Protein Environment: A Crucial Triggering Factor in Josephin Domain Aggregation: The Role of 2,2,2-Trifluoroethanol

 , and
, and 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
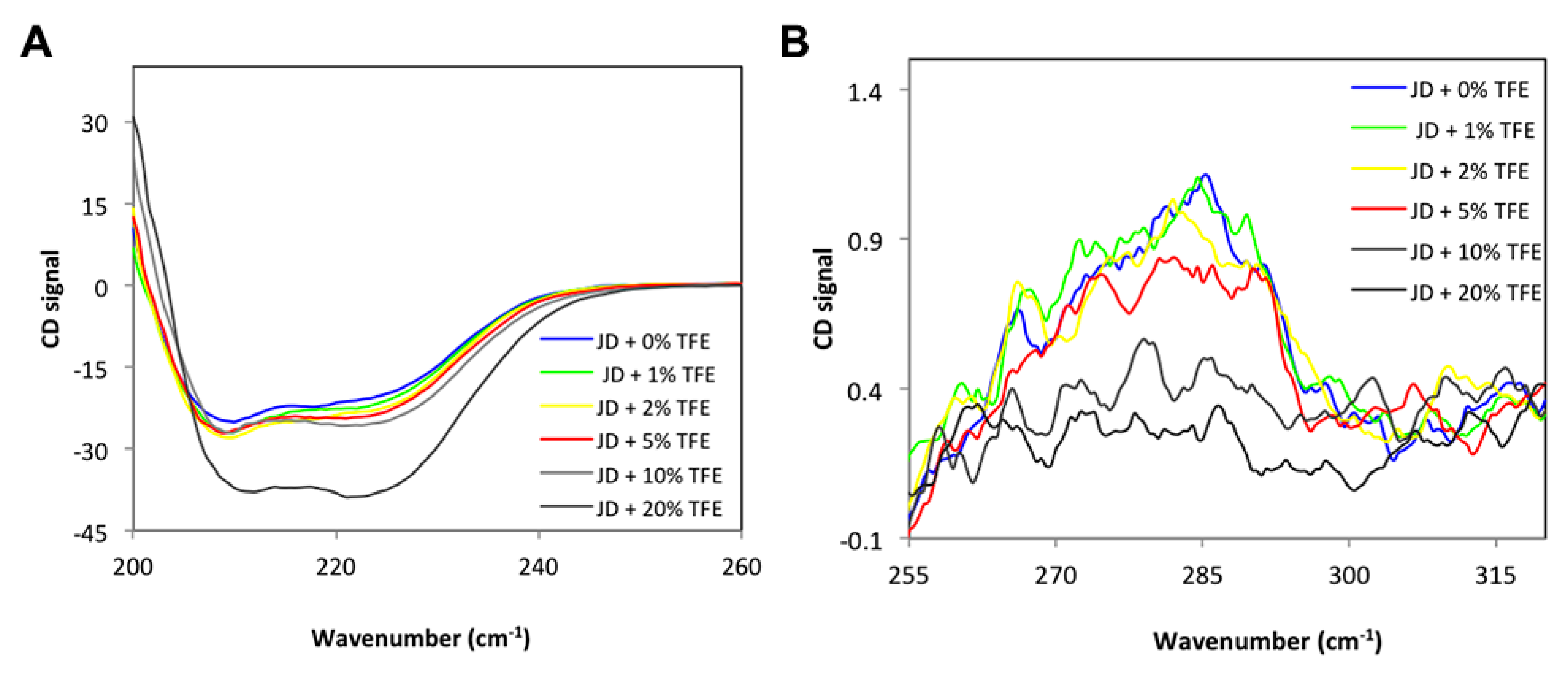
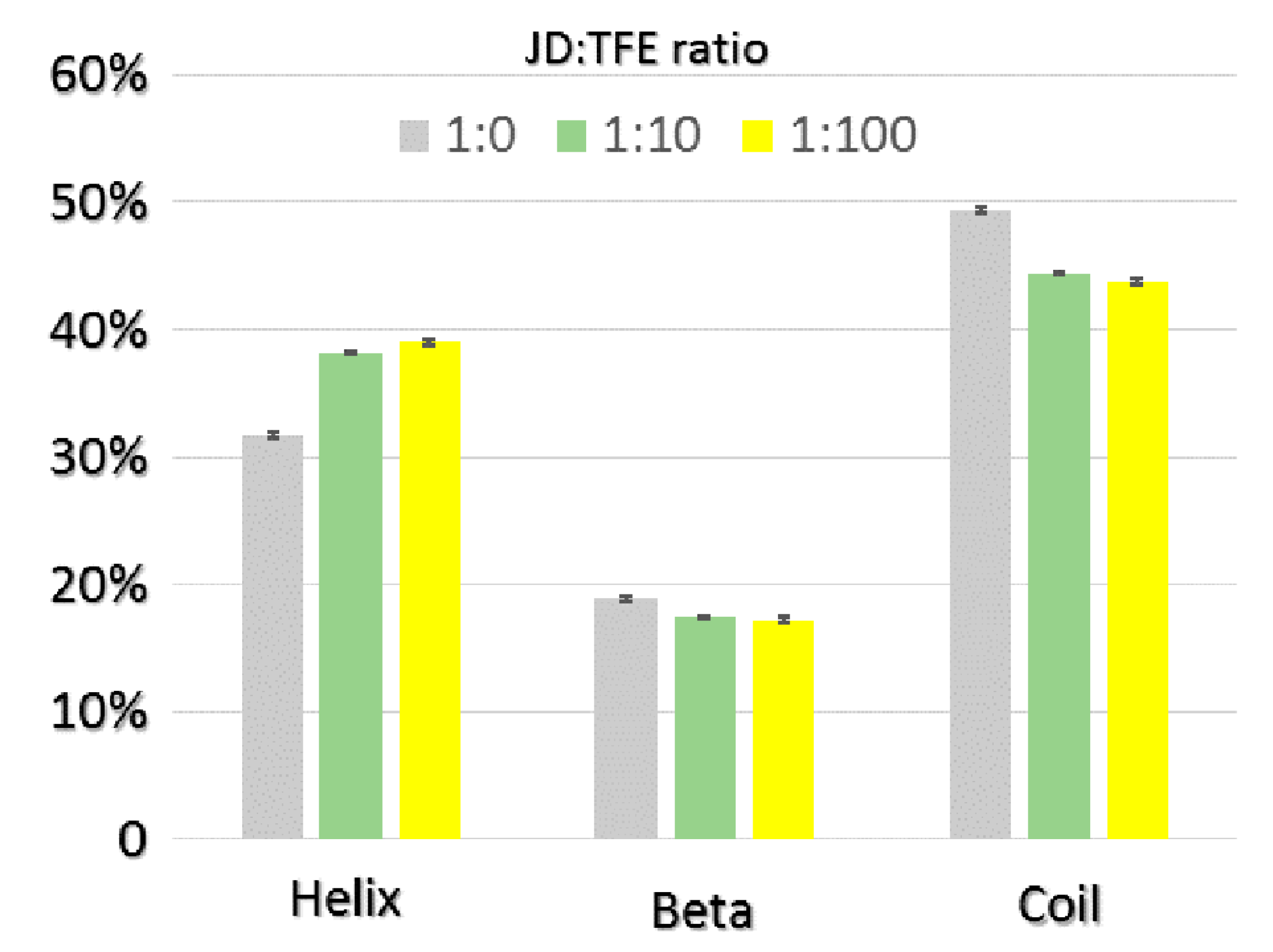
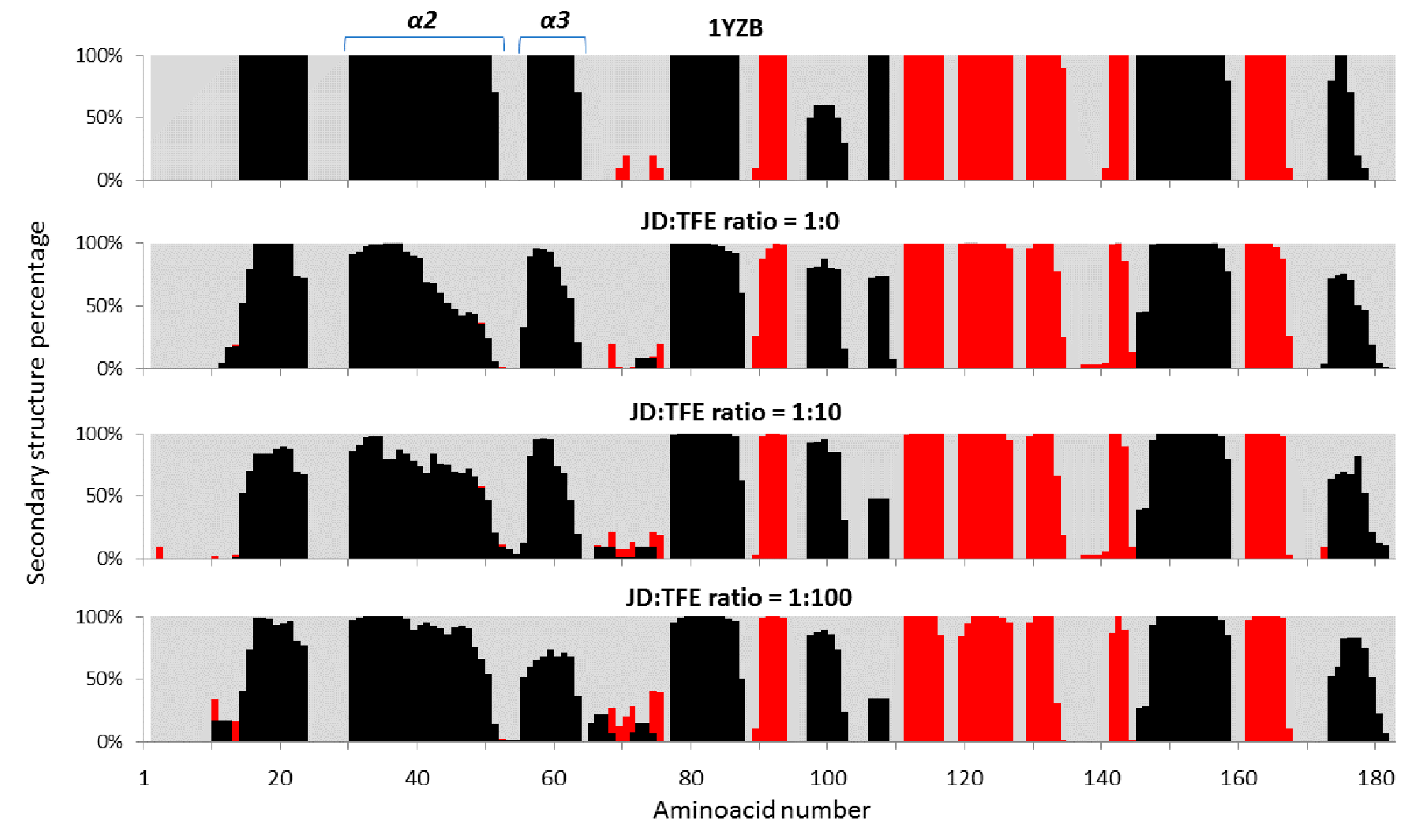
2.1. TFE Increases the α-Helix Content of the Josephin Domain
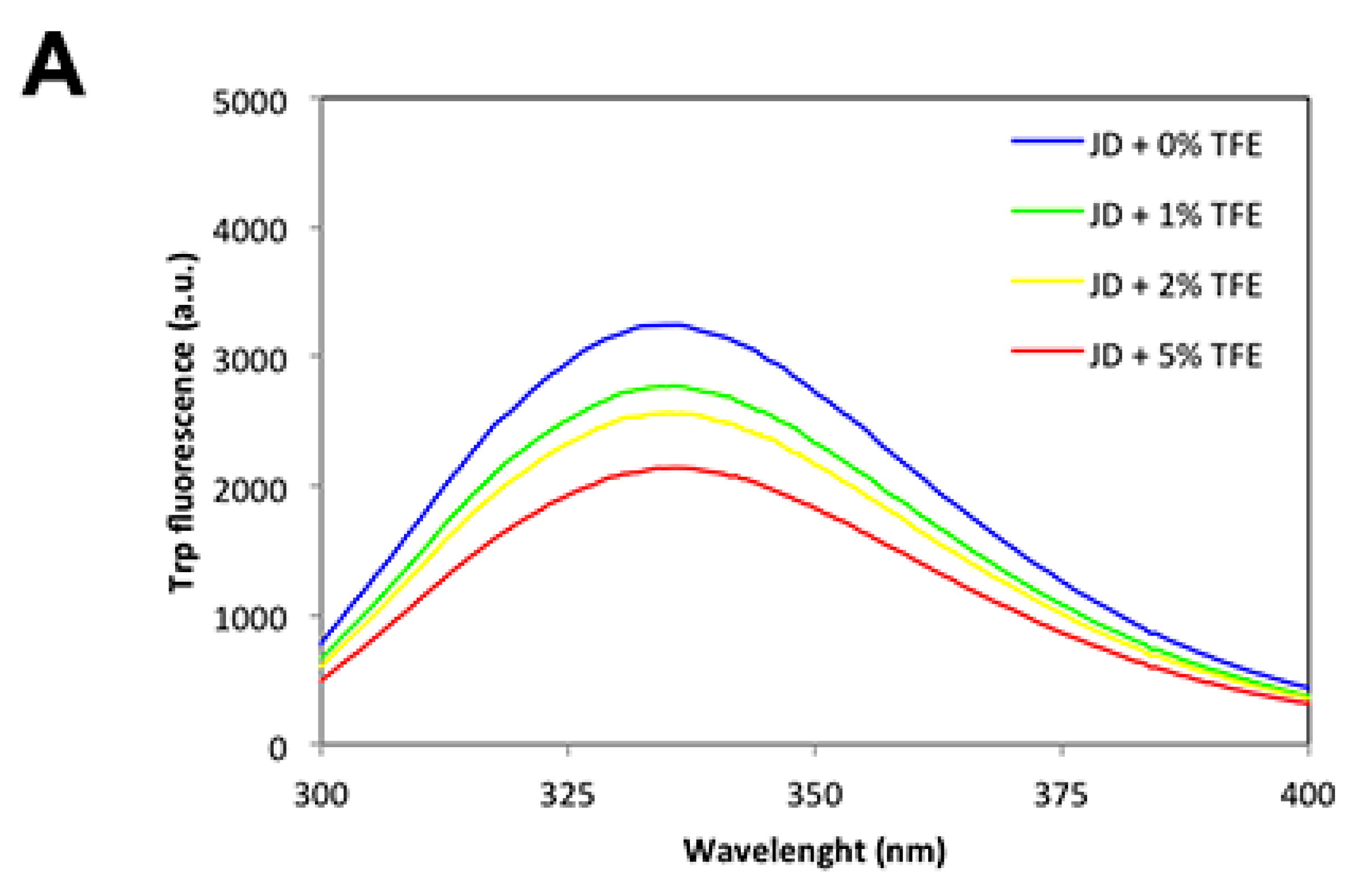
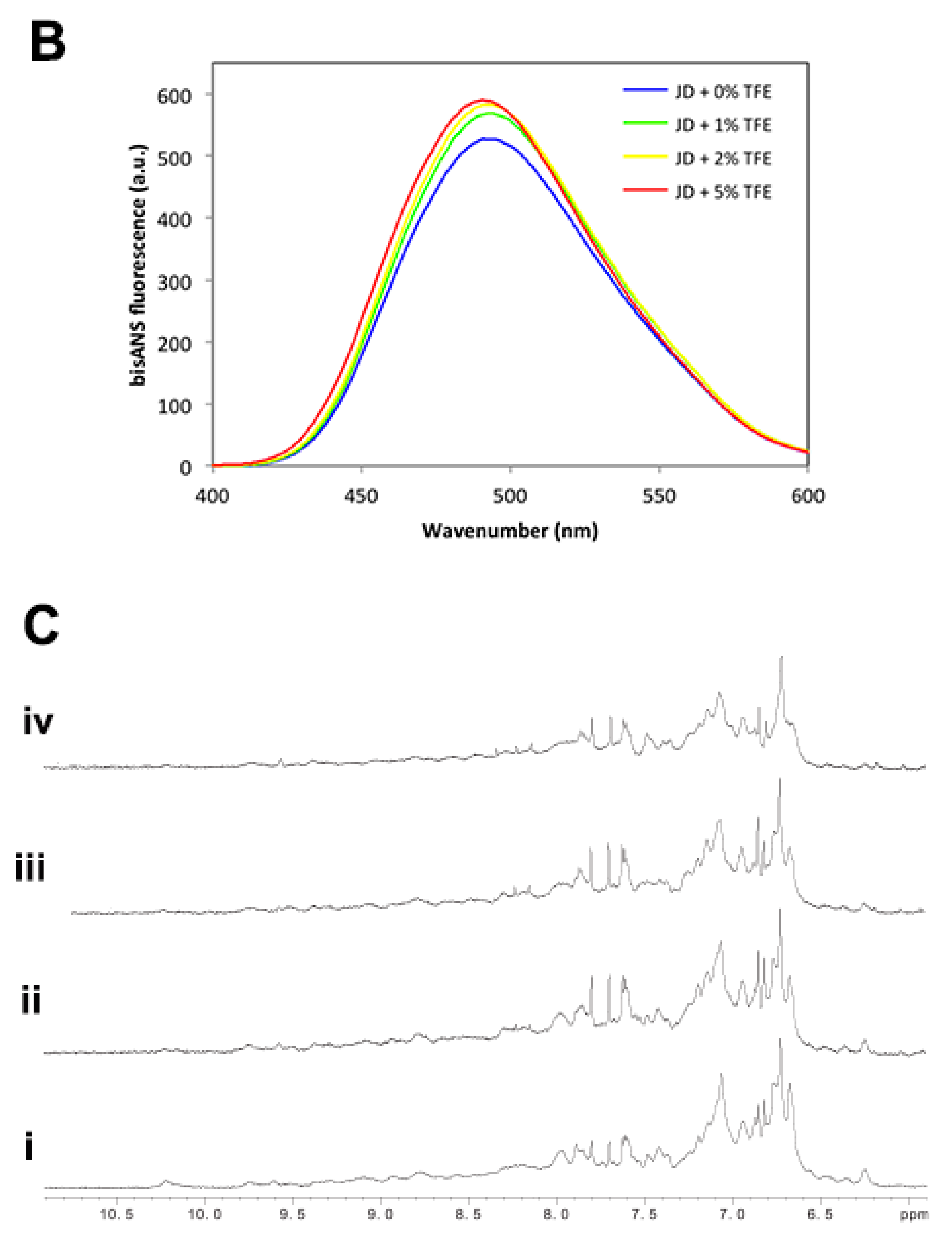
2.2. Low TFE Concentrations Do Not Significantly Alter JD Structure
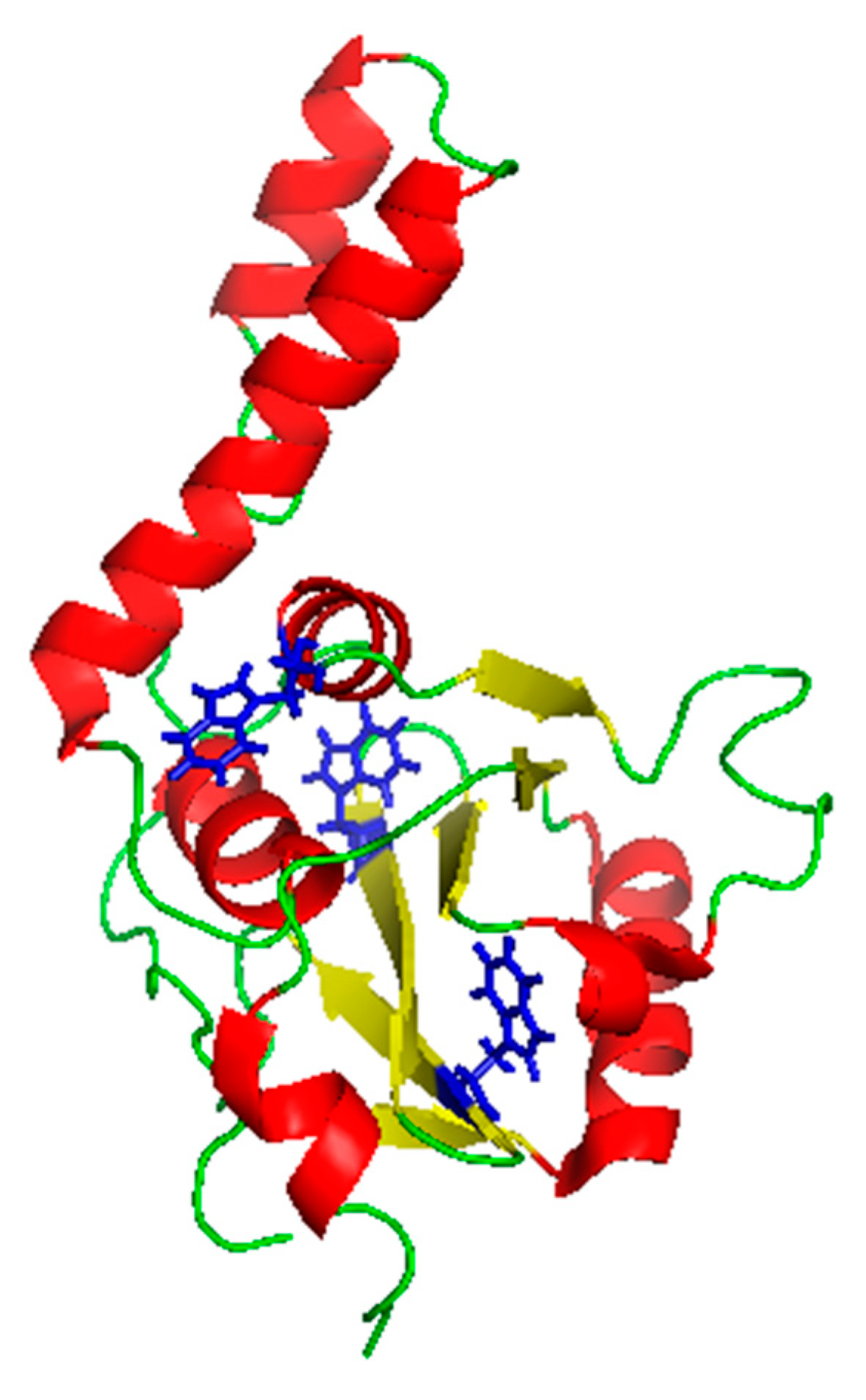
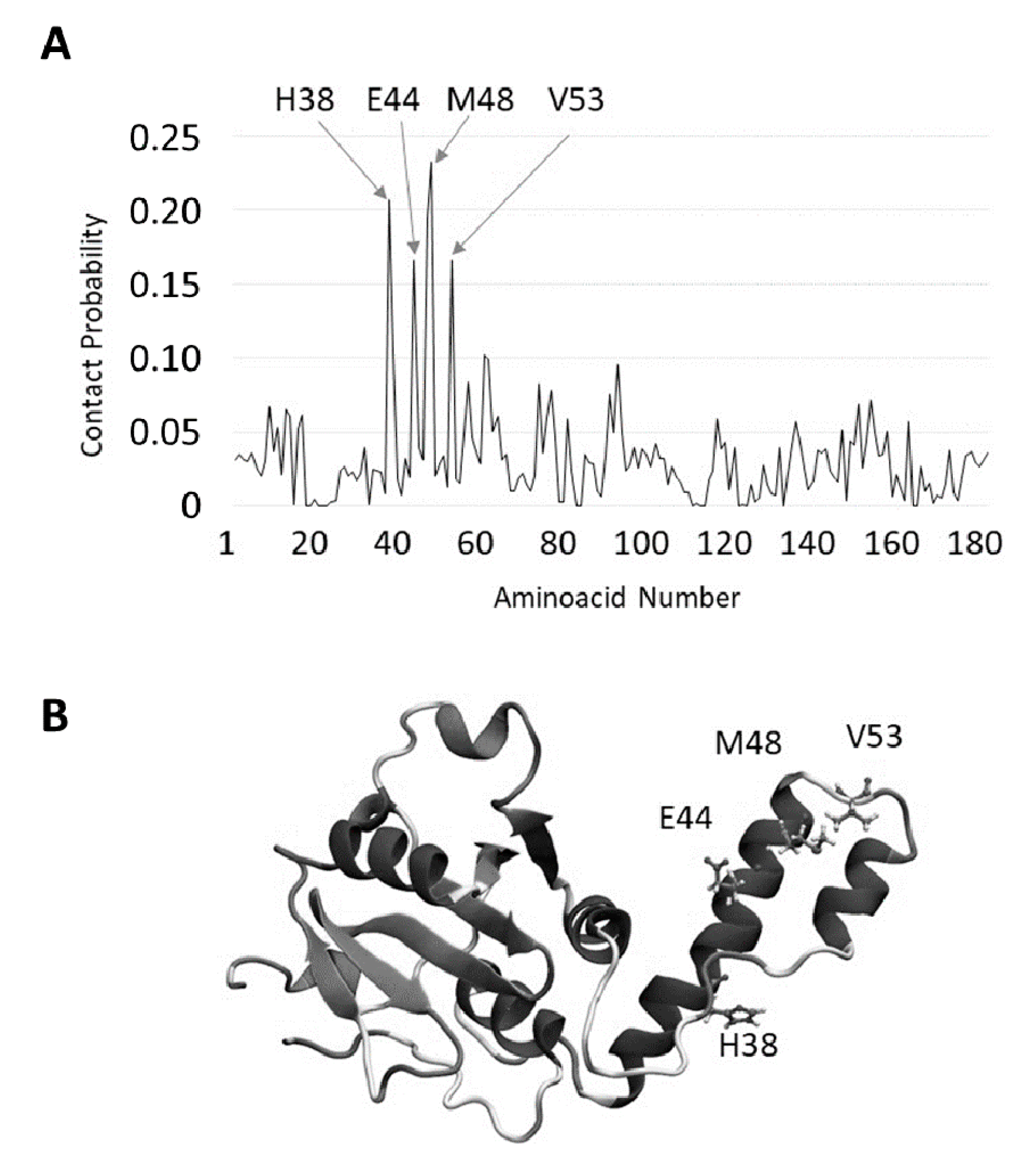
2.3. JD-TFE Binding Sites
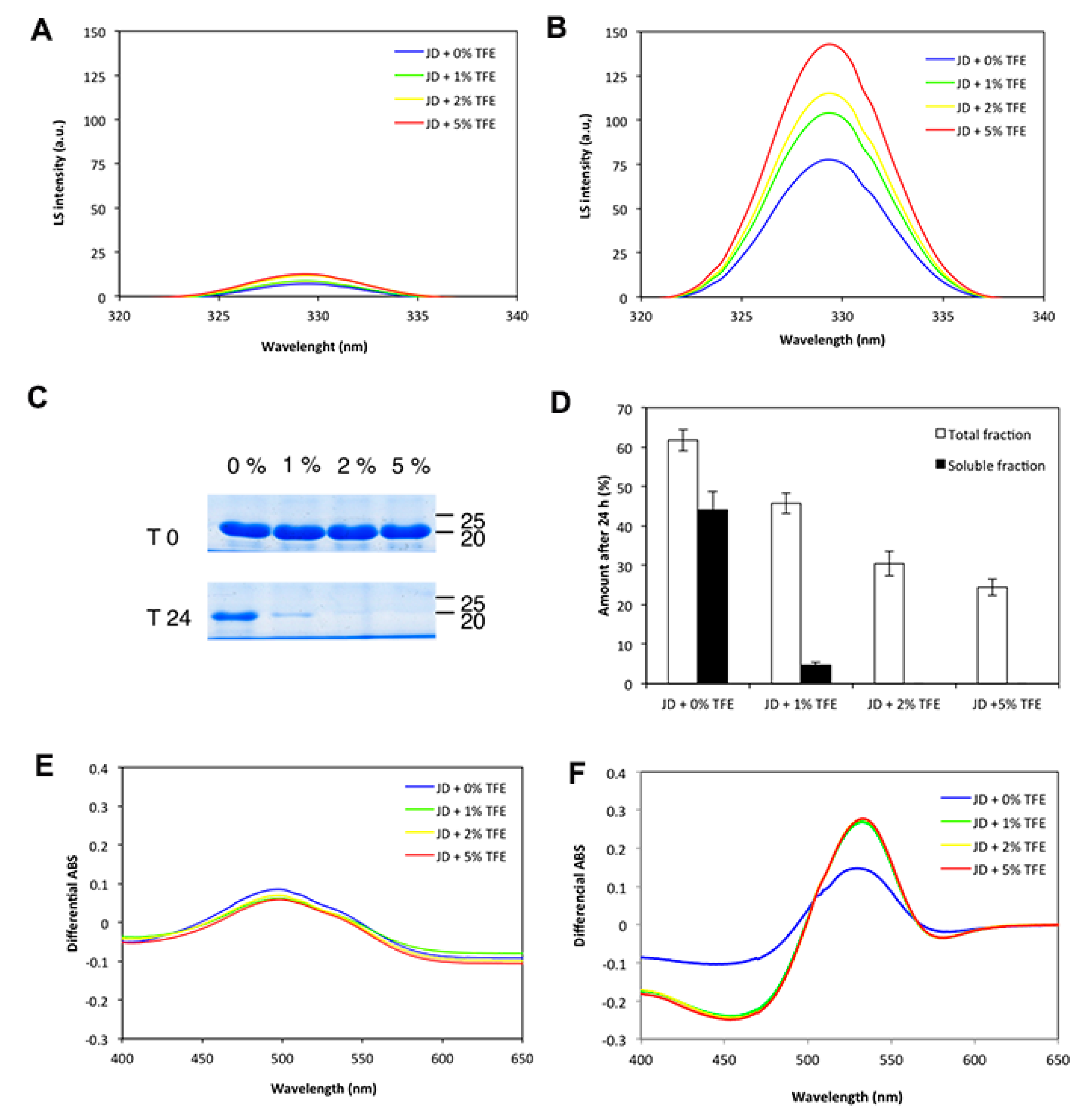
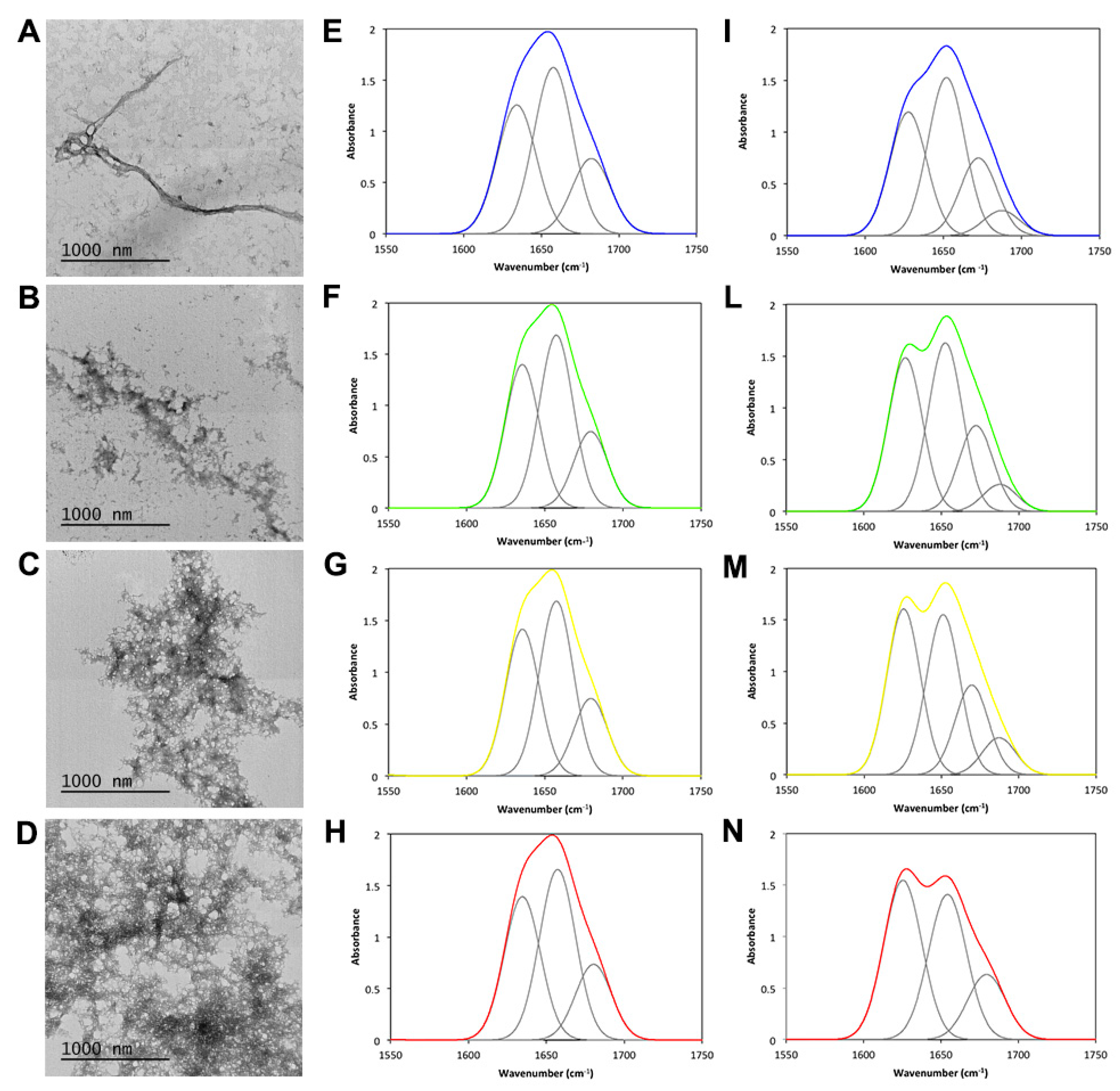
2.4. TFE Promotes JD Aggregation
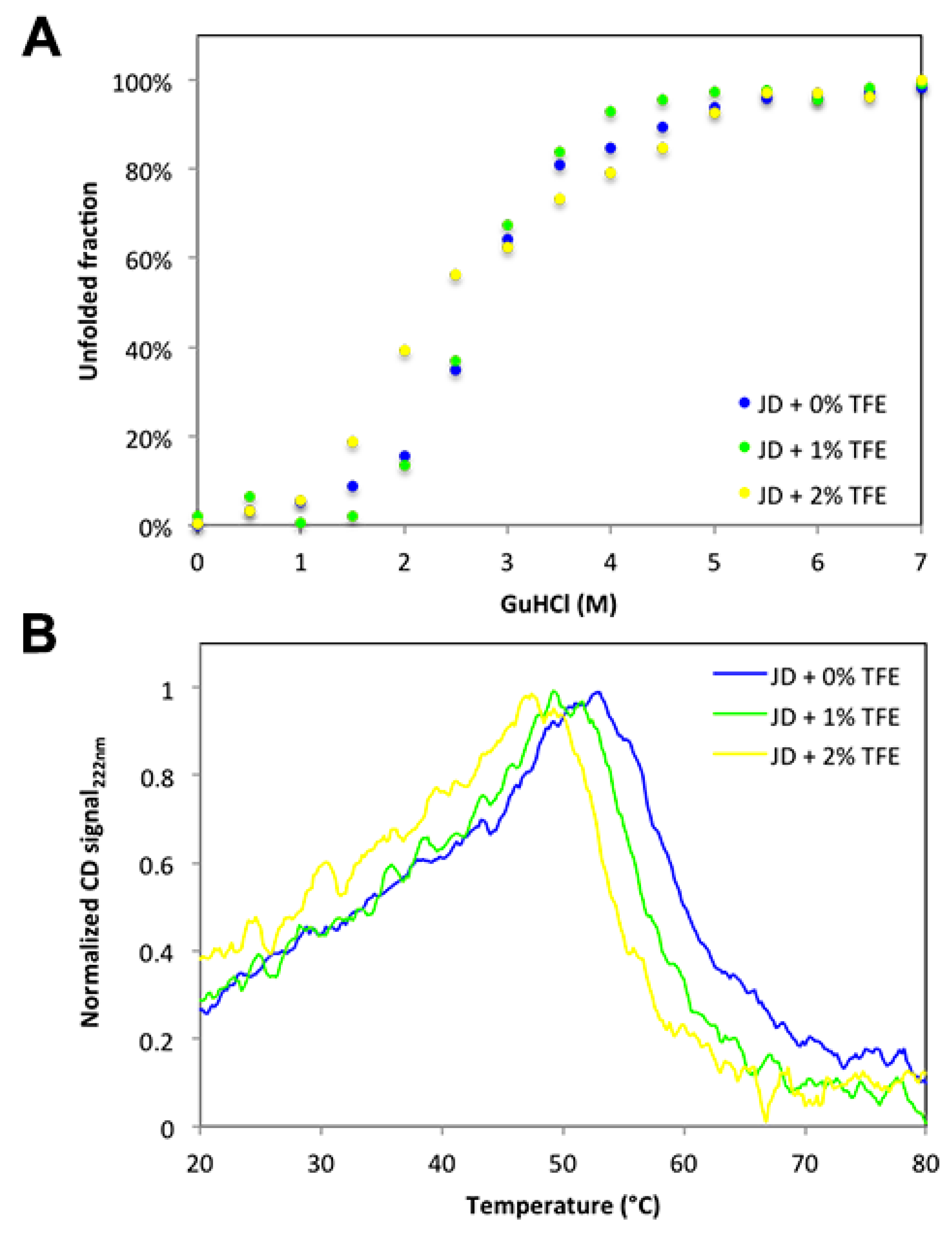
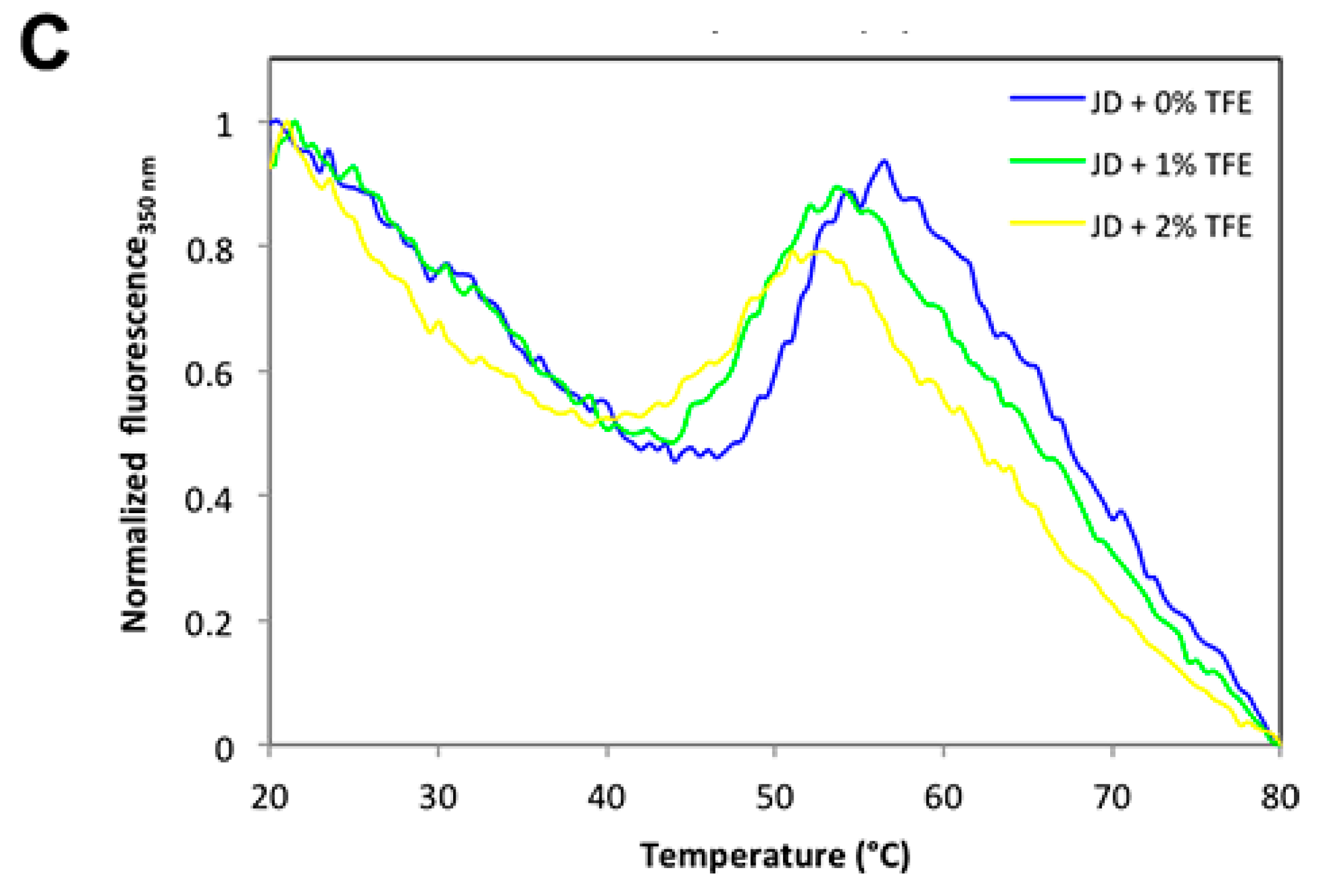
2.5. TFE Impacts on JD Thermal Stability
3. Discussion
4. Materials and Methods
4.1. Josephin Domain Purification
4.2. Josephin Domain Aggregation and Soluble Fraction Analysis
4.3. Spectroscopic Methods
4.4. Congo Red Binding
4.5. bis-ANS Binding
4.6. Aggregation Kinetics
4.7. Fourier Transform Infrared Spectroscopy
4.8. Transmission Electron Microscopy
4.9. Molecular Dynamics
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Cummings, C.J.; Zoghbi, H.Y. Trinucleotide repeats: Mechanisms and pathophysiology. Annu. Rev. Genom. Hum. Genet. 2000, 1, 281–328. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A.; Poirier, M.A. Opinion: What is the role of protein aggregation in neurodegeneration? Nat. Rev. Mol. Cell Biol. 2005, 6, 891–898. [Google Scholar] [CrossRef] [PubMed]
