Targeting Amyloid Aggregation: An Overview of Strategies and Mechanisms



Abstract
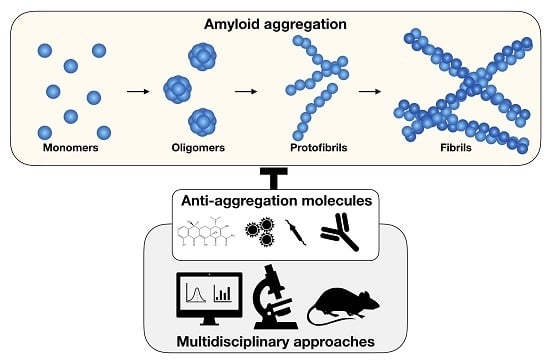
:
1. Introduction
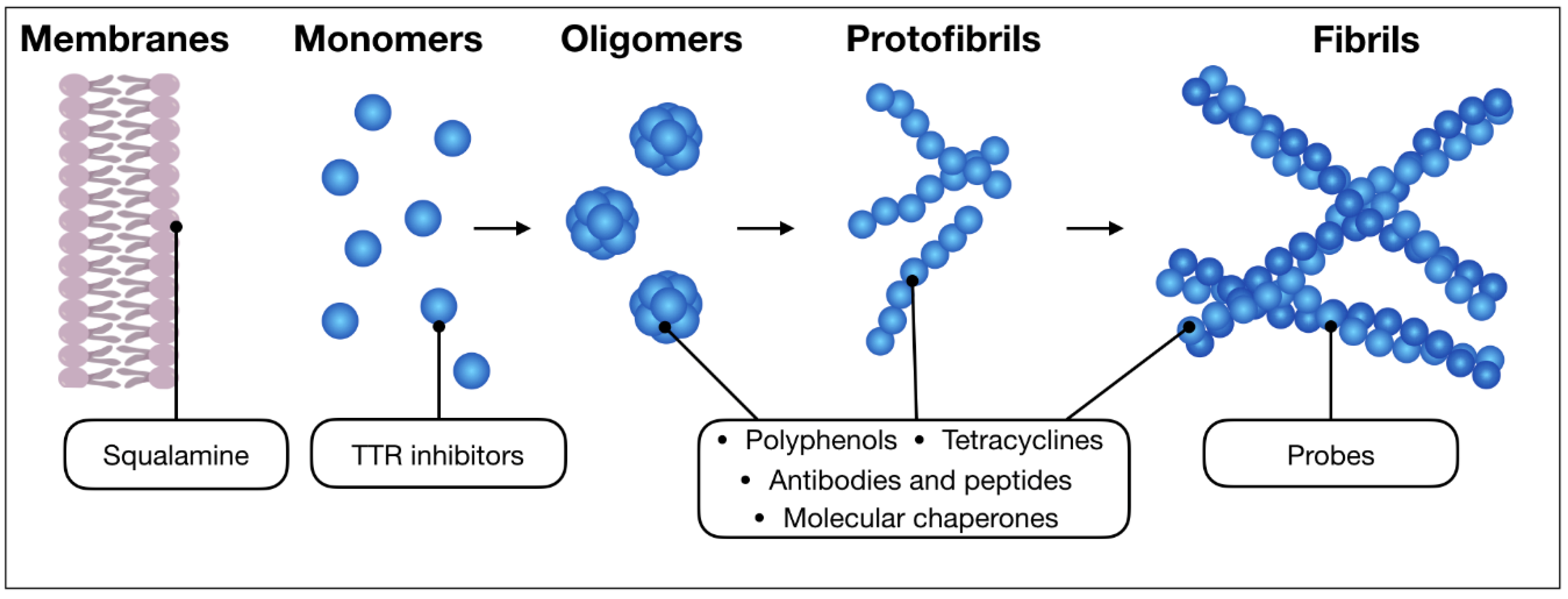
2. The Main Classes of Anti-Amyloid Compounds
2.1. Probes and Diagnostic Molecules
2.2. Anthracyclines and Tetracyclines
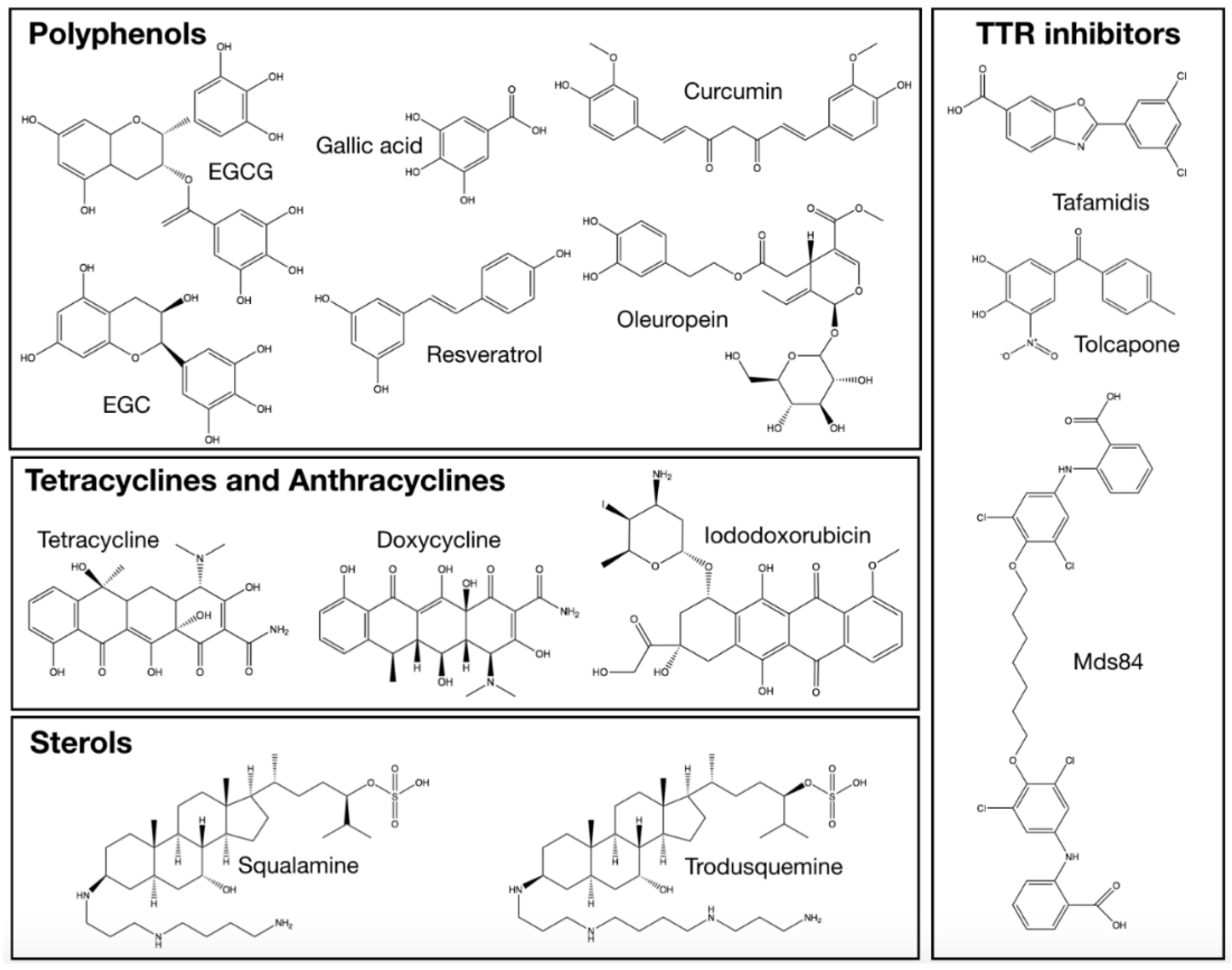
2.3. Sterols
2.4. Peptides and Engineered Antibodies
2.5. Polyphenols
2.5.1. (−)-Epigallocatechin-gallate (EGCG) and Related Compounds
2.5.2. Resveratrol
2.5.3. Curcumin
2.5.4. Oleuropein
2.6. Compounds Retarding Transthyretin Aggregation
2.7. Nanoparticles
3. Lipid-Modulated Amyloid Aggregation and Antiamyloid Drugs
4. The Contribution of the Computational Approaches
5. Final Remarks and Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AA | serum amyloid A amyloidosis |
| Aβ | amyloid-beta |
| Aβ2-m | β2-microglobulin related amyloidosis |
| AD | Alzheimer’s disease |
| AL | immunoglobulin light chain amyloidosis |
| APrP | Prp amyloidosis |
| α-syn | alpha-synuclein |
| ATX3 | ataxin-3 |
| ATTR | transthyretin amyloidosis |
| β2-m | β2-microglobulin |
| CH | corea of Hungtington |
| CR | Congo red |
| cryo-EM | cryo electron microscopy |
| DOX | doxycycline |
| DRA | dialysis-related amyloidosis |
| EGC | (−)-epigallocatechin |
| EGCG | (−)-Epigallocatechin-gallate |
| GA | gallic acid |
| GM1 | Monosialotetrahexosylganglioside 1 |
| HEWL | egg-white lysozyme |
| htt | huntingtin |
| IAPP | amylin |
| IDOX | 4′-iodo-4′-deoxy-doxorubicin |
| LC | immunoglobulin light chain |
| MD | molecular dynamics |
| PD | Parkinson’s disease |
| polyQ | polyglutamine |
| PrP | prion protein |
| RES | resveratrol (3,5,4′-trihydroxy-trans-stilbene) |
| QSAR | quantitative structure-activity relationship |
| SAA | serum amyloid A |
| SSNMR | solid-state nuclear magnetic resonance |
| TTR | transthyretin |
References
- Dobson, C.M. Protein folding and misfolding. Nature 2003, 426, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Hartl, F.U. Protein misfolding diseases. Annu. Rev. Biochem. 2017, 86, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Chiti, F.; Dobson, C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006, 75, 333–366. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.F.; Knowles, T.P.; Dobson, C.M.; MacPhee, C.E.; Welland, M.E. Characterization of the nanoscale properties of individual amyloid fibrils. Proc. Natl. Acad. Sci. USA 2006, 103, 15806–15811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morel, B.; Varela, L.; Conejero-Lara, F. The thermodynamic stability of amyloid fibrils studied by differential scanning calorimetry. J. Phys. Chem. B 2010, 114, 4010–4019. [Google Scholar] [CrossRef] [PubMed]
- Knauer, M.F.; Soreghan, B.; Burdick, D.; Kosmoski, J.; Glabe, C.G. Intracellular accumulation and resistance to degradation of the Alzheimer amyloid A4/beta protein. Proc. Natl. Acad. Sci. USA 1992, 89, 7437–7441. [Google Scholar] [CrossRef] [PubMed]
- Meier, B.H.; Riek, R.; Böckmann, A. Emerging structural understanding of amyloid fibrils by solid-state NMR. Trends Biochem. Sci. 2017, 42, 777–787. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, A.W.; Falcon, B.; He, S.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Crowther, R.A.; Ghetti, B.; Goedert, M.; Scheres, S.H. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 2017, 547, 185–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartl, F.U.; Hayer-Hartl, M. Converging concepts of protein folding in vitro and in vivo. Nat. Struct. Mol. Biol. 2009, 16, 574–581. [Google Scholar] [CrossRef] [PubMed]
- Blancas-Mejía, L.M.; Ramirez-Alvarado, M. Systemic amyloidoses. Annu. Rev. Biochem. 2013, 82, 745–774. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.I.; Vendruscolo, M.; Dobson, C.M.; Knowles, T.P. From macroscopic measurements to microscopic mechanisms of protein aggregation. J. Mol. Biol. 2012, 421, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Knowles, T.P.; Waudby, C.A.; Devlin, G.L.; Cohen, S.I.; Aguzzi, A.; Vendruscolo, M.; Terentjev, E.M.; Welland, M.E.; Dobson, C.M. An analytical solution to the kinetics of breakable filament assembly. Science 2009, 326, 1533–1537. [Google Scholar] [CrossRef] [PubMed]
- Bucciantini, M.; Giannoni, E.; Chiti, F.; Baroni, F.; Formigli, L.; Zurdo, J.; Taddei, N.; Ramponi, G.; Dobson, C.M.; Stefani, M. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 2002, 416, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Cremades, N.; Cohen, S.I.; Deas, E.; Abramov, A.Y.; Chen, A.Y.; Orte, A.; Sandal, M.; Clarke, R.W.; Dunne, P.; Aprile, F.A.; et al. Direct observation of the interconversion of normal and toxic forms of α-synuclein. Cell 2012, 149, 1048–1059. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.W.; Drakulic, S.; Deas, E.; Ouberai, M.; Aprile, F.A.; Arranz, R.; Ness, S.; Roodveldt, C.; Guilliams, T.; De-Genst, E.J.; et al. Structural characterization of toxic oligomers that are kinetically trapped during α-synuclein fibril formation. Proc. Natl. Acad. Sci. USA 2015, 112, E1994–E2003. [Google Scholar] [CrossRef] [PubMed]
- Fusco, G.; Chen, S.W.; Williamson, P.T.; Cascella, R.; Perni, M.; Jarvis, J.A.; Cecchi, C.; Vendruscolo, M.; Chiti, F.; Cremades, N.; et al. Structural basis of membrane disruption and cellular toxicity by α-synuclein oligomers. Science 2017, 358, 1440–1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsia, A.Y.; Masliah, E.; McConlogue, L.; Yu, G.-Q.; Tatsuno, G.; Hu, K.; Kholodenko, D.; Malenka, R.C.; Nicoll, R.A.; Mucke, L. Plaque-independent disruption of neural circuits in Alzheimer’s disease mouse models. Proc. Natl. Acad. Sci. USA 1999, 96, 3228–3233. [Google Scholar] [CrossRef] [PubMed]
- Mucke, L.; Masliah, E.; Yu, G.-Q.; Mallory, M.; Rockenstein, E.M.; Tatsuno, G.; Hu, K.; Kholodenko, D.; Johnson-Wood, K.; McConlogue, L. High-level neuronal expression of Aβ1–42 in wild-type human amyloid protein precursor transgenic mice: Synaptotoxicity without plaque formation. J. Neurosci. 2000, 20, 4050–4058. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.M.; Klyubin, I.; Fadeeva, J.V.; Cullen, W.K.; Anwyl, R.; Wolfe, M.S.; Rowan, M.J.; Selkoe, D.J. Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature 2002, 416, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Kamenetz, F.; Tomita, T.; Hsieh, H.; Seabrook, G.; Borchelt, D.; Iwatsubo, T.; Sisodia, S.; Malinow, R. APP processing and synaptic function. Neuron 2003, 37, 925–937. [Google Scholar] [CrossRef]
