A One-Step Extraction and Luminescence Assay for Quantifying Glucose and ATP Levels in Cultured HepG2 Cells

 ,
,  and
and
Abstract
:1. Introduction
2. Results
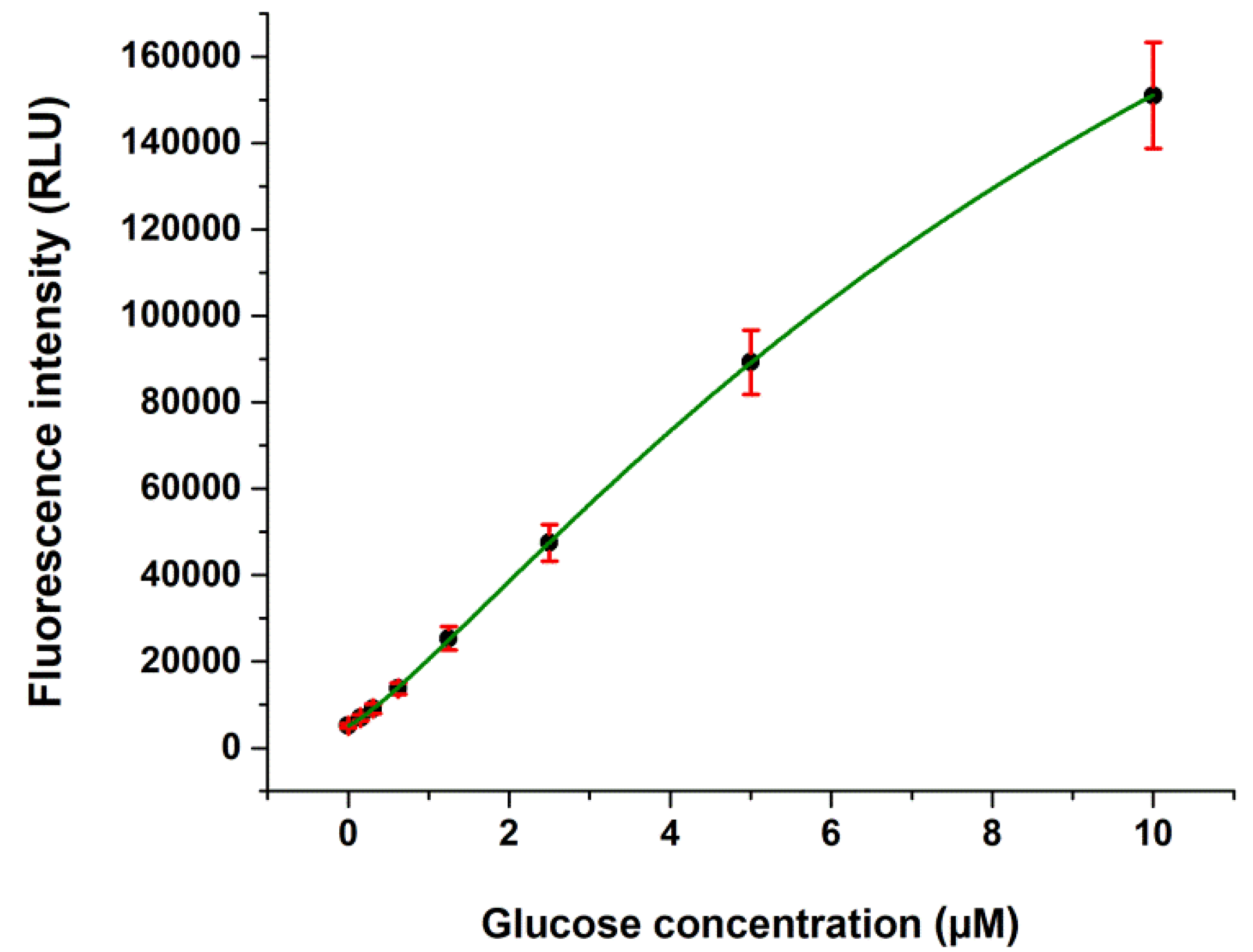
2.1. Validation Data
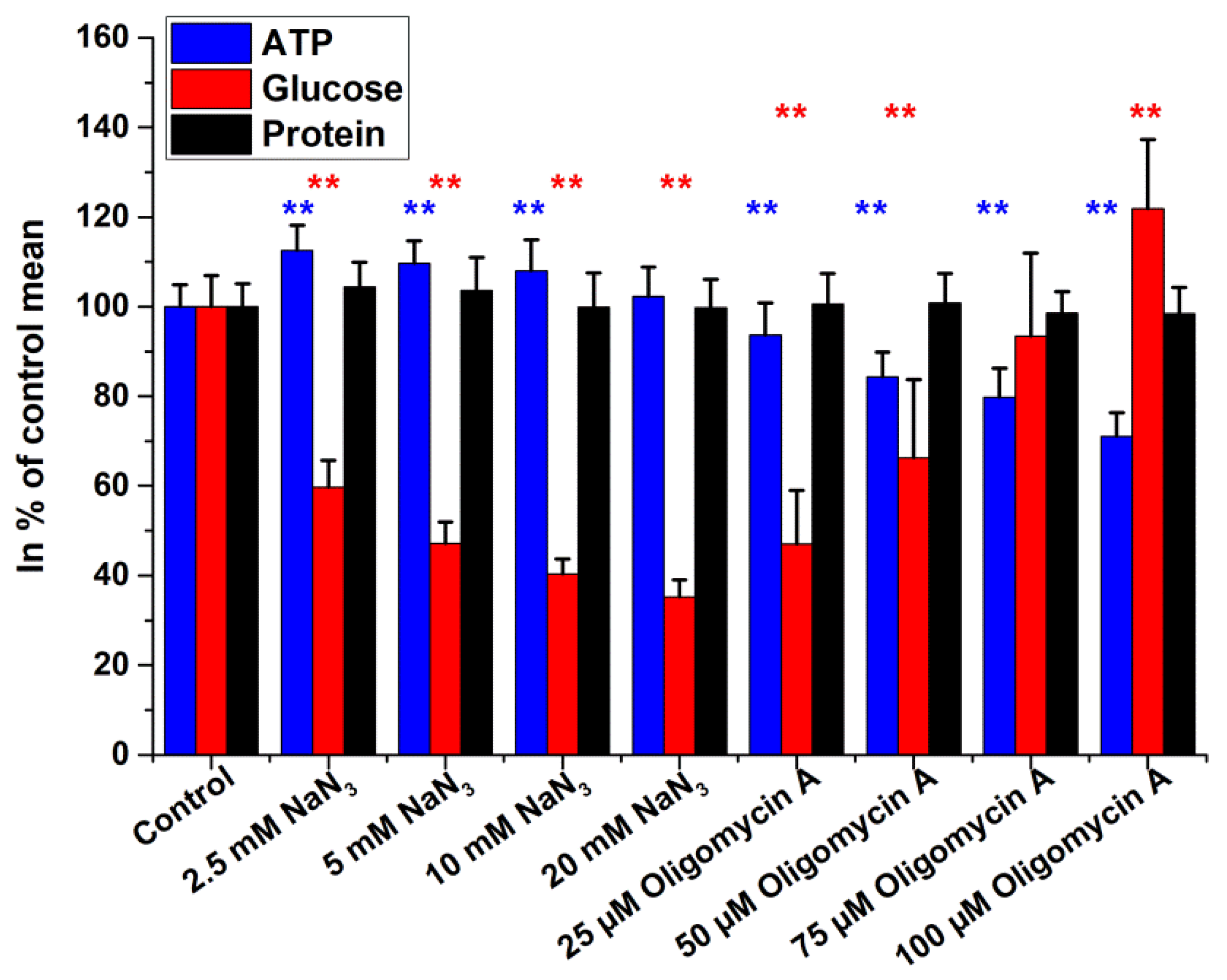
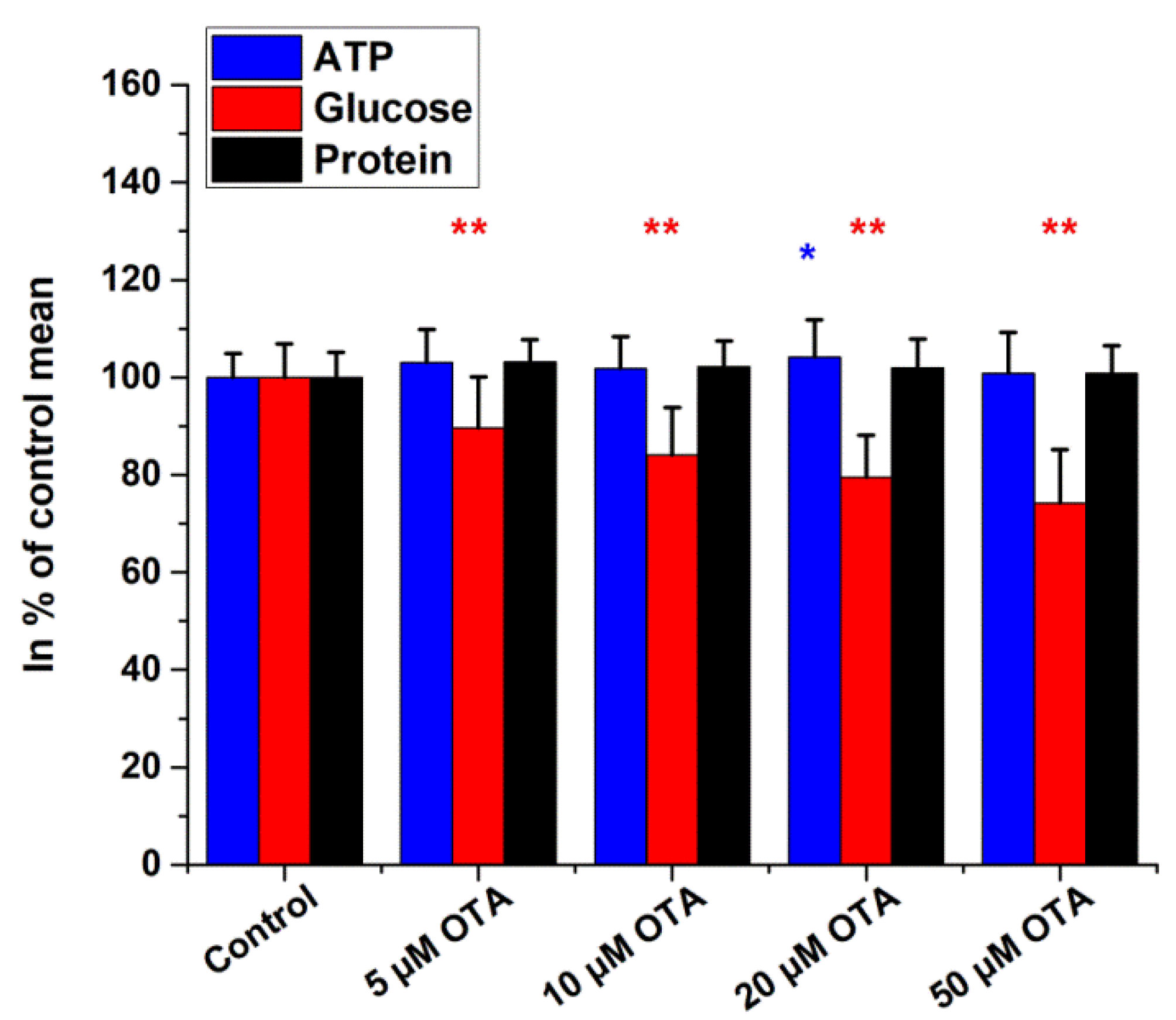
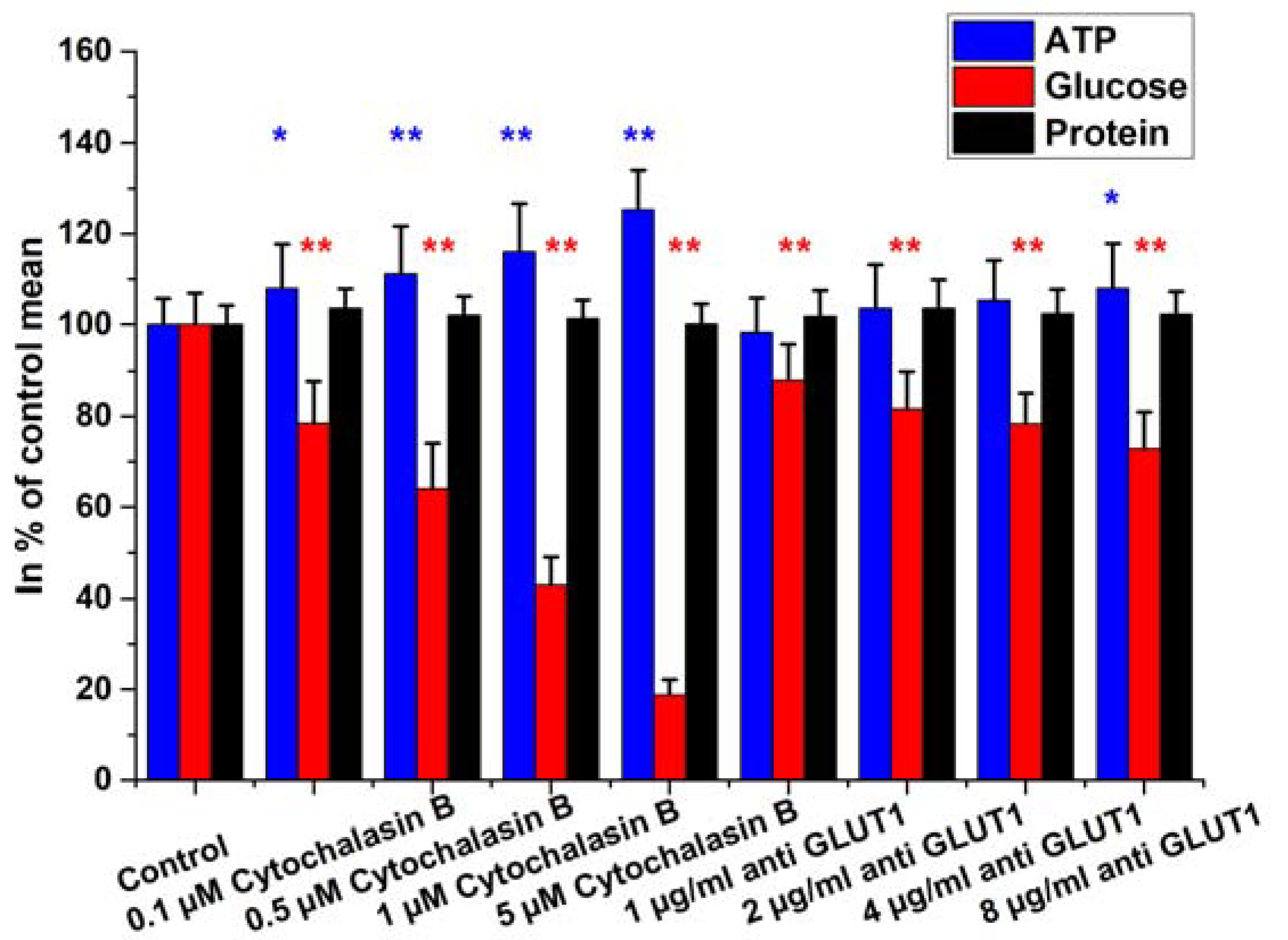
2.2. Intracellular Glucose, ATP, and Protein Measurements
2.3. Extracellular Lactate Levels
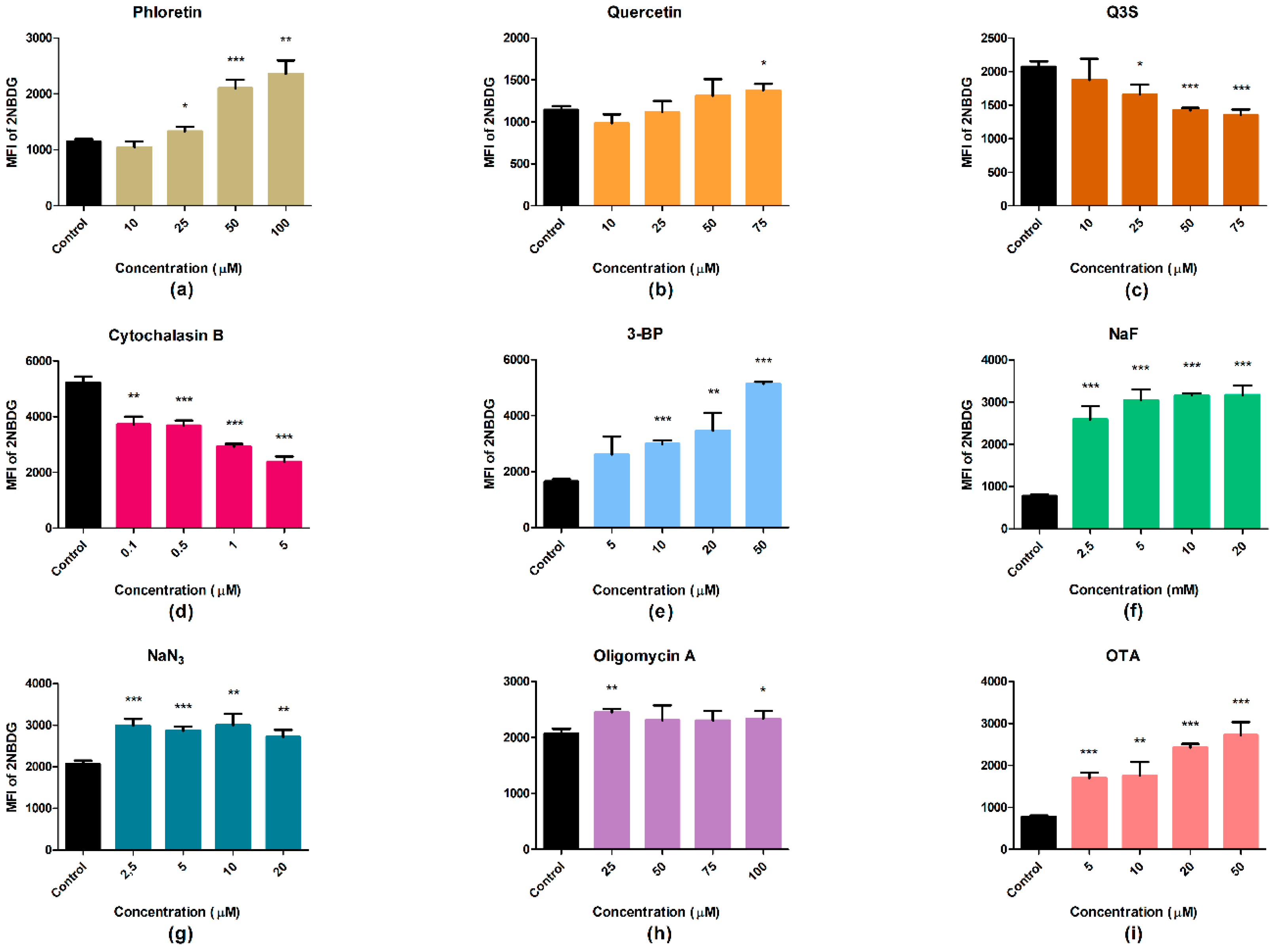
2.4. Glucose Uptake
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cell Culture and Treatments
4.3. Glucose/ATP/Protein Extraction and Measuring Reagents
4.4. Validation of Glucose Assay
4.5. Intracellular Glucose, ATP, and Protein Assay
4.6. Measurement of Lactate Production
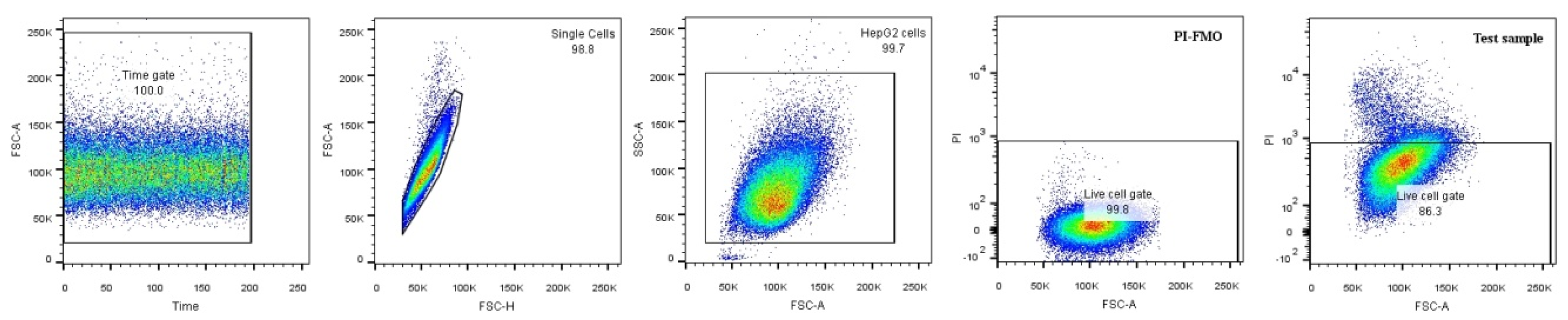
4.7. 2-NBDG Uptake by Flow Cytometry
4.8. Statistical Evaluation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Yuen, V.G.; McNeill, J.H. Comparison of the glucose oxidase method for glucose determination by manual assay and automated analyzer. J. Pharmacol. Toxicol. Methods 2000, 44, 543–546. [Google Scholar] [CrossRef]
- Hall, M.B.; Keuler, N.S. Factors affecting accuracy and time requirements of a glucose oxidase-peroxidase assay for determination of glucose. J. AOAC Int. 2009, 92, 50–60. [Google Scholar] [PubMed]
- Galant, A.L.; Kaufman, R.C.; Wilson, J.D. Glucose: Detection and analysis. Food Chem. 2015, 188, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.F.; Qiang, L.; Ren, J.; Ren, X.L.; Tang, F.Q.; Meng, X.W. Fluorescence turn-off detection of hydrogen peroxide and glucose directly using carbon nanodots as probes. Anal. Methods 2014, 6, 1922–1927. [Google Scholar] [CrossRef]
- Pang, Y.; Huang, Z.; Yang, Y.; Long, Y.; Zheng, H. Colorimetric detection of glucose based on ficin with peroxidase-like activity. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2018, 189, 510–515. [Google Scholar] [CrossRef] [PubMed]