- Todd, T.W.; Lim, J. Aggregation formation in the polyglutamine diseases: Protection at a cost? Mol. Cells 2013, 36, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, Y.; Okamoto, T.; Taniwaki, M.; Aizawa, M.; Inoue, M.; Katayama, S.; Kawakami, H.; Nakamura, S.; Nishimura, M.; Akiguchi, I. CAG expansions in a novel gene for Machado-Joseph disease at chromosome 14q32.1. Nat. Genet. 1994, 8, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Masino, L.; Musi, V.; Menon, R.P.; Fusi, P.; Kelly, G.; Frenkiel, T.A.; Trottier, Y.; Pastore, A. Domain architecture of the polyglutamine protein ataxin-3: A globular domain followed by a flexible tail. FEBS Lett. 2003, 549, 21–25. [Google Scholar] [CrossRef]
- Ellisdon, A.M.; Pearce, M.C.; Bottomley, S.P. Mechanisms of ataxin-3 misfolding and fibril formation: Kinetic analysis of a disease-associated polyglutamine protein. J. Mol. Biol. 2007, 368, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Ellisdon, A.M.; Thomas, B.; Bottomley, S.P. The two-stage pathway of ataxin-3 fibrillogenesis involves a polyglutamine-independent step. J. Biol. Chem. 2006, 281, 16888–16896. [Google Scholar] [CrossRef] [PubMed]
- Saunders, H.M.; Gilis, D.; Rooman, M.; Dehouck, Y.; Robertson, A.L.; Bottomley, S.P. Flanking domain stability modulates the aggregation kinetics of a polyglutamine disease protein. Protein Sci. 2011, 20, 1675–1681. [Google Scholar] [CrossRef] [PubMed]
- Masino, L.; Nicastro, G.; Menon, R.P.; Dal Piaz, F.; Calder, L.; Pastore, A. Characterization of the structure and the amyloidogenic properties of the Josephin domain of the polyglutamine-containing protein ataxin-3. J. Mol. Biol. 2004, 344, 1021–1035. [Google Scholar] [CrossRef] [PubMed]
