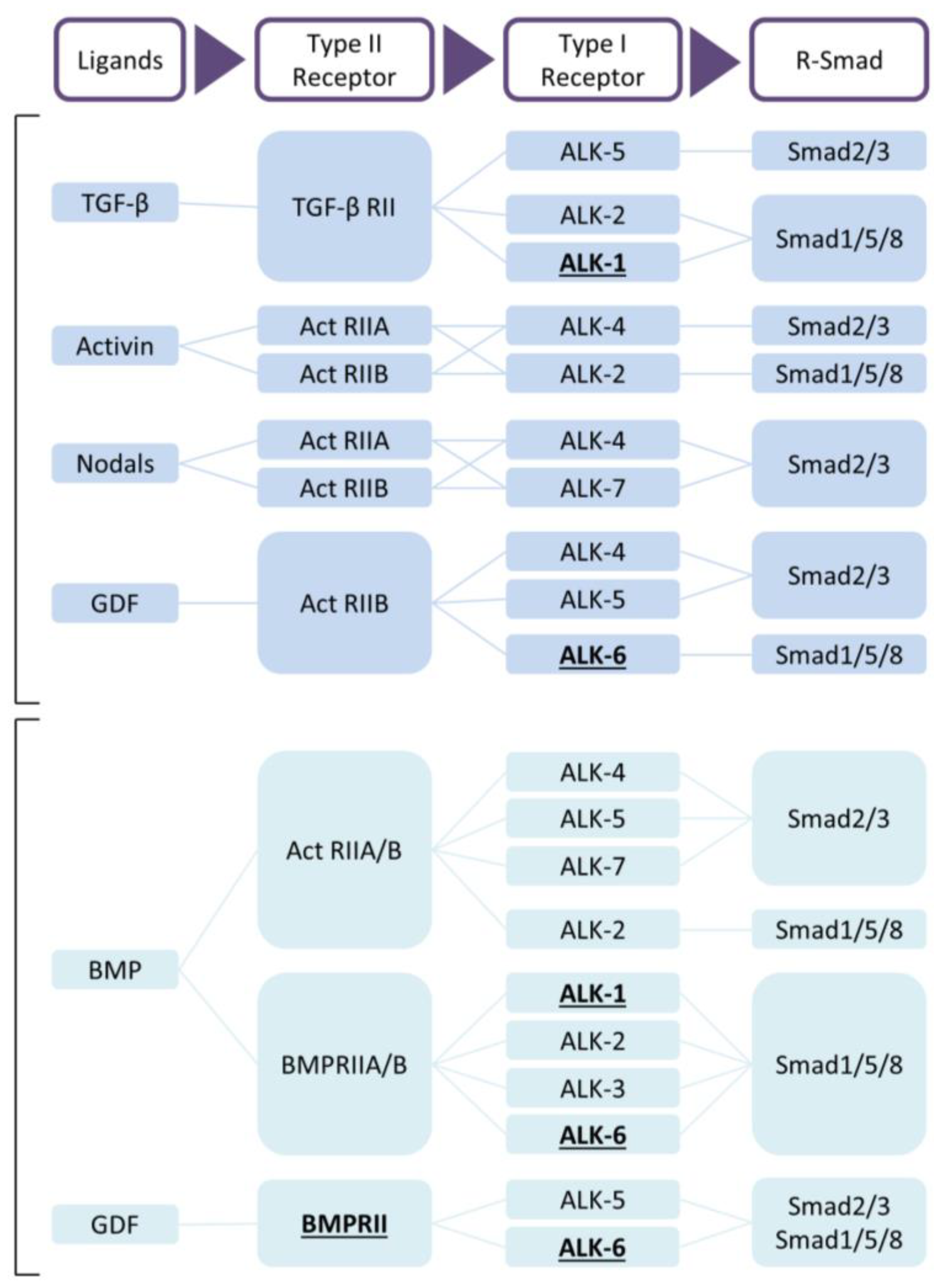{kind=link}
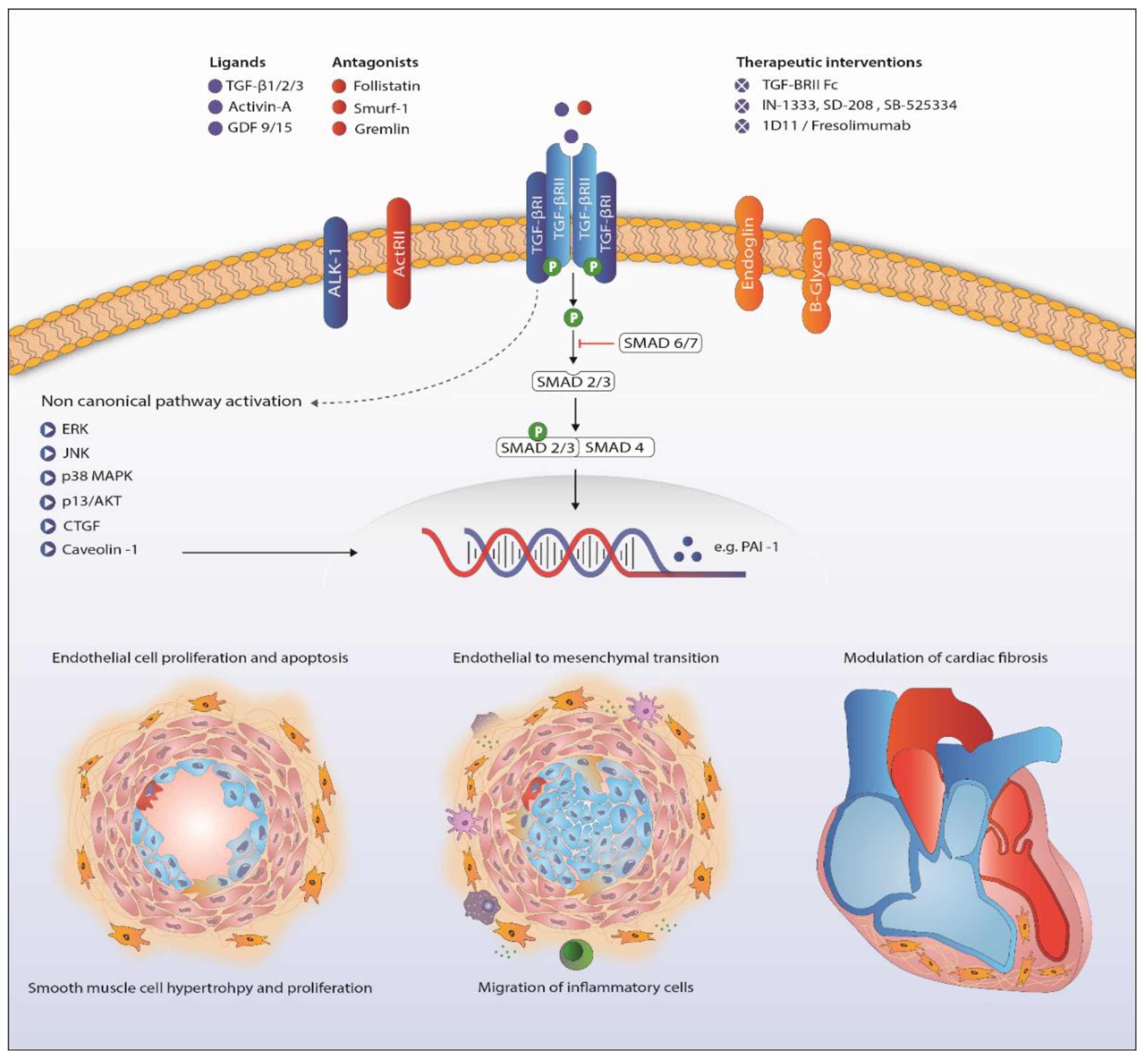{kind=link}
| Serum | Lung Tissue/Vessel | Heart Tissue | EC | SMC | References | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ligands | ||||||||||||
| TGF-β1 | mRNA | = | = | ↑ a,b,d | ↑ b | = | ↑ | [12,18,19,20,21,22,23,24,25,26] | ||||
| Protein | ↑ | ↑ | ↑ a,b,d | ↑ b | ||||||||
| TGF-β2 | mRNA | ↓ b | [12,20] | |||||||||
| Protein | = | |||||||||||
| TGF-β3 | mRNA | ↓ | ↑ | [12,20,27,28,29,30] | ||||||||
| Protein | ↑ | = b,c ↑ a | ||||||||||
| Activin A | mRNA | [31] | ||||||||||
| Protein | ↑ | ↑ a | ||||||||||
| GDF 9/15 | mRNA | ↑ | [32,33,34] | |||||||||
| Protein | ↑ | ↑ | ||||||||||
| Type I receptors | ||||||||||||
| ALK1 | mRNA | ↑ | ↑ | = | [19,35,36,37] | |||||||
| Protein | ↑ | ↑↓ b | ↑ | = | ||||||||
| ALK5 | mRNA | =↑ | = | = | = | [19,21,38,39,40,41] | ||||||
| Protein | = | = | ||||||||||
| Type II receptors | ||||||||||||
| TGFBRII | mRNA | ↓ b↑ a,c | [25,42,43,44] | |||||||||