- Volkenhoff, A.; Hirrlinger, J.; Kappel, J.M.; Klambt, C.; Schirmeier, S. Live imaging using a fret glucose sensor reveals glucose delivery to all cell types in the drosophila brain. J. Insect. Physiol. 2018, 106, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Semaan, D.G.; Igoli, J.O.; Young, L.; Gray, A.I.; Rowan, E.G.; Marrero, E. In vitro anti-diabetic effect of flavonoids and pheophytins from Allophylus cominia Sw. on the glucose uptake assays by HepG2, L6, 3T3-L1 and fat accumulation in 3T3-L1 adipocytes. J. Ethnopharmacol. 2018, 216, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Dawson, N.; Ferrington, L.; Olverman, H.J.; Kelly, P.A. Novel analysis for improved validity in semi-quantitative 2-deoxyglucose autoradiographic imaging. J. Neurosci. Methods 2008, 175, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Zou, C.H.; Wang, Y.J.; Shen, Z.F. 2-NBDG as a fluorescent indicator for direct glucose uptake measurement. J. Biochem. Biophys. Methods 2005, 64, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Blodgett, A.B.; Kothinti, R.K.; Kamyshko, I.; Petering, D.H.; Kumar, S.; Tabatabai, N.M. A fluorescence method for measurement of glucose transport in kidney cells. Diabetes Technol. Ther. 2011, 13, 743–751. [Google Scholar] [CrossRef] [PubMed]
- Hassanein, M.; Weidow, B.; Koehler, E.; Bakane, N.; Garbett, S.; Shyr, Y.; Quaranta, V. Development of high-throughput quantitative assays for glucose uptake in cancer cell lines. Mol. Imaging Biol. 2011, 13, 840–852. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.W.; Peng, F.Y. 2-NBDG fluorescence imaging of hypermetabolic circulating tumor cells in mouse xenograft model of breast cancer. J. Fluoresc. 2013, 23, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Kamalian, L.; Chadwick, A.E.; Bayliss, M.; French, N.S.; Monshouwer, M.; Snoeys, J.; Park, B.K. The utility of HepG2 cells to identify direct mitochondrial dysfunction in the absence of cell death. Toxicol. In Vitro 2015, 29, 732–740. [Google Scholar] [CrossRef] [PubMed]