- Lupton, C.J.; Steer, D.L.; Wintrode, P.L.; Bottomley, S.P.; Hughes, V.A.; Ellisdon, A.M. Enhanced Molecular Mobility of Ordinarily Structured Regions Drives Polyglutamine Disease. J. Biol. Chem. 2015, 290, 24190–24200. [Google Scholar] [CrossRef] [PubMed]
- Deriu, M.A.; Grasso, G.; Licandro, G.; Danani, A.; Gallo, D.; Tuszynski, J.A.; Morbiducci, U. Investigation of the Josephin Domain Protein-Protein Interaction by Molecular Dynamics. PLoS ONE 2014, 9, e108677. [Google Scholar] [CrossRef] [PubMed]
- Deriu, M.A.; Grasso, G.; Tuszynski, J.A.; Gallo, D.; Morbiducci, U.; Danani, A. Josephin Domain Structural Conformations Explored by Metadynamics in Essential Coordinates. PLoS Comput. Biol. 2016, 12, e1004699. [Google Scholar] [CrossRef] [PubMed]
- Grasso, G.; Tuszynski, J.A.; Morbiducci, U.; Licandro, G.; Danani, A.; Deriu, M.A. Thermodynamic and kinetic stability of the Josephin Domain closed arrangement: Evidences from replica exchange molecular dynamics. Biol. Direct 2017, 12, 2. [Google Scholar] [CrossRef] [PubMed]
- Apicella, A.; Soncini, M.; Deriu, M.A.; Natalello, A.; Bonanomi, M.; Dellasega, D.; Tortora, P.; Regonesi, M.E.; Casari, C.S. A hydrophobic gold surface triggers misfolding and aggregation of the amyloidogenic Josephin domain in monomeric form, while leaving the oligomers unaffected. PLoS ONE 2013, 8, e58794. [Google Scholar] [CrossRef] [PubMed]
- Fioroni, M.; Diaz, M.D.; Burger, K.; Berger, S. Solvation phenomena of a tetrapeptide in water/trifluoroethanol and water/ethanol mixtures: A diffusion NMR, intermolecular NOE, and molecular dynamics study. J. Am. Chem. Soc. 2002, 124, 7737–7744. [Google Scholar] [CrossRef] [PubMed]