- Shankar, G.M.; Bloodgood, B.L.; Townsend, M.; Walsh, D.M.; Selkoe, D.J.; Sabatini, B.L. Natural oligomers of the Alzheimer amyloid-β protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J. Neurosci. 2007, 27, 2866–2875. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Hong, S.; Shepardson, N.E.; Walsh, D.M.; Shankar, G.M.; Selkoe, D. Soluble oligomers of amyloid β protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron 2009, 62, 788–801. [Google Scholar] [CrossRef] [PubMed]
- Flagmeier, P.; De, S.; Wirthensohn, D.C.; Lee, S.F.; Vincke, C.; Muyldermans, S.; Knowles, T.P.; Gandhi, S.; Dobson, C.M.; Klenerman, D. Ultrasensitive measurement of Ca2+ influx into lipid vesicles induced by protein aggregates. Angew. Chem.-Int. Ed. 2017, 56, 7750–7754. [Google Scholar] [CrossRef] [PubMed]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, I.; Bajouco, L.; Mota, S.; Auberson, Y.; Oliveira, C.; Rego, A. Amyloid beta peptide 1–42 disturbs intracellular calcium homeostasis through activation of GluN2B-containing N-methyl-d-aspartate receptors in cortical cultures. Cell Calcium 2012, 51, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Eckert, A.; Hauptmann, S.; Scherping, I.; Meinhardt, J.; Rhein, V.; Dröse, S.; Brandt, U.; Fändrich, M.; Müller, W.E.; Götz, J. Oligomeric and fibrillar species of β-amyloid (Aβ42) both impair mitochondrial function in P301L tau transgenic mice. J. Mol. Med. 2008, 86, 1255–1267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salminen, A.; Ojala, J.; Kauppinen, A.; Kaarniranta, K.; Suuronen, T. Inflammation in Alzheimer's disease: Amyloid-β oligomers trigger innate immunity defence via pattern recognition receptors. Progr. Neurobiol. 2009, 87, 181–194. [Google Scholar] [CrossRef]
- Bellotti, V.; Chiti, F. Amyloidogenesis in its biological environment: Challenging a fundamental issue in protein misfolding diseases. Curr. Opin. Struct. Biol. 2008, 18, 771–779. [Google Scholar] [CrossRef] [PubMed]
- Kummer, M.P.; Heneka, M.T. Truncated and modified amyloid-beta species. Alzheimer Res. Ther. 2014, 6, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galvagnion, C.; Buell, A.K.; Meisl, G.; Michaels, T.C.; Vendruscolo, M.; Knowles, T.P.; Dobson, C.M. Lipid vesicles trigger α-synuclein aggregation by stimulating primary nucleation. Nat. Chem. Biol. 2015, 11, 229–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merlini, G.; Bellotti, V. Molecular mechanisms of amyloidosis. N. Engl. J. Med. 2003, 349, 583–596. [Google Scholar] [CrossRef] [PubMed]
- Stoppini, M.; Bellotti, V. Systemic amyloidosis: lessons from β2-microglobulin. J. Biol. Chem. 2015, 290, 9951–9958. [Google Scholar] [CrossRef] [PubMed]
- Valleix, S.; Gillmore, J.D.; Bridoux, F.; Mangione, P.P.; Dogan, A.; Nedelec, B.; Boimard, M.; Touchard, G.; Goujon, J.-M.; Lacombe, C.; et al. Hereditary systemic amyloidosis due to Asp76Asn variant β2-microglobulin. N. Engl. J. Med. 2012, 366, 2276–2283. [Google Scholar] [CrossRef] [PubMed]
- Marcoux, J.; Mangione, P.P.; Porcari, R.; Degiacomi, M.T.; Verona, G.; Taylor, G.W.; Giorgetti, S.; Raimondi, S.; Sanglier-Cianférani, S.; Benesch, J.L.; et al. A novel mechano-enzymatic cleavage mechanism underlies transthyretin amyloidogenesis. EMBO Mol. Med. 2015, 7, 1337–1349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perni, M.; Galvagnion, C.; Maltsev, A.; Meisl, G.; Müller, M.B.; Challa, P.K.; Kirkegaard, J.B.; Flagmeier, P.; Cohen, S.I.; Cascella, R.; et al. A natural product inhibits the initiation of α-synuclein aggregation and suppresses its toxicity. Proc. Natl. Acad. Sci. USA 2017, 114, E1009–E1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perni, M.; Flagmeier, P.; Limbocker, R.; Cascella, R.; Aprile, F.A.; Galvagnion, C.; Heller, G.T.; Meisl, G.; Chen, S.W.; Kumita, J.R.; et al. Multistep inhibition of α-synuclein aggregation and toxicity in vitro and in vivo by trodusquemine. ACS Chem. Biol. 2018, 13, 2308–2319. [Google Scholar] [CrossRef] [PubMed]
- Forloni, G.; Colombo, L.; Girola, L.; Tagliavini, F.; Salmona, M. Anti-amyloidogenic activity of tetracyclines: Studies in vitro. FEBS Lett. 2001, 487, 404–407. [Google Scholar] [CrossRef]
- Airoldi, C.; Colombo, L.; Manzoni, C.; Sironi, E.; Natalello, A.; Doglia, S.M.; Forloni, G.; Tagliavini, F.; Del Favero, E.; Cantu, L.; et al. Tetracycline prevents Aβ oligomer toxicity through an atypical supramolecular interaction. Org. Biomol. Chem. 2011, 9, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Marcinko, T.M.; Kiefer, P.A.; Vachet, R.W. Using covalent labeling and mass spectrometry to study protein binding sites of amyloid inhibiting molecules. Anal. Chem. 2017, 89, 11583–11591. [Google Scholar] [CrossRef] [PubMed]
- Giorgetti, S.; Raimondi, S.; Pagano, K.; Relini, A.; Bucciantini, M.; Corazza, A.; Fogolari, F.; Codutti, L.; Salmona, M.; Mangione, P.; et al. Effect of tetracyclines on the dynamics of formation and destructuration of β2-microglobulin amyloid fibrils. J. Biol. Chem. 2011, 286, 2121–2131. [Google Scholar] [CrossRef] [PubMed]
- Marcinko, T.M.; Dong, J.; LeBlanc, R.; Daborowski, K.V.; Vachet, R.W. Small molecule-mediated inhibition of β-2-microglobulin amyloid fibril formation. J. Biol. Chem. 2017, 292, 10630–10638. [Google Scholar] [CrossRef] [PubMed]
- Obici, L.; Cortese, A.; Lozza, A.; Lucchetti, J.; Gobbi, M.; Palladini, G.; Perlini, S.; Saraiva, M.J.; Merlini, G. Doxycycline plus tauroursodeoxycholic acid for transthyretin amyloidosis: a phase II study. Amyloid 2012, 19, 34–36. [Google Scholar] [CrossRef] [PubMed]
- Montagna, G.; Cazzulani, B.; Obici, L.; Uggetti, C.; Giorgetti, S.; Porcari, R.; Ruggiero, R.; Mangione, P.P.; Brambilla, M.; Lucchetti, J.; et al. Benefit of doxycycline treatment on articular disability caused by dialysis related amyloidosis. Amyloid 2013, 20, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Piccoli, G.B.; Hachemi, M.; Molfino, I.; Coindre, J.P.; Boursot, C. Doxycycline treatment in dialysis related amyloidosis: Discrepancy between antalgic effect and inflammation, studied with FDG-positron emission tomography: A case report. BMC Nephrol. 2017, 18, 285. [Google Scholar] [CrossRef] [PubMed]
- Wechalekar, A.; Whelan, C. Encouraging impact of doxycycline on early mortality in cardiac light chain (AL) amyloidosis. Blood Cancer J. 2017, 7, e546. [Google Scholar] [CrossRef] [PubMed]
- Gianni, L.; Bellotti, V.; Gianni, A.M.; Merlini, G. New drug therapy of amyloidoses: resorption of AL-type deposits with 4′-iodo-4′-deoxydoxorubicin. Blood 1995, 86, 855–861. [Google Scholar] [PubMed]
- Merlini, G.; Ascari, E.; Amboldi, N.; Bellotti, V.; Arbustini, E.; Perfetti, V.; Ferrari, M.; Zorzoli, I.; Marinone, M.G.; Garini, P.; et al. Interaction of the anthracycline 4′-iodo-4′-deoxydoxorubicin with amyloid fibrils: Inhibition of amyloidogenesis. Proc. Natl. Acad. Sci. USA 1995, 92, 2959–2963. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Abedini, A.; Plesner, A.; Middleton, C.T.; Potter, K.J.; Zanni, M.T.; Verchere, C.B.; Raleigh, D.P. The sulfated triphenyl methane derivative acid fuchsin is a potent inhibitor of amyloid formation by human islet amyloid polypeptide and protects against the toxic effects of amyloid formation. J. Mol. Biol. 2010, 400, 555–566. [Google Scholar] [CrossRef] [PubMed]