- Koszegi, T.; Petrik, J.; Vladimir-Knezevic, S.; Nagy, S. Co-determination of ATP and proteins in triton X 100 non-ionic detergent-opened monolayer cultured cells. Luminescence 2007, 22, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Sali, N.; Nagy, S.; Poor, M.; Koszegi, T. Multiparametric luminescent cell viability assay in toxicology models: A critical evaluation. J. Pharmacol. Toxicol. Methods 2016, 79, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Geciova, J.; Bury, D.; Jelen, P. Methods for disruption of microbial cells for potential use in the dairy industry—A review. Int. Dairy J. 2002, 12, 541–553. [Google Scholar] [CrossRef]
- Mattei, B.; Franca, A.D.; Riske, K.A. Solubilization of binary lipid mixtures by the detergent triton X-100: The role of cholesterol. Langmuir 2015, 31, 378–386. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Diwu, Z.; Panchuk-Voloshina, N.; Haugland, R.P. A stable nonfluorescent derivative of resorufin for the fluorometric determination of trace hydrogen peroxide: Applications in detecting the activity of phagocyte nadph oxidase and other oxidases. Anal. Biochem. 1997, 253, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Csepregi, R.; Temesfoi, V.; Poor, M.; Faust, Z.; Koszegi, T. Green fluorescent protein-based viability assay in a multiparametric configuration. Molecules 2018, 23, 1575. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Hou, X.; Zhang, B.; Wang, Y.; He, L. The analysis of carbohydrates in milk powder by a new “heart-cutting” two-dimensional liquid chromatography method. J. Pharm. Biomed. Anal. 2014, 91, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Gangola, M.P.; Jaiswal, S.; Khedikar, Y.P.; Chibbar, R.N. A reliable and rapid method for soluble sugars and RFO analysis in chickpea using HPAEC-PAD and its comparison with HPLC-RI. Food Chem. 2014, 154, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Shanmugavelan, P.; Kim, S.Y.; Kim, J.B.; Kim, H.W.; Cho, S.M.; Kim, S.N.; Kim, S.Y.; Cho, Y.S.; Kim, H.R. Evaluation of sugar content and composition in commonly consumed korean vegetables, fruits, cereals, seed plants, and leaves by HPLC-ELSD. Carbohydr. Res. 2013, 380, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Behjousiar, A.; Kontoravdi, C.; Polizzi, K.M. In situ monitoring of intracellular glucose and glutamine in cho cell culture. PLoS ONE 2012, 7, e34512. [Google Scholar] [CrossRef] [PubMed]