- Wei, G.; Shea, J.-E. Effects of solvent on the structure of the Alzheimer amyloid-beta(25–35) peptide. Biophys. J. 2006, 91, 1638–1647. [Google Scholar] [CrossRef] [PubMed]
- Schönbrunner, N.; Wey, J.; Engels, J.; Georg, H.; Kiefhaber, T. Native-like β-structure in a Trifluoroethanol-induced Partially Folded State of the All-β-sheet Protein Tendamistat. J. Mol. Biol. 1996, 260, 432–445. [Google Scholar] [CrossRef] [PubMed]
- Buck, M. Trifluoroethanol and colleagues: Cosolvents come of age. Recent studies with peptides and proteins. Q. Rev. Biophys. 1998, 31, 297–355. [Google Scholar] [CrossRef] [PubMed]
- Cammers-Goodwin, A.; Allen, T.J.; Oslick, S.L.; McClure, K.F.; Lee, J.H.; Kemp, D.S. Mechanism of Stabilization of Helical Conformations of Polypeptides by Water Containing Trifluoroethanol. J. Am. Chem. Soc. 1996, 118, 3082–3090. [Google Scholar] [CrossRef]
- Myers, J.K.; Nick Pace, C.; Martin Scholtz, J. Trifluoroethanol effects on helix propensity and electrostatic interactions in the helical peptide from ribonuclease T1. Protein Sci. 2008, 7, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Srisailam, S.; Kumar, T.K.S.; Rajalingam, D.; Kathir, K.M.; Sheu, H.-S.; Jan, F.-J.; Chao, P.-C.; Yu, C. Amyloid-like fibril formation in an all beta-barrel protein. Partially structured intermediate state(s) is a precursor for fibril formation. J. Biol. Chem. 2003, 278, 17701–17709. [Google Scholar] [CrossRef] [PubMed]