- How, S.C.; Yang, S.M.; Hsin, A.; Tseng, C.P.; Hsueh, S.S.; Lin, M.S.; Chen, R.P.; Chou, W.L.; Wang, S.S. Examining the inhibitory potency of food additive fast green FCF against amyloid fibrillogenesis under acidic conditions. Food Funct. 2016, 7, 4898–4907. [Google Scholar] [CrossRef] [PubMed]
- Chang, E.; Congdon, E.E.; Honson, N.S.; Duff, K.E.; Kuret, J. Structure–activity relationship of cyanine tau aggregation inhibitors. J. Med. Chem. 2009, 52, 3539–3547. [Google Scholar] [CrossRef] [PubMed]
- Necula, M.; Chirita, C.N.; Kuret, J. Cyanine dye N744 inhibits tau fibrillization by blocking filament extension: Implications for the treatment of tauopathic neurodegenerative diseases. Biochemistry 2005, 44, 10227–10237. [Google Scholar] [CrossRef] [PubMed]
- Frid, P.; Anisimov, S.V.; Popovic, N. Congo red and protein aggregation in neurodegenerative diseases. Brain Res. Rev. 2007, 53, 135–160. [Google Scholar] [CrossRef] [PubMed]
- Lendel, C.; Bolognesi, B.; Wahlström, A.; Dobson, C.M.; Gräslund, A. Detergent-like interaction of Congo red with the amyloid β peptide. Biochemistry 2010, 49, 1358–1360. [Google Scholar] [CrossRef] [PubMed]
- Stefani, M.; Rigacci, S. Protein folding and aggregation into amyloid: the interference by natural phenolic compounds. Int. J. Mol. Sci. 2013, 14, 12411–12457. [Google Scholar] [CrossRef] [PubMed]
- Cisek, K.; Cooper, G.L.; Huseby, C.J.; Kuret, J. Structure and mechanism of action of tau aggregation inhibitors. Curr. Alzheimer Res. 2014, 11, 918–927. [Google Scholar] [CrossRef] [PubMed]
- Razavi, H.; Palaninathan, S.K.; Powers, E.T.; Wiseman, R.L.; Purkey, H.E.; Mohamedmohaideen, N.N.; Deechongkit, S.; Chiang, K.P.; Dendle, M.T.; Sacchettini, J.C.; et al. Benzoxazoles as transthyretin amyloid fibril inhibitors: Synthesis, evaluation, and mechanism of action. Angew. Chem.-Int. Ed. 2003, 115, 2864–2867. [Google Scholar] [CrossRef]
- Verona, G.; Mangione, P.P.; Raimondi, S.; Giorgetti, S.; Faravelli, G.; Porcari, R.; Corazza, A.; Gillmore, J.D.; Hawkins, P.N.; Pepys, M.B.; et al. Inhibition of the mechano-enzymatic amyloidogenesis of transthyretin: role of ligand affinity, binding cooperativity and occupancy of the inner channel. Sci. Rep. 2017, 7, 182. [Google Scholar] [CrossRef] [PubMed]
- Sievers, S.A.; Karanicolas, J.; Chang, H.W.; Zhao, A.; Jiang, L.; Zirafi, O.; Stevens, J.T.; Münch, J.; Baker, D.; Eisenberg, D. Structure-based design of non-natural amino-acid inhibitors of amyloid fibril formation. Nature 2011, 475, 96–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sivanesam, K.; Shu, I.; Huggins, K.N.; Tatarek-Nossol, M.; Kapurniotu, A.; Andersen, N.H. Peptide inhibitors of the amyloidogenesis of IAPP: Verification of the hairpin-binding geometry hypothesis. FEBS Lett. 2016, 590, 2575–2583. [Google Scholar] [CrossRef] [PubMed]
- Andreetto, E.; Malideli, E.; Yan, L.M.; Kracklauer, M.; Farbiarz, K.; Tatarek-Nossol, M.; Rammes, G.; Prade, E.; Neumüller, T.; Caporale, A.; et al. A hot-segment-based approach for the design of cross-amyloid interaction surface mimics as inhibitors of amyloid self-assembly. Angew. Chem.-Int. Ed. 2015, 54, 13095–13100. [Google Scholar]
- Yan, L.M.; Velkova, A.; Tatarek-Nossol, M.; Rammes, G.; Sibaev, A.; Andreetto, E.; Kracklauer, M.; Bakou, M.; Malideli, E.; Göke, B.; et al. Selectively N-methylated soluble IAPP mimics as potent IAPP receptor agonists and nanomolar inhibitors of cytotoxic self-assembly of both IAPP and Aβ40. Angew. Chem.-Int. Ed. 2013, 52, 10378–10383. [Google Scholar] [CrossRef] [PubMed]
- Truex, N.L.; Wang, Y.; Nowick, J.S. Assembly of peptides derived from β-sheet regions of β-amyloid. J. Am. Chem. Soc. 2016, 138, 13882–13890. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.-H.; Pardon, E.; Menzer, L.; De Genst, E.; Kumita, J.R.; Christodoulou, J.; Saerens, D.; Brans, A.; Bouillenne, F.; Archer, D.B.; et al. Engineering a camelid antibody fragment that binds to the active site of human lysozyme and inhibits its conversion into amyloid fibrils. Biochemistry 2008, 47, 11041–11054. [Google Scholar] [CrossRef] [PubMed]
- El-Turk, F.; Newby, F.N.; De Genst, E.; Guilliams, T.; Sprules, T.; Mittermaier, A.; Dobson, C.M.; Vendruscolo, M. Structural effects of two camelid nanobodies directed to distinct C-terminal epitopes on α-synuclein. Biochemistry 2016, 55, 3116–3122. [Google Scholar] [CrossRef] [PubMed]
- Drews, A.; Flint, J.; Shivji, N.; Jönsson, P.; Wirthensohn, D.; De Genst, E.; Vincke, C.; Muyldermans, S.; Dobson, C.; Klenerman, D. Individual aggregates of amyloid beta induce temporary calcium influx through the cell membrane of neuronal cells. Sci. Rep. 2016, 6, 31910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raimondi, S.; Porcari, R.; Mangione, P.P.; Verona, G.; Marcoux, J.; Giorgetti, S.; Taylor, G.W.; Ellmerich, S.; Ballico, M.; Zanini, S.; et al. A specific nanobody prevents amyloidogenesis of D76N β 2-microglobulin in vitro and modifies its tissue distribution in vivo. Sci. Rep. 2017, 7, 46711. [Google Scholar] [CrossRef] [PubMed]
- Perchiacca, J.M.; Ladiwala, A.R.A.; Bhattacharya, M.; Tessier, P.M. Structure-based design of conformation-and sequence-specific antibodies against amyloid β. Proc. Natl. Acad. Sci. USA 2012, 109, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Sormanni, P.; Aprile, F.A.; Vendruscolo, M. Rational design of antibodies targeting specific epitopes within intrinsically disordered proteins. Proc. Natl. Acad. Sci. USA 2015, 112, 9902–9907. [Google Scholar] [CrossRef] [PubMed]
- Aprile, F.A.; Sormanni, P.; Perni, M.; Arosio, P.; Linse, S.; Knowles, T.P.; Dobson, C.M.; Vendruscolo, M. Selective targeting of primary and secondary nucleation pathways in Aβ42 aggregation using a rational antibody scanning method. Sci. Adv. 2017, 3, e1700488. [Google Scholar] [CrossRef] [PubMed]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Tucker, S.; Möller, C.; Tegerstedt, K.; Lord, A.; Laudon, H.; Sjödahl, J.; Söderberg, L.; Spens, E.; Sahlin, C.; Waara, E.R.; et al. The murine version of BAN2401 (mAb158) selectively reduces amyloid-β protofibrils in brain and cerebrospinal fluid of tg-ArcSwe mice. J. Alzheimers Dis. 2015, 43, 575–588. [Google Scholar] [CrossRef] [PubMed]
- Adolfsson, O.; Pihlgren, M.; Toni, N.; Varisco, Y.; Buccarello, A.L.; Antoniello, K.; Lohmann, S.; Piorkowska, K.; Gafner, V.; Atwal, J.K.; et al. An effector-reduced anti-β-amyloid (Aβ) antibody with unique aβ binding properties promotes neuroprotection and glial engulfment of Aβ. J. Neurosci. 2012, 32, 9677–9689. [Google Scholar] [CrossRef] [PubMed]