- Berrone, E.; Beltramo, E.; Solimine, C.; Ape, A.U.; Porta, M. Regulation of intracellular glucose and polyol pathway by thiamine and benfotiamine in vascular cells cultured in high glucose. J. Biol. Chem. 2006, 281, 9307–9313. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Minchin, R.F.; Butcher, N.J. Arylamine n-acetyltransferase 1 protects against reactive oxygen species during glucose starvation: Role in the regulation of p53 stability. PLoS ONE 2018, 13, e0193560. [Google Scholar] [CrossRef] [PubMed]
- Watabe, S.; Sakamoto, Y.; Morikawa, M.; Okada, R.; Miura, T.; Ito, E. Highly sensitive determination of hydrogen peroxide and glucose by fluorescence correlation spectroscopy. PLoS ONE 2011, 6, e22955. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Su, F.; Buizer, S.; Kong, X.; Lee, F.; Day, K.; Tian, Y.; Meldrum, D.R. A polymer-based ratiometric intracellular glucose sensor. Chem. Commun. 2014, 50, 6920–6922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strobel, P.; Allard, C.; Perez-Acle, T.; Calderon, R.; Aldunate, R.; Leighton, F. Myricetin, quercetin and catechin-gallate inhibit glucose uptake in isolated rat adipocytes. Biochem. J. 2005, 386, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Xintaropoulou, C.; Ward, C.; Wise, A.; Marston, H.; Turnbull, A.; Langdon, S.P. A comparative analysis of inhibitors of the glycolysis pathway in breast and ovarian cancer cell line models. Oncotarget 2015, 6, 25677–25695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feron, O. Pyruvate into lactate and back: From the warburg effect to symbiotic energy fuel exchange in cancer cells. Radiother. Oncol. 2009, 92, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.S.; Lotspeich, W.D. Comparative effects of phlorizin and phloretin on glucose transport in the cat kidney. Am. J. Physiol. 1962, 203, 975–979. [Google Scholar] [CrossRef] [PubMed]