- Pallarès, I.; Vendrell, J.; Avilés, F.X.; Ventura, S. Amyloid fibril formation by a partially structured intermediate state of alpha-chymotrypsin. J. Mol. 2004, 342, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Marinelli, P.; Castillo, V.; Ventura, S. Trifluoroethanol Modulates Amyloid Formation by the All α-Helical URN1 FF Domain. Int. J. Mol. Sci. 2013, 14, 17830–17844. [Google Scholar] [CrossRef] [PubMed]
- Povey, J.F.; Smales, C.M.; Hassard, S.J.; Howard, M.J. Comparison of the effects of 2,2,2-trifluoroethanol on peptide and protein structure and function. J. Struct. Biol. 2007, 157, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Nicastro, G.; Menon, R.P.; Masino, L.; Knowles, P.P.; McDonald, N.Q.; Pastore, A. The solution structure of the Josephin domain of ataxin-3: Structural determinants for molecular recognition. Proc. Natl. Acad. Sci. USA 2005, 102, 10493–10498. [Google Scholar] [CrossRef] [PubMed]
- Scarff, C.A.; Almeida, B.; Fraga, J.; Macedo-Ribeiro, S.; Radford, S.E.; Ashcroft, A.E. Examination of Ataxin-3 (atx-3) Aggregation by Structural Mass Spectrometry Techniques: A Rationale for Expedited Aggregation upon Polyglutamine (polyQ) Expansion. Mol. Cell. Proteom. 2015, 14, 1241–1253. [Google Scholar] [CrossRef] [PubMed]
- Perham, M.; Liao, J.; Wittung-Stafshede, P. Differential Effects of Alcohols on Conformational Switchovers in α-Helical and β-Sheet Protein Models. Biochemistry 2006, 45, 7740–7749. [Google Scholar] [CrossRef] [PubMed]
- Masino, L.; Nicastro, G.; De Simone, A.; Calder, L.; Molloy, J.; Pastore, A. The Josephin domain determines the morphological and mechanical properties of ataxin-3 fibrils. Biophys. J. 2011, 100, 2033–2042. [Google Scholar] [CrossRef] [PubMed]