- Bohrmann, B.; Baumann, K.; Benz, J.; Gerber, F.; Huber, W.; Knoflach, F.; Messer, J.; Oroszlan, K.; Rauchenberger, R.; Richter, W.F.; et al. Gantenerumab: A novel human anti-Aβ antibody demonstrates sustained cerebral amyloid-β binding and elicits cell-mediated removal of human amyloid-β. J. Alzheimers Dis. 2012, 28, 49–69. [Google Scholar] [CrossRef] [PubMed]
- Lannfelt, L.; Relkin, N.R.; Siemers, E.R. Amyloid-β-directed immunotherapy for Alzheimer's disease. J. Intern. Med. 2014, 275, 284–295. [Google Scholar] [CrossRef]
- Wang, Q.; Yu, X.; Patal, K.; Hu, R.; Chuang, S.; Zhang, G.; Zheng, J. Tanshinones inhibit amyloid aggregation by amyloid-β peptide, disaggregate amyloid fibrils, and protect cultured cells. ACS Chem. Neurosci. 2013, 4, 1004–1015. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhu, M.; Manning-Bog, A.B.; Di Monte, D.A.; Fink, A.L. Dopamine and L-dopa disaggregate amyloid fibrils: Implications for Parkinson's and Alzheimer's disease. FASEB J. 2004, 18, 962–964. [Google Scholar] [CrossRef] [PubMed]
- Saunders, J.C.; Young, L.M.; Mahood, R.A.; Jackson, M.P.; Revill, C.H.; Foster, R.J.; Smith, D.A.; Ashcroft, A.E.; Brockwell, D.J.; Radford, S.E. An in vivo platform for identifying inhibitors of protein aggregation. Nat. Chem. Biol. 2016, 12, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Cavaliere, P.; Torrent, J.; Prigent, S.; Granata, V.; Pauwels, K.; Pastore, A.; Rezaei, H.; Zagari, A. Binding of methylene blue to a surface cleft inhibits the oligomerization and fibrillization of prion protein. Biochim. Biophys. Acta 2013, 1832, 20–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Necula, M.; Breydo, L.; Milton, S.; Kayed, R.; van der Veer, W.E.; Tone, P.; Glabe, C.G. Methylene blue inhibits amyloid Aβ oligomerization by promoting fibrillization. Biochemistry 2007, 46, 8850–8860. [Google Scholar] [CrossRef] [PubMed]
- Heller, G.T.; Aprile, F.A.; Vendruscolo, M. Methods of probing the interactions between small molecules and disordered proteins. Cell. Mol. Life Sci. 2017, 74, 3225–3243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Younan, N.D.; Viles, J.H. A comparison of three fluorophores for the detection of amyloid fibers and prefibrillar oligomeric assemblies. ThT (thioflavin T); ANS (1-anilinonaphthalene-8-sulfonic acid); and bisANS (4, 4′-dianilino-1, 1′-binaphthyl-5, 5′-disulfonic acid). Biochemistry 2015, 54, 4297–4306. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Vastl, J.; Gao, J. Highly sensitive amyloid detection enabled by thioflavin T dimers. Mol. Biosyst. 2010, 6, 1791–1795. [Google Scholar] [CrossRef] [PubMed]
- Nesterov, E.E.; Skoch, J.; Hyman, B.T.; Klunk, W.E.; Bacskai, B.J.; Swager, T.M. In vivo optical imaging of amyloid aggregates in brain: Design of fluorescent markers. Angew. Chem.-Int. Ed. 2005, 44, 5452–5456. [Google Scholar] [CrossRef] [PubMed]
- Tagliavini, F.; Forloni, G.; Colombo, L.; Rossi, G.; Girola, L.; Canciani, B.; Angeretti, N.; Giampaolo, L.; Peressini, E.; Awan, T.; et al. Tetracycline affects abnormal properties of synthetic PrP peptides and PrPSc in vitro. J. Mol. Biol. 2000, 300, 1309–1322. [Google Scholar] [CrossRef] [PubMed]
- Cosentino, U.; Varí, M.R.; Saracino, A.G.; Pitea, D.; Moro, G.; Salmona, M. Tetracycline and its analogues as inhibitors of amyloid fibrils: searching for a geometrical pharmacophore by theoretical investigation of their conformational behavior in aqueous solution. J. Mol. Model. 2005, 11, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, D.S.; Sawaya, M.R. Structural studies of amyloid proteins at the molecular level. Annu. Rev. Biochem. 2017, 86, 69–95. [Google Scholar] [CrossRef] [PubMed]
- Bonanomi, M.; Natalello, A.; Visentin, C.; Pastori, V.; Penco, A.; Cornelli, G.; Colombo, G.; Malabarba, M.G.; Doglia, S.M.; Relini, A.; et al. Epigallocatechin-3-gallate and tetracycline differently affect ataxin-3 fibrillogenesis and reduce toxicity in spinocerebellar ataxia type 3 model. Hum. Mol. Genet. 2014, 23, 6542–6552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonanomi, M.; Visentin, C.; Natalello, A.; Spinelli, M.; Vanoni, M.; Airoldi, C.; Regonesi, M.E.; Tortora, P. How epigallocatechin-3-gallate and tetracycline interact with the josephin domain of ataxin-3 and alter its aggregation mode. Chem.-Eur. J. 2015, 21, 18383–18393. [Google Scholar] [CrossRef] [PubMed]
- Rao, M.N.; Shinnar, A.E.; Noecker, L.A.; Chao, T.L.; Feibush, B.; Snyder, B.; Sharkansky, I.; Sarkahian, A.; Zhang, X.; Jones, S.R.; et al. Aminosterols from the dogfish shark Squalus acanthias. J. Nat. Prod. 2000, 63, 631–635. [Google Scholar] [CrossRef] [PubMed]
- Sills, A.K.; Williams, J.I.; Tyler, B.M.; Epstein, D.S.; Sipos, E.P.; Davis, J.D.; McLane, M.P.; Pitchford, S.; Cheshire, K.; Gannon, F.H.; et al. Squalamine inhibits angiogenesis and solid tumor growth in vivo and perturbs embryonic vasculature. Cancer Res. 1998, 58, 2784–2792. [Google Scholar] [PubMed]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.-Y.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. α-Synuclein in lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Young, L.M.; Ashcroft, A.E.; Radford, S.E. Small molecule probes of protein aggregation. Curr. Opin. Chem. Biol. 2017, 39, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Viet, M.H.; Ngo, S.T.; Lam, N.S.; Li, M.S. Inhibition of aggregation of amyloid peptides by beta-sheet breaker peptides and their binding affinity. J. Phys. Chem. B 2011, 115, 7433–7446. [Google Scholar] [CrossRef] [PubMed]
- Soto, C.; Sigurdsson, E.M.; Morelli, L.; Kumar, R.A.; Castaño, E.M.; Frangione, B. β-sheet breaker peptides inhibit fibrillogenesis in a rat brain model of amyloidosis: Implications for Alzheimer's therapy. Nat. Med. 1998, 4, 822–826. [Google Scholar] [CrossRef] [PubMed]
- Poduslo, J.F.; Curran, G.L.; Kumar, A.; Frangione, B.; Soto, C. β-Sheet breaker peptide inhibitor of Alzheimer's amyloidogenesis with increased blood–brain barrier permeability and resistance to proteolytic degradation in plasma. J. Neurobiol. 1999, 39, 371–382. [Google Scholar] [CrossRef]
- Adessi, C.; Soto, C. Converting a peptide into a drug: strategies to improve stability and bioavailability. Curr. Med. Chem. 2002, 9, 963–978. [Google Scholar] [CrossRef] [PubMed]
- Jha, A.; Kumar, M.G.; Gopi, H.N.; Paknikar, K.M. Inhibition of β-amyloid aggregation through a designed β-hairpin peptide. Langmuir 2018, 34, 1591–1600. [Google Scholar] [CrossRef] [PubMed]