| Protein | ↓ b | = | = | |||||||||
| ActRII | mRNA | [31] | ||||||||||
| Protein | ↑ a | |||||||||||
| Co-receptors | ||||||||||||
| β-glycan | mRNA | = | ↓ | [28,38] | ||||||||
| Protein | ||||||||||||
| Endoglin | mRNA | = | ↓ b | ↑ | = | [19,43] | ||||||
| Protein | ↑ | ↑ | = | |||||||||
| Canonical signaling | ||||||||||||
| Smad2 | mRNA | ↓ | [12,27,31,37,39,40,41,43,44,45,46] | |||||||||
| Protein | ↑↓= a,b | ↓ | ↑ | |||||||||
| Smad3 | mRNA | ↓ | ↓ b | [12,13,27,31,41,43] | ||||||||
| Protein | ↑ a,b↓ b | |||||||||||
| Smad4 | mRNA | ↓ | ↓ b | [27,37,39,43,47,48] | ||||||||
| Protein | ↓ b | = | = | |||||||||
| Smad6/7 | mRNA | ↑ a↓ b | [37,44,48] | |||||||||
| Protein | ↑ a | |||||||||||
| PAI-1 | mRNA | ↑ | ↓ | ↑ b | [12,13,49,50] | |||||||
| Protein | ↓ | ↑ b | ||||||||||
| Non-canonical signaling | ||||||||||||
| MAPKs | mRNA | ↑ | [51] | |||||||||
| Protein | ||||||||||||
| Cav1 | mRNA | [36,37,52] | ||||||||||
| Protein | ↓ | = b | ||||||||||
| CTGF | mRNA | ↓ b | ↑ c,e | [43,53,54] | ||||||||