- Curto, L.M.; Angelani, C.R.; Caramelo, J.J.; Delfino, J.M. Truncation of a β-barrel scaffold dissociates intrinsic stability from its propensity to aggregation. Biophys. J. 2012, 103, 1929–1939. [Google Scholar] [CrossRef] [PubMed]
- Bemporad, F.; Chiti, F. ‘Native-like aggregation’ of the acylphosphatase from Sulfolobus solfataricus and its biological implications. FEBS Lett. 2009, 583, 2630–2638. [Google Scholar] [CrossRef] [PubMed]
- Rezaei-Ghaleh, N.; Ebrahim-Habibi, A.; Moosavi-Movahedi, A.A.; Nemat-Gorgani, M. Effect of polyamines on the structure, thermal stability and 2,2,2-trifluoroethanol-induced aggregation of alpha-chymotrypsin. Int. J. Biol. Macromol. 2007, 41, 597–604. [Google Scholar] [CrossRef] [PubMed]
- Anderson, V.L.; Webb, W.W. A desolvation model for trifluoroethanol-induced aggregation of enhanced green fluorescent protein. Biophys. J. 2012, 102, 897–906. [Google Scholar] [CrossRef] [PubMed]
- Luo, P.; Baldwin, R.L. Mechanism of helix induction by trifluoroethanol: A framework for extrapolating the helix-forming properties of peptides from trifluoroethanol/water mixtures back to water. Biochemistry 1997, 36, 8413–8421. [Google Scholar] [CrossRef] [PubMed]
- Ruggeri, F.S.; Longo, G.; Faggiano, S.; Lipiec, E.; Pastore, A.; Dietler, G. Infrared nanospectroscopy characterization of oligomeric and fibrillar aggregates during amyloid formation. Nat. Commun. 2015, 6, 7831. [Google Scholar] [CrossRef] [PubMed]
- Chiti, F.; Dobson, C.M. Amyloid formation by globular proteins under native conditions. Nat. Chem. Biol. 2009, 5, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Nicastro, G.; Habeck, M.; Masino, L.; Svergun, D.I.; Pastore, A. Structure validation of the Josephin domain of ataxin-3: Conclusive