- Hoyer, W.; Grönwall, C.; Jonsson, A.; Ståhl, S.; Härd, T. Stabilization of a β-hairpin in monomeric Alzheimer's amyloid-β peptide inhibits amyloid formation. Proc. Natl. Acad. Sci. USA 2008, 105, 5099–5104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirecka, E.A.; Shaykhalishahi, H.; Gauhar, A.; Akgül, Ş.; Lecher, J.; Willbold, D.; Stoldt, M.; Hoyer, W. Sequestration of a β-hairpin for control of α-synuclein aggregation. Angew. Chem.-Int. Ed. 2014, 53, 4227–4230. [Google Scholar] [CrossRef] [PubMed]
- Shaykhalishahi, H.; Mirecka, E.A.; Gauhar, A.; Grüning, C.S.; Willbold, D.; Härd, T.; Stoldt, M.; Hoyer, W. A β-Hairpin-Binding protein for three different disease-related amyloidogenic proteins. ChemBioChem 2015, 16, 411–414. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Carranza, D.L.; Sengupta, U.; Guerrero-Muñoz, M.J.; Lasagna-Reeves, C.A.; Gerson, J.E.; Singh, G.; Estes, D.M.; Barrett, A.D.; Dineley, K.T.; Jackson, G.R.; et al. Passive immunization with Tau oligomer monoclonal antibody reverses tauopathy phenotypes without affecting hyperphosphorylated neurofibrillary tangles. J. Neurosci. 2014, 34, 4260–4272. [Google Scholar] [CrossRef] [PubMed]
- Guilliams, T.; El-Turk, F.; Buell, A.K.; O'Day, E.M.; Aprile, F.A.; Esbjörner, E.K.; Vendruscolo, M.; Cremades, N.; Pardon, E.; Wyns, L. Nanobodies raised against monomeric α-synuclein distinguish between fibrils at different maturation stages. J. Mol. Biol. 2013, 425, 2397–2411. [Google Scholar] [CrossRef] [PubMed]
- Arosio, P.; Michaels, T.C.; Linse, S.; Månsson, C.; Emanuelsson, C.; Presto, J.; Johansson, J.; Vendruscolo, M.; Dobson, C.M.; Knowles, T.P. Kinetic analysis reveals the diversity of microscopic mechanisms through which molecular chaperones suppress amyloid formation. Nat. Commun. 2016, 7, 10948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aprile, F.A.; Arosio, P.; Fusco, G.; Chen, S.W.; Kumita, J.R.; Dhulesia, A.; Tortora, P.; Knowles, T.P.; Vendruscolo, M.; Dobson, C.M. Inhibition of α-synuclein fibril elongation by Hsp70 is governed by a kinetic binding competition between α-synuclein species. Biochemistry 2017, 56, 1177–1180. [Google Scholar] [CrossRef] [PubMed]
- Aprile, F.A.; Sormanni, P.; Vendruscolo, M. A rational design strategy for the selective activity enhancement of a molecular chaperone toward a target substrate. Biochemistry 2015, 54, 5103–5112. [Google Scholar] [CrossRef] [PubMed]
- Bongiovanni, M.N.; Aprile, F.A.; Sormanni, P.; Vendruscolo, M. A rationally designed Hsp70 variant rescues the aggregation-associated toxicity of human IAPP in cultured pancreatic islet β-cells. Int. J. Mol. Sci. 2018, 19, 1443. [Google Scholar] [CrossRef] [PubMed]
- Bu, X.-L.; Rao, P.P.; Wang, Y.-J. Anti-amyloid aggregation activity of natural compounds: Implications for Alzheimer’s drug discovery. Mol. Neurobiol. 2016, 53, 3565–3575. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Pukala, T.L.; Musgrave, I.F.; Williams, D.M.; Dehle, F.C.; Carver, J.A. Gallic acid is the major component of grape seed extract that inhibits amyloid fibril formation. Bioorg. Med. Chem. Lett. 2013, 23, 6336–6340. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.E.; Rhoo, K.Y.; Lee, S.; Lee, J.T.; Park, J.H.; Bhak, G.; Paik, S.R. EGCG-mediated protection of the membrane disruption and cytotoxicity caused by the ‘active Oligomer’of α-synuclein. Sci. Rep. 2017, 7, 17945. [Google Scholar] [CrossRef] [PubMed]
- Young, L.M.; Cao, P.; Raleigh, D.P.; Ashcroft, A.E.; Radford, S.E. Ion mobility spectrometry–mass spectrometry defines the oligomeric intermediates in amylin amyloid formation and the mode of action of inhibitors. J. Am. Chem. Soc. 2014, 136, 660–670. [Google Scholar] [CrossRef] [PubMed]
- Andrich, K.; Hegenbart, U.; Kimmich, C.; Kedia, N.; Bergen, H.R.; Schönland, S.; Wanker, E.E.; Bieschke, J. Aggregation of full-length immunoglobulin light chains from systemic light chain amyloidosis (AL) patients is remodeled by epigallocatechin-3-gallate. J. Biol. Chem. 2017, 292, 2328–2344. [Google Scholar] [CrossRef] [PubMed]
- Ehrnhoefer, D.E.; Bieschke, J.; Boeddrich, A.; Herbst, M.; Masino, L.; Lurz, R.; Engemann, S.; Pastore, A.; Wanker, E.E. EGCG redirects amyloidogenic polypeptides into unstructured, off-pathway oligomers. Nat. Struct. Mol. Biol. 2008, 15, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Visentin, C.; Pellistri, F.; Natalello, A.; Vertemara, J.; Bonanomi, M.; Gatta, E.; Penco, A.; Relini, A.; De Gioia, L.; Airoldi, C.; et al. Epigallocatechin-3-gallate and related phenol compounds redirect the amyloidogenic aggregation pathway of ataxin-3 towards non-toxic aggregates and prevent toxicity in neural cells and Caenorhabditis elegans animal model. Hum. Mol. Genet. 2017, 26, 3271–3284. [Google Scholar] [CrossRef] [PubMed]
- Mo, Y.; Lei, J.; Sun, Y.; Zhang, Q.; Wei, G. Conformational ensemble of hIAPP dimer: insight into the molecular mechanism by which a green tea extract inhibits hIAPP aggregation. Sci. Rep. 2016, 6, 33076. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-H.; Dong, X.-Y.; Sun, Y. Thermodynamic analysis of the molecular interactions between amyloid β-protein fragments and (−)-epigallocatechin-3-gallate. J. Phys. Chem. B 2012, 116, 5803–5809. [Google Scholar] [CrossRef] [PubMed]
- Palhano, F.L.; Lee, J.; Grimster, N.P.; Kelly, J.W. Toward the molecular mechanism (s) by which EGCG treatment remodels mature amyloid fibrils. J. Am Chem. Soc. 2013, 135, 7503–7510. [Google Scholar] [CrossRef] [PubMed]
- Severino, J.F.; Goodman, B.A.; Kay, C.W.; Stolze, K.; Tunega, D.; Reichenauer, T.G.; Pirker, K.F. Free radicals generated during oxidation of green tea polyphenols: electron paramagnetic resonance spectroscopy combined with density functional theory calculations. Free Radic. Biol. Med. 2009, 46, 1076–1088. [Google Scholar] [CrossRef] [PubMed]
- An, T.-T.; Feng, S.; Zeng, C.-M. Oxidized epigallocatechin gallate inhibited lysozyme fibrillation more strongly than the native form. Redox Biol. 2017, 11, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Carver, J.A.; Calabrese, A.N.; Pukala, T.L. Gallic acid interacts with α-synuclein to prevent the structural collapse necessary for its aggregation. Biochim. Biophys. Acta 2014, 1844, 1481–1485. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Liu, Z.; Huo, X.; Wang, C.; Meng, Q.; Liu, Q.; Sun, H.; Sun, P.; Yang, X.; Shu, X.; et al. Enhancement effect of resveratrol on the intestinal absorption of bestatin by regulating PEPT1, MDR1 and MRP2 in vivo and in vitro. Int. J. Pharm. 2015, 495, 588–598. [Google Scholar] [CrossRef] [PubMed]
- Pallàs, M.; Porquet, D.; Vicente, A.; Sanfeliu, C. Resveratrol: New avenues for a natural compound in neuroprotection. Curr. Pharm. Des. 2013, 19, 6726–6731. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Miranda, V.; Soto-Montenegro, M.; Vera, G.; Herradon, E.; Desco, M.; Abalo, R. Resveratrol: A neuroprotective polyphenol in the mediterranean diet. Rev. Neurol. 2012, 54, 349–356. [Google Scholar] [PubMed]