| Protein | ↓ b | ↑ c,e | ||||||||||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rol, N.; Kurakula, K.B.; Happé, C.; Bogaard, H.J.; Goumans, M.-J. TGF-β and BMPR2 Signaling in PAH: Two Black Sheep in One Family. Int. J. Mol. Sci. 2018, 19, 2585. https://doi.org/10.3390/ijms19092585
Rol N, Kurakula KB, Happé C, Bogaard HJ, Goumans M-J. TGF-β and BMPR2 Signaling in PAH: Two Black Sheep in One Family. International Journal of Molecular Sciences. 2018; 19(9):2585. https://doi.org/10.3390/ijms19092585
Chicago/Turabian StyleRol, Nina, Konda Babu Kurakula, Chris Happé, Harm Jan Bogaard, and Marie-José Goumans. 2018. "TGF-β and BMPR2 Signaling in PAH: Two Black Sheep in One Family" International Journal of Molecular Sciences 19, no. 9: 2585. https://doi.org/10.3390/ijms19092585
APA StyleRol, N., Kurakula, K. B., Happé, C., Bogaard, H. J., & Goumans, M. -J. (2018). TGF-β and BMPR2 Signaling in PAH: Two Black Sheep in One Family. International Journal of Molecular Sciences, 19(9), 2585. https://doi.org/10.3390/ijms19092585