- Mullen, W.; Edwards, C.A.; Crozier, A. Absorption, excretion and metabolite profiling of methyl-, glucuronyl-, glucosyl- and sulpho-conjugates of quercetin in human plasma and urine after ingestion of onions. Br. J. Nutr. 2006, 96, 107–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janisch, K.M.; Williamson, G.; Needs, P.; Plumb, G.W. Properties of quercetin conjugates: Modulation of ldl oxidation and binding to human serum albumin. Free Radic. Res. 2004, 38, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Poor, M.; Boda, G.; Needs, P.W.; Kroon, P.A.; Lemli, B.; Bencsik, T. Interaction of quercetin and its metabolites with warfarin: Displacement of warfarin from serum albumin and inhibition of CYP2C9 enzyme. Biomed. Pharmacother. 2017, 88, 574–581. [Google Scholar] [CrossRef] [PubMed]
- Kurokawa, T.; Yoshida, Y.; Kawahara, K.; Tsuchida, T.; Okazawa, H.; Fujibayashi, Y.; Yonekura, Y.; Kotsuji, F. Expression of GLUT-1 glucose transfer, cellular proliferation activity and grade of tumor correlate with [F-18]-fluorodeoxyglucose uptake by positron emission tomography in epithelial tumors of the ovary. Int. J. Cancer 2004, 109, 926–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapoor, K.; Finer-Moore, J.S.; Pedersen, B.P.; Caboni, L.; Waight, A.; Hillig, R.C.; Bringmann, P.; Heisler, I.; Muller, T.; Siebeneicher, H.; et al. Mechanism of inhibition of human glucose transporter GLUT1 is conserved between cytochalasin B and phenylalanine amides. Proc. Natl. Acad. Sci. USA 2016, 113, 4711–4716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higashi, T.; Tamaki, N.; Torizuka, T.; Nakamoto, Y.; Sakahara, H.; Kimura, T.; Honda, T.; Inokuma, T.; Katsushima, S.; Ohshio, G.; et al. FDG uptake, GLUT-1 glucose transporter and cellularity in human pancreatic tumors. J. Nucl. Med. 1998, 39, 1727–1735. [Google Scholar] [PubMed]
- Rastogi, S.; Banerjee, S.; Chellappan, S.; Simon, G.R. Glut-1 antibodies induce growth arrest and apoptosis in human cancer cell lines. Cancer Lett. 2007, 257, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Omerovic, J.; Hammond, D.E.; Clague, M.J.; Prior, I.A. Ras isoform abundance and signalling in human cancer cell lines. Oncogene 2008, 27, 2754–2762. [Google Scholar] [CrossRef] [PubMed]
- Barbier, O.; Arreola-Mendoza, L.; Del Razo, L.M. Molecular mechanisms of fluoride toxicity. Chem.-Biol. Intteract. 2010, 188, 319–333. [Google Scholar] [CrossRef] [PubMed]