- Feng, Y.; Wang, X.-P.; Yang, S.-G.; Wang, Y.-J.; Zhang, X.; Du, X.-T.; Sun, X.-X.; Zhao, M.; Huang, L.; Liu, R.-T. Resveratrol inhibits beta-amyloid oligomeric cytotoxicity but does not prevent oligomer formation. Neurotoxicology 2009, 30, 986–995. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Aucoin, D.; Ahmed, M.; Ziliox, M.; Van Nostrand, W.E.; Smith, S.O. Capping of Aβ42 oligomers by small molecule inhibitors. Biochemistry 2014, 53, 7893–7903. [Google Scholar] [CrossRef] [PubMed]
- Evers, F.; Jeworrek, C.; Tiemeyer, S.; Weise, K.; Sellin, D.; Paulus, M.; Struth, B.; Tolan, M.; Winter, R. Elucidating the mechanism of lipid membrane-induced IAPP fibrillogenesis and its inhibition by the red wine compound resveratrol: A synchrotron X-ray reflectivity study. J. Am. Chem. Soc. 2009, 131, 9516–9521. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Jiang, P.; Xu, W.; Li, H.; Zhang, H.; Yan, L.; Chan-Park, M.B.; Liu, X.-W.; Tang, K.; Mu, Y.; et al. The molecular basis of distinct aggregation pathways of islet amyloid polypeptide. J. Biol. Chem. 2011, 286, 6291–6300. [Google Scholar] [CrossRef] [PubMed]
- Monroy, A.; Lithgow, G.J.; Alavez, S. Curcumin and neurodegenerative diseases. Biofactors 2013, 39, 122–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatcher, H.; Planalp, R.; Cho, J.; Torti, F.; Torti, S. Curcumin: from ancient medicine to current clinical trials. Cell. Mol. Life Sci. 2008, 65, 1631–1652. [Google Scholar] [CrossRef] [PubMed]
- Endo, H.; Nikaido, Y.; Nakadate, M.; Ise, S.; Konno, H. Structure activity relationship study of curcumin analogues toward the amyloid-beta aggregation inhibitor. Bioorg. Med. Chem. Lett. 2014, 24, 5621–5626. [Google Scholar] [CrossRef] [PubMed]
- Thapa, A.; Jett, S.D.; Chi, E.Y. Curcumin attenuates amyloid-β aggregate toxicity and modulates amyloid-β aggregation pathway. ACS Chem. Neurosci. 2016, 7, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Liang, F.; Wan, Y.; Schaak, D.; Ward, J.; Shen, X.; Tanzi, R.E.; Zhang, C.; Quan, Q. Nanoplasmonic fiber tip probe detects significant reduction of intracellular Alzheimer’s disease-related oligomers by curcumin. Sci. Rep. 2017, 7, 5722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahsan, N.; Mishra, S.; Jain, M.K.; Surolia, A.; Gupta, S. Curcumin Pyrazole and its derivative (N-(3-Nitrophenylpyrazole) curcumin inhibit aggregation, disrupt fibrils and modulate toxicity of wild type and mutant α-synuclein. Sci. Rep. 2015, 5, 9862. [Google Scholar] [CrossRef] [PubMed]
- Okuda, M.; Fujita, Y.; Hijikuro, I.; Wada, M.; Uemura, T.; Kobayashi, Y.; Waku, T.; Tanaka, N.; Nishimoto, T.; Izumi, Y.; et al. PE859, A novel curcumin derivative, inhibits amyloid-β and Tau aggregation, and ameliorates cognitive dysfunction in senescence-accelerated mouse prone 8. J. Alzheimers Dis. 2017, 59, 313–328. [Google Scholar] [CrossRef] [PubMed]
- Rane, J.S.; Bhaumik, P.; Panda, D. Curcumin inhibits tau aggregation and disintegrates preformed tau filaments in vitro. J. Alzheimer Dis. 2017, 60, 999–1014. [Google Scholar] [CrossRef] [PubMed]
- Hafner-Bratkovič, I.; Gašperšič, J.; Šmid, L.M.; Bresjanac, M.; Jerala, R. Curcumin binds to the α-helical intermediate and to the amyloid form of prion protein—A new mechanism for the inhibition of PrPSc accumulation. J. Neurochem. 2008, 104, 1553–1564. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Lim, G.P.; Begum, A.N.; Ubeda, O.J.; Simmons, M.R.; Ambegaokar, S.S.; Chen, P.P.; Kayed, R.; Glabe, C.G.; Frautschy, S.A. Curcumin inhibits formation of amyloid β oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J. Biol. Chem. 2005, 280, 5892–5901. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, D.; Taguchi, H.; Morikawa, S.; Kato, T.; Hirao, K.; Shirai, N.; Tooyama, I. Novel curcumin derivatives as potent inhibitors of amyloid β aggregation. Biochem. Biophys. Rep. 2015, 4, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Orteca, G.; Tavanti, F.; Bednarikova, Z.; Gazova, Z.; Rigillo, G.; Imbriano, C.; Basile, V.; Asti, M.; Rigamonti, L.; Saladini, M.; et al. Curcumin derivatives and Aβ-fibrillar aggregates: An interactions’ study for diagnostic/therapeutic purposes in neurodegenerative diseases. Bioorg. Med. Chem. 2018, 26, 4288–4300. [Google Scholar] [CrossRef] [PubMed]
- Sherif, I.O.; Al-Gayyar, M.M. Oleuropein potentiates anti-tumor activity of cisplatin against HepG2 through affecting proNGF/NGF balance. Life Sci. 2018, 198, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Qabaha, K.; Al-Rimawi, F.; Qasem, A.; Naser, S.A. Oleuropein is responsible for the major anti-inflammatory effects of olive leaf extract. J. Med. Food. 2018, 21, 302–305. [Google Scholar] [CrossRef] [PubMed]
- Umeno, A.; Takashima, M.; Murotomi, K.; Nakajima, Y.; Koike, T.; Matsuo, T.; Yoshida, Y. Radical-scavenging activity and antioxidative effects of olive leaf components oleuropein and hydroxytyrosol in comparison with homovanillic alcohol. J. Oleo Sci. 2015, 64, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Rigacci, S.; Guidotti, V.; Bucciantini, M.; Nichino, D.; Relini, A.; Berti, A.; Stefani, M. Aβ (1–42) aggregates into non-toxic amyloid assemblies in the presence of the natural polyphenol oleuropein aglycon. Curr. Alzheimer Res. 2011, 8, 841–852. [Google Scholar] [CrossRef] [PubMed]
- Palazzi, L.; Bruzzone, E.; Bisello, G.; Leri, M.; Stefani, M.; Bucciantini, M.; Polverino de Laureto, P. Oleuropein aglycone stabilizes the monomeric α-synuclein and favours the growth of non-toxic aggregates. Sci. Rep. 2018, 8, 8337. [Google Scholar] [CrossRef] [PubMed]
- Leri, M.; Oropesa-Nuñez, R.; Canale, C.; Raimondi, S.; Giorgetti, S.; Bruzzone, E.; Bellotti, V.; Stefani, M.; Bucciantini, M. Oleuropein aglycone: A polyphenol with different targets against amyloid toxicity. Biochim. Biophys. Acta 2018, 1862, 1432–1442. [Google Scholar] [CrossRef] [PubMed]
- Leri, M.; Nosi, D.; Natalello, A.; Porcari, R.; Ramazzotti, M.; Chiti, F.; Bellotti, V.; Doglia, S.M.; Stefani, M.; Bucciantini, M. The polyphenol oleuropein aglycone hinders the growth of toxic transthyretin amyloid assemblies. J. Nutr. Biochem. 2016, 30, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Rigacci, S.; Guidotti, V.; Bucciantini, M.; Parri, M.; Nediani, C.; Cerbai, E.; Stefani, M.; Berti, A. Oleuropein aglycon prevents cytotoxic amyloid aggregation of human amylin. J. Nutr. Biochem. 2010, 21, 726–735. [Google Scholar] [CrossRef] [PubMed]
- Daccache, A.; Lion, C.; Sibille, N.; Gerard, M.; Slomianny, C.; Lippens, G.; Cotelle, P. Oleuropein and derivatives from olives as Tau aggregation inhibitors. Neurochem. Int. 2011, 58, 700–707. [Google Scholar] [CrossRef] [PubMed]