evidence for an open conformation. J. Biomol. NMR 2006, 36, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Nicastro, G.; Masino, L.; Esposito, V.; Menon, R.P.; De Simone, A.; Fraternali, F.; Pastore, A. Josephin domain of ataxin-3 contains two distinct ubiquitin-binding sites. Biopolymers 2009, 91, 1203–1214. [Google Scholar] [CrossRef] [PubMed]
- Sanfelice, D.; De Simone, A.; Cavalli, A.; Faggiano, S.; Vendruscolo, M.; Pastore, A. Characterization of the conformational fluctuations in the Josephin domain of ataxin-3. Biophys. J. 2014, 107, 2932–2940. [Google Scholar] [CrossRef] [PubMed]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 2006, 65, 712–725. [Google Scholar] [CrossRef] [PubMed]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [PubMed]
- Lindorff-Larsen, K.; Maragakis, P.; Piana, S.; Eastwood, M.P.; Dror, R.O.; Shaw, D.E. Systematic validation of protein force fields against experimental data. PLoS ONE 2012, 7, e32131. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, G.; Patra, N.; Barua, P.; Jayaram, B. A fast empirical GAFF compatible partial atomic charge assignment scheme for modeling interactions of small molecules with biomolecular targets. J. Comput. Chem. 2011, 32, 893–907. [Google Scholar] [CrossRef] [PubMed]
- Sousa da Silva, A.W.; Vranken, W.F. ACPYPE—AnteChamber PYthon Parser interfacE. BMC Res. Notes 2012, 5, 367. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926. [Google Scholar] [CrossRef]
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar]