- Lomakina, G.Y.; Modestova, Y.A.; Ugarova, N.N. Bioluminescence assay for cell viability. Biochemistry (Mosc.) 2015, 80, 701–713. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, D.A.; Simmons, T.D.; Slater, K.J.; Crouch, S.P.M. Measurement of the ADP: ATP ratio in human leukaemic cell lines can be used as an indicator of cell viability, necrosis and apoptosis. J. Immunol. Methods 2000, 240, 79–92. [Google Scholar] [CrossRef]
- Ramio-Lluch, L.; Yeste, M.; Fernandez-Novell, J.M.; Estrada, E.; Rocha, L.; Cebrian-Perez, J.A.; Muino-Blanco, T.; Concha, I.I.; Ramirez, A.; Rodriguez-Gil, J.E. Oligomycin a-induced inhibition of mitochondrial atp-synthase activity suppresses boar sperm motility and in vitro capacitation achievement without modifying overall sperm energy levels. Reproduct. Fertil. Dev. 2014, 26, 883–897. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.F.; Xie, S.Y.; Xu, F.F.; Liu, A.M.; Wang, Y.X.; Chen, D.M.; Pan, Y.H.; Huang, L.L.; Peng, D.P.; Wang, X.; et al. Ochratoxin a: Toxicity, oxidative stress and metabolism. Food Chem. Toxicol. 2018, 112, 320–331. [Google Scholar] [CrossRef] [PubMed]
- Locasale, J.W. New concepts in feedback regulation of glucose metabolism. Curr. Opin. Syst. Biol. 2018, 8, 32–38. [Google Scholar] [CrossRef]
- James, D.E.; Strube, M.; Mueckler, M. Molecular cloning and characterization of an insulin-regulatable glucose transporter. Nature 1989, 338, 83–87. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, P.E.; Joseph, J.W.; Rorsman, P. Glucose-sensing mechanisms in pancreatic β-cells. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2005, 360, 2211–2225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macheda, M.L.; Rogers, S.; Best, J.D. Molecular and cellular regulation of glucose transporter (GLUT) proteins in cancer. J. Cell. Physiol. 2005, 202, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Vega, M.; Riera, A.; Fernandez-Cid, A.; Herrero, P.; Moreno, F. Hexokinase 2 is an intracellular glucose sensor of yeast cells that maintains the structure and activity of Mig1 protein repressor complex. J. Biol. Chem. 2016, 291, 7267–7285. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, N.; Ueda-Wakagi, M.; Sato, T.; Kawasaki, K.; Sawada, K.; Kawabata, K.; Akagawa, M.; Ashida, H. Measurement of glucose uptake in cultured cells. Curr. Protoc. Pharmacol. 2015, 71. [Google Scholar] [CrossRef]