- Colon, W.; Kelly, J.W. Partial denaturation of transthyretin is sufficient for amyloid fibril formation in vitro. Biochemistry 1992, 31, 8654–8660. [Google Scholar] [CrossRef] [PubMed]
- Mangione, P.P.; Porcari, R.; Gillmore, J.D.; Pucci, P.; Monti, M.; Porcari, M.; Giorgetti, S.; Marchese, L.; Raimondi, S.; Serpell, L.C.; et al. Proteolytic cleavage of Ser52Pro variant transthyretin triggers its amyloid fibrillogenesis. Proc. Natl. Acad. Sci. USA 2014, 111, 1539–1544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangione, P.P.; Verona, G.; Corazza, A.; Marcoux, J.; Canetti, D.; Giorgetti, S.; Raimondi, S.; Stoppini, M.; Esposito, M.; Relini, A.; et al. Plasminogen activation triggers transthyretin amyloidogenesis in vitro. J. Biol. Chem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, S.; Chang, C.-W.; Chou, H.-H. Gold nanoparticles as amyloid-like fibrillogenesis inhibitors. Colloids Surf. B Biointerfaces 2013, 112, 525–529. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.H.; Chang, Y.J.; Yoshiike, Y.; Chang, Y.C.; Chen, Y.R. Negatively charged gold nanoparticles inhibit Alzheimer's amyloid-β fibrillization, induce fibril dissociation, and mitigate neurotoxicity. Small 2012, 8, 3631–3639. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Sun, Y.; Luo, Y.; Zhu, Y.; Liu, Y.; Li, H. Exploring the mechanism of inhibition of Au nanoparticles on the aggregation of amyloid-β (16-22) peptides at the atom level by all-atom molecular dynamics. Int. J. Mol. Sci. 2018, 19, 1815. [Google Scholar] [CrossRef] [PubMed]
- Debnath, K.; Pradhan, N.; Singh, B.K.; Jana, N.R. Poly(trehalose) nanoparticles prevent amyloid aggregation and suppress polyglutamine aggregation in a Huntington’s Disease model mouse. ACS Appl. Mater. Interfaces 2017, 9, 24126–24139. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Kakinen, A.; Pilkington, E.H.; Davis, T.P.; Ke, P.C. Differential effects of silver and iron oxide nanoparticles on IAPP amyloid aggregation. Biomater. Sci. 2017, 5, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Bokvist, M.; Lindström, F.; Watts, A.; Gröbner, G. Two types of Alzheimer's β-amyloid (1–40) peptide membrane interactions: Aggregation preventing transmembrane anchoring versus accelerated surface fibril formation. J. Mol. Biol. 2004, 335, 1039–1049. [Google Scholar] [CrossRef] [PubMed]
- Galvagnion, C.; Brown, J.W.; Ouberai, M.M.; Flagmeier, P.; Vendruscolo, M.; Buell, A.K.; Sparr, E.; Dobson, C.M. Chemical properties of lipids strongly affect the kinetics of the membrane-induced aggregation of α-synuclein. Proc. Natl. Acad. Sci. USA 2016, 113, 7065–7070. [Google Scholar] [CrossRef] [PubMed]
- Al Kayal, T.; Russo, E.; Pieri, L.; Caminati, G.; Berti, D.; Bucciantini, M.; Stefani, M.; Baglioni, P. Interactions of lysozyme with phospholipid vesicles: effects of vesicle biophysical features on protein misfolding and aggregation. Soft Matter 2012, 8, 9115–9126. [Google Scholar] [CrossRef]
- Matsuzaki, K.; Kato, K.; Yanagisawa, K. Aβ polymerization through interaction with membrane gangliosides. Biochim. Biophys. Acta 2010, 1801, 868–877. [Google Scholar] [CrossRef] [PubMed]
- Bucciantini, M.; Nosi, D.; Forzan, M.; Russo, E.; Calamai, M.; Pieri, L.; Formigli, L.; Quercioli, F.; Soria, S.; Pavone, F. Toxic effects of amyloid fibrils on cell membranes: The importance of ganglioside GM1. FASEB J. 2012, 26, 818–831. [Google Scholar] [CrossRef] [PubMed]
- Cecchi, C.; Nichino, D.; Zampagni, M.; Bernacchioni, C.; Evangelisti, E.; Pensalfini, A.; Liguri, G.; Gliozzi, A.; Stefani, M.; Relini, A. A protective role for lipid raft cholesterol against amyloid-induced membrane damage in human neuroblastoma cells. Biochim. Biophys. Acta 2009, 1788, 2204–2216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seghezza, S.; Diaspro, A.; Canale, C.; Dante, S. Cholesterol drives Aβ (1–42) interaction with lipid rafts in model membranes. Langmuir 2014, 30, 13934–13941. [Google Scholar] [CrossRef] [PubMed]
- Habchi, J.; Chia, S.; Galvagnion, C.; Michaels, T.C.; Bellaiche, M.M.; Ruggeri, F.S.; Sanguanini, M.; Idini, I.; Kumita, J.R.; Sparr, E.; et al. Cholesterol catalyses Aβ42 aggregation through a heterogeneous nucleation pathway in the presence of lipid membranes. Nat. Chem. 2018, 10, 673–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenzen, N.; Nielsen, S.B.; Yoshimura, Y.; Vad, B.S.; Andersen, C.B.; Betzer, C.; Kaspersen, J.D.; Christiansen, G.; Pedersen, J.S.; Jensen, P.H.; et al. How epigallocatechin gallate can inhibit α-synuclein oligomer toxicity in vitro. J. Biol. Chem. 2014, 289, 21299–21310. [Google Scholar] [CrossRef] [PubMed]
- Scarff, C.A.; Almeida, B.; Fraga, J.; Macedo-Ribeiro, S.; Radford, S.E.; Ashcroft, A.E. Examination of ataxin-3 aggregation by structural mass spectrometry techniques: a rationale for expedited aggregation upon polyglutamine expansion. Mol. Cell. Proteom. 2015, 14, 1241–1253. [Google Scholar] [CrossRef] [PubMed]
- Lupton, C.J.; Steer, D.L.; Wintrode, P.L.; Bottomley, S.P.; Hughes, V.A.; Ellisdon, A.M. Enhanced molecular mobility of ordinarily structured regions drives polyglutamine disease. J. Biol. Chem. 2015, 290, 24190–24200. [Google Scholar] [CrossRef] [PubMed]
- Masino, L.; Nicastro, G.; Calder, L.; Vendruscolo, M.; Pastore, A. Functional interactions as a survival strategy against abnormal aggregation. FASEB J. 2011, 25, 45–54. [Google Scholar] [PubMed] [Green Version]
- Zhang, T.; Zhang, J.; Derreumaux, P.; Mu, Y. Molecular mechanism of the inhibition of EGCG on the Alzheimer Aβ1–42 dimer. J. Phys. Chem. B 2013, 117, 3993–4002. [Google Scholar] [CrossRef] [PubMed]
- Tarus, B.; Nguyen, P.H.; Berthoumieu, O.; Faller, P.; Doig, A.J.; Derreumaux, P. Molecular structure of the NQTrp inhibitor with the alzheimer Aβ1-28 monomer. Eur. J. Med. Chem. 2015, 91, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Doig, A.J.; Derreumaux, P. Inhibition of protein aggregation and amyloid formation by small molecules. Curr. Opin. Struct. Biol. 2015, 30, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Chebaro, Y.; Jiang, P.; Zang, T.; Mu, Y.; Nguyen, P.H.; Mousseau, N.; Derreumaux, P. Structures of Aβ17–42 trimers in isolation and with five small-molecule drugs using a hierarchical computational procedure. J. Phys. Chem. B 2012, 116, 8412–8422. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Murakami, K.; Uno, M.; Nakagawa, Y.; Katayama, S.; Akagi, K.-I.; Masuda, Y.; Takegoshi, K.; Irie, K. Site-specific inhibitory mechanism for amyloid-β42 aggregation by catechol-type flavonoids targeting the Lys residues. J. Biol. Chem. 2013, 288, 23212–23224. [Google Scholar] [CrossRef] [PubMed]
- Ronga, L.; Langella, E.; Palladino, P.; Marasco, D.; Tizzano, B.; Saviano, M.; Pedone, C.; Improta, R.; Ruvo, M. Does tetracycline bind helix 2 of prion? An integrated spectroscopical and computational study of the interaction between the antibiotic and α helix 2 human prion protein fragments. Proteins 2007, 66, 707–715. [Google Scholar] [CrossRef] [PubMed]
- Stoilova, T.; Colombo, L.; Forloni, G.; Tagliavini, F.; Salmona, M. A new face for old antibiotics: Tetracyclines in treatment of amyloidoses. J. Med. Chem. 2013, 56, 5987–6006. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Liu, C.; Leibly, D.; Landau, M.; Zhao, M.; Hughes, M.P.; Eisenberg, D.S. Structure-based discovery of fiber-binding compounds that reduce the cytotoxicity of amyloid beta. elife 2013, 2, e00857. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Liang, G.; Zhang, M.; Zhao, J.; Patel, K.; Yu, X.; Zhao, C.; Ding, B.; Zhang, G.; Zhou, F.; et al. De novo design of self-assembled hexapeptides as β-amyloid (Aβ) peptide inhibitors. ACS Chem. Neurosci. 2014, 5, 972–981. [Google Scholar] [CrossRef] [PubMed]
- Habchi, J.; Chia, S.; Limbocker, R.; Mannini, B.; Ahn, M.; Perni, M.; Hansson, O.; Arosio, P.; Kumita, J.R.; Challa, P.K.; et al. Systematic development of small molecules to inhibit specific microscopic steps of Aβ42 aggregation in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, E200–E208. [Google Scholar] [CrossRef] [PubMed]
- Habchi, J.; Arosio, P.; Perni, M.; Costa, A.R.; Yagi-Utsumi, M.; Joshi, P.; Chia, S.; Cohen, S.I.; Müller, M.B.; Linse, S.; et al. An anticancer drug suppresses the primary nucleation reaction that initiates the production of the toxic Aβ42 aggregates linked with Alzheimer’s disease. Sci. Adv. 2016, 2, e1501244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scherzer-Attali, R.; Pellarin, R.; Convertino, M.; Frydman-Marom, A.; Egoz-Matia, N.; Peled, S.; Levy-Sakin, M.; Shalev, D.E.; Caflisch, A.; Gazit, E. Complete phenotypic recovery of an Alzheimer's disease model by a quinone-tryptophan hybrid aggregation inhibitor. PLoS ONE 2010, 5, e11101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minicozzi, V.; Chiaraluce, R.; Consalvi, V.; Giordano, C.; Narcisi, C.; Punzi, P.; Rossi, G.C.; Morante, S. Computational and experimental studies on β-sheet breakers targeting Aβ1–40 fibrils. J. Biol. Chem. 2014, 289, 11242–11252. [Google Scholar] [CrossRef] [PubMed]
- Kokotidou, C.; Jonnalagadda, S.V.R.; Orr, A.A.; Seoane-Blanco, M.; Apostolidou, C.P.; van Raaij, M.J.; Kotzabasaki, M.; Chatzoudis, A.; Jakubowski, J.M.; Mossou, E.; et al. A novel amyloid designable scaffold and potential inhibitor inspired by GAIIG of amyloid beta and the HIV-1 V3 loop. FEBS Lett. 2018, 592, 1777–1788. [Google Scholar] [CrossRef] [PubMed]
- Frydman-Marom, A.; Convertino, M.; Pellarin, R.; Lampel, A.; Shaltiel-Karyo, R.; Segal, D.; Caflisch, A.; Shalev, D.E.; Gazit, E. Structural basis for inhibiting β-amyloid oligomerization by a non-coded β-breaker-substituted endomorphin analogue. ACS Chem. Biol. 2011, 6, 1265–1276. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
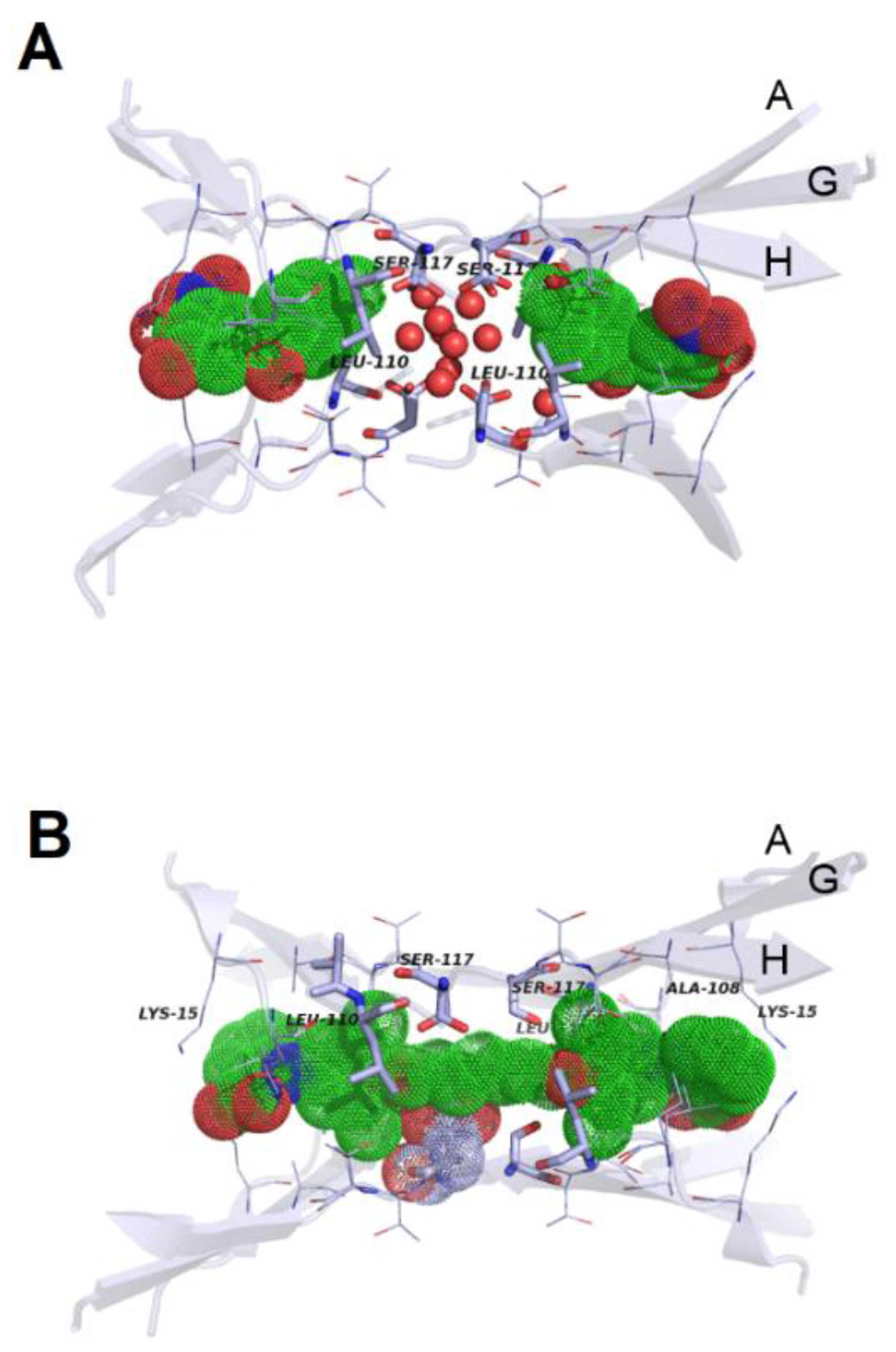{kind=link}
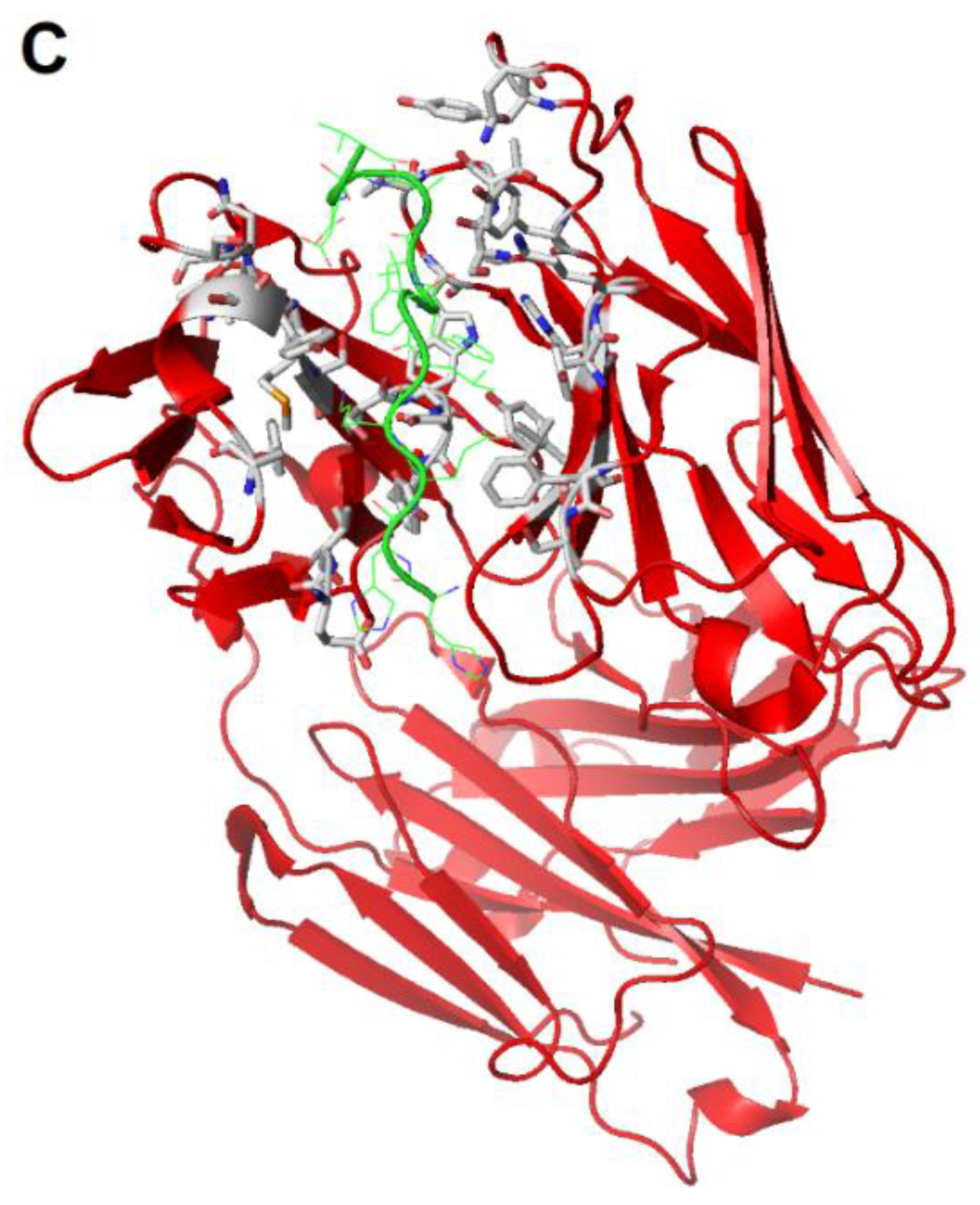{kind=link}
| Molecule | Class | Target Protein | Disease | Key References |
|---|---|---|---|---|
| Squalamine * | Sterol | α-syn | PD | [35] |
| Trodusquemine | Sterol | α-syn | PD | [36] |
| Tetracycline | Tetracyclines | Prp/Aβ | APrP/AD | [37] |
| Doxycycline | Tetracyclines | Aβ/PrP/β2-m/TTR/LC | AD/Aβ2-m/ATTR/AL | [37,38,39,40,41,42,43,44,45] |
| 4′Iodo-4′-doxorubicin | Anthracyclines | AL/SAA/TTR/Aβ/PrP | AD/AL/AA/ATTR/Aβ2-m | [46,47] |
| Acid fuchsin | Triarylmethane dye | IAPP | AD/diabetes | [48] |
| Fast Green FCF | Triarylmethane dye | IAPP | AD/diabetes | [49] |
| Crystal violet | Triarylmethine dye | tau | AD | [50] |
| N744 | Cyanine dye | tau | AD | [51] |
| Congo red | Azo dye | Aβ/casein/PrP/α-syn | AD/systemic amyloidosis/prion disease/PD | [52,53] |
| Resveratrol | Polyphenol | Aβ/IAPP | AD/diabetes | [54] |
| Curcumin | Polyphenol | Aβ/tau/α-syn /htt/PrP | AD/PD/CH | [54] |
| EGCG | Polyphenol | Aβ/α-syn/htt/TTR/IAPP/PAP248–286/HEWL/k-casein and calcitonin/polyQ proteins | AD/PD/CH/HIV infectivity | [54] |
| Quercetin and myricetin | Polyphenol | Aβ/α-syn/insulin/IAPP | AD/PD/diabetes | [54] |
| Olive oil phenols | Polyphenol | Aβ/IAPP | AD/diabetes | [54] |
| Oleuropein | Polyphenol | |||
| Baicalein (quinone) ** | Polyphenol | tau | AD | [55] |
| Tafamidis (Vyndaqel) | Benzoxazole | TTR | ATTR | [56] |
| Tolcapone | Benzophenone | TTR | ATTR | [57] |
| Mds84 | Palindromic ligand | TTR | ATTR | [57] |
| Oleocanthal ** | Aldehyde | tau | AD | [55] |
| Cinnamaldehyde ** | Aldehyde | tau | AD | [55] |
| Asperbenzaldehyde ** | Aldehyde | tau | AD | [55] |
| β-Breakers | Peptide | Aβ | AD | [58] |
| β-Breakers | Peptide | IAPP | AD/diabetes | [59] |
| β-Breakers | Peptide | IAPP | AD/diabetes | [60] |
| β-Breakers | Peptide | Aβ/IAPP | AD/diabetes | [61] |
| (Bi)Cyclic peptides | Peptide | Aβ | AD | [62] |
| Nanobodies | Single domain antibodies | α-syn/Aβ/lysozyme/β2-m | AD/PD/systemic | [63,64,65,66] |
| Rationally designed antibodies | Single domain antibodies | Aβ/α-syn/IAPP | AD/PD/diabetes | [67,68,69] |
| Aducanumab | Monoclonal antibody | Aβ | AD | [70] |
| mAb158 *** (BAN2401) | Monoclonal antibody | Aβ | AD | [71] |
| Crenezumab | Monoclonal antibody | Aβ | AD | [72] |
| Gantenerumab | Monoclonal antibody | Aβ | AD | [73] |
| Solanezumab **** | Monoclonal antibody | Aβ | AD | [74] |
| Tanshinones | Diterpene | Aβ | AD | [75] |
| Dopamine and l-dopa | Neurotransmitter | Aβ/α-syn/IAPP | AD/PD/diabetes | [76,77] |
| Methylene Blue | Thiazine dye | tau/PrP/ Aβ | AD | [55,78,79] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giorgetti, S.; Greco, C.; Tortora, P.; Aprile, F.A. Targeting Amyloid Aggregation: An Overview of Strategies and Mechanisms. Int. J. Mol. Sci. 2018, 19, 2677. https://doi.org/10.3390/ijms19092677
Giorgetti S, Greco C, Tortora P, Aprile FA. Targeting Amyloid Aggregation: An Overview of Strategies and Mechanisms. International Journal of Molecular Sciences. 2018; 19(9):2677. https://doi.org/10.3390/ijms19092677
Chicago/Turabian StyleGiorgetti, Sofia, Claudio Greco, Paolo Tortora, and Francesco Antonio Aprile. 2018. "Targeting Amyloid Aggregation: An Overview of Strategies and Mechanisms" International Journal of Molecular Sciences 19, no. 9: 2677. https://doi.org/10.3390/ijms19092677
APA StyleGiorgetti, S., Greco, C., Tortora, P., & Aprile, F. A. (2018). Targeting Amyloid Aggregation: An Overview of Strategies and Mechanisms. International Journal of Molecular Sciences, 19(9), 2677. https://doi.org/10.3390/ijms19092677