- Saito, K.; Lee, S.; Shiuchi, T.; Toda, C.; Kamijo, M.; Inagaki-Ohara, K.; Okamoto, S.; Minokoshi, Y. An enzymatic photometric assay for 2-deoxyglucose uptake in insulin-responsive tissues and 3T3-L1 adipocytes. Anal. Biochem. 2011, 412, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Valley, M.P.; Karassina, N.; Aoyama, N.; Carlson, C.; Cali, J.J.; Vidugiriene, J. A bioluminescent assay for measuring glucose uptake. Anal. Biochem. 2016, 505, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Needs, P.W.; Kroon, P.A. Convenient syntheses of metabolically important quercetin glucuronides and sulfates. Tetrahedron 2006, 62, 6862–6868. [Google Scholar] [CrossRef]
- Magnusson, B.; Örnemark, U. The Fitness for Purpose of Analytical Methods—A Laboratory Guide to Method Validation and Related Topics, 2nd ed.; Eurachem: Torino, Italy, 2014; p. 57. [Google Scholar]
- Armbruster, D.A.; Pry, T. Limit of blank, limit of detection and limit of quantitation. Clin. Biochem. Rev. 2008, 29 (Suppl. 1), S49–S52. [Google Scholar] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Intra-Assay (n = 32) | Interassay (n = 160) | |||||
|---|---|---|---|---|---|---|
| Glucose (µM) | Mean ± SD (µM) | CV (%) | Recovery (%) | Mean ± SD (µM) | CV (%) | Recovery (%) |
| 0.5 a | 0.47 ± 0.02 | 4.26 | 94.06 | 0.47 ± 0.04 | 8.51 | 95.04 |
| 2.5 a | 2.55 ± 0.11 | 4.31 | 102.09 | 2.52 ± 0.13 | 5.16 | 100.80 |
| 1.0 b | 0.99 ± 0.04 | 4.04 | 99.00 | 0.97 ± 0.08 | 8.24 | 97.00 |
| 5.0 b | 4.59 ± 0.12 | 2.61 | 91.80 | 4.49 ± 0.25 | 5.56 | 89.80 |
| Intra-Assay (n = 32) | Interassay (n = 160) | |||||||
|---|---|---|---|---|---|---|---|---|
| Mean ± SD (µM) | CV (%) | Recovery (µM) | Recovery (%) | Mean ± SD (µM) | CV (%) | Recovery (µM) | Recovery (%) | |
| HepG2 cells | 0.95 ± 0.07 | 8.06 | n.a. | n.a. | 0.94 ± 0.11 | 12.11 | n.a. | n.a. |
| HepG2 + 0.5 µM glucose | 1.38 ± 0.13 | 9.45 | 0.43 | 91.81 | 1.37 ± 0.15 | 11.03 | 0.43 | 90.41 |
| HepG2 + 2.5 µM glucose | 3.00 ± 0.21 | 7.25 | 2.06 | 83.70 | 3.03 ± 0.24 | 8.03 | 2.10 | 83.70 |
| Mode of Action | Treatment | Lactate ± SD (%) |
|---|---|---|
| DMEM | - | 57 ± 7 |
| Control cells in DMEM | - | 100 ± 29 |
| Inhibitors of glucose transporters | 10 µM PHL | 78 ± 19 |
| 100 µM PHL | 98 ± 20 | |
| 10 µM Que | 104 ± 30 | |
| 75 µM Que | 91 ± 24 | |
| 10 µM Q3′S | 105 ± 30 | |
| 75 µM Q3′S | 100 ± 28 | |
| Inhibitors of glycolysis | 2.5 mM NaF | 65 ± 12 |
| 20 mM NaF | 27 ± 8 | |
| 5 µM 3-BP | 76 ± 19 | |
| 50 µM 3-BP | 71 ± 18 | |
| Inhibitors of terminal oxidation | 2.5 mM NaN3 | 114 ± 28 |
| 20 mM NaN3 | 138 ± 33 | |
| 25 µM oligomycin A | 113 ± 23 | |
| 100 µM oligomycin A | 93 ± 29 | |
| Complex effect | 5 µM OTA | 106 ± 27 |
| 50 µM OTA | 95 ± 30 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Csepregi, R.; Temesfői, V.; Sali, N.; Poór, M.; W. Needs, P.; A. Kroon, P.; Kőszegi, T. A One-Step Extraction and Luminescence Assay for Quantifying Glucose and ATP Levels in Cultured HepG2 Cells. Int. J. Mol. Sci. 2018, 19, 2670. https://doi.org/10.3390/ijms19092670
Csepregi R, Temesfői V, Sali N, Poór M, W. Needs P, A. Kroon P, Kőszegi T. A One-Step Extraction and Luminescence Assay for Quantifying Glucose and ATP Levels in Cultured HepG2 Cells. International Journal of Molecular Sciences. 2018; 19(9):2670. https://doi.org/10.3390/ijms19092670
Chicago/Turabian StyleCsepregi, Rita, Viktória Temesfői, Nikolett Sali, Miklós Poór, Paul W. Needs, Paul A. Kroon, and Tamás Kőszegi. 2018. "A One-Step Extraction and Luminescence Assay for Quantifying Glucose and ATP Levels in Cultured HepG2 Cells" International Journal of Molecular Sciences 19, no. 9: 2670. https://doi.org/10.3390/ijms19092670
APA StyleCsepregi, R., Temesfői, V., Sali, N., Poór, M., W. Needs, P., A. Kroon, P., & Kőszegi, T. (2018). A One-Step Extraction and Luminescence Assay for Quantifying Glucose and ATP Levels in Cultured HepG2 Cells. International Journal of Molecular Sciences, 19(9), 2670. https://doi.org/10.3390/ijms19092670