- Korendovych, I.V.; Kulp, D.W.; Wu, Y.; Cheng, H.; Roder, H.; DeGrado, W.F. Design of a switchable eliminase. Proc. Natl. Acad. Sci. USA 2011, 108, 6823–6827. [Google Scholar] [CrossRef] [PubMed]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; Postma, J.P.M.; Van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Nosé, S.; Klein, M.L. Constant pressure molecular dynamics for molecular systems. Mol. Phys. 1983, 50, 1055–1076. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Heinig, M.; Frishman, D. STRIDE: A web server for secondary structure assignment from known atomic coordinates of proteins. Nucleic Acids Res. 2004, 32, W500–W502. [Google Scholar] [CrossRef] [PubMed]
- Deriu, M.A.; Grasso, G.; Tuszynski, J.A.; Massai, D.; Gallo, D.; Morbiducci, U.; Danani, A. Characterization of the AXH domain of Ataxin-1 using enhanced sampling and functional mode analysis. Proteins Struct. Funct. Bioinform. 2016, 84, 666–673. [Google Scholar] [CrossRef] [PubMed]
- Grasso, G.; Deriu, M.A.; Tuszynski, J.A.; Gallo, D.; Morbiducci, U.; Danani, A. Conformational fluctuations of the AXH monomer of Ataxin-1. Proteins Struct. Funct. Bioinform. 2016, 84, 52–59. [Google Scholar] [CrossRef] [PubMed]












© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Visentin, C.; Navarro, S.; Grasso, G.; Regonesi, M.E.; Deriu, M.A.; Tortora, P.; Ventura, S. Protein Environment: A Crucial Triggering Factor in Josephin Domain Aggregation: The Role of 2,2,2-Trifluoroethanol. Int. J. Mol. Sci. 2018, 19, 2151. https://doi.org/10.3390/ijms19082151
Visentin C, Navarro S, Grasso G, Regonesi ME, Deriu MA, Tortora P, Ventura S. Protein Environment: A Crucial Triggering Factor in Josephin Domain Aggregation: The Role of 2,2,2-Trifluoroethanol. International Journal of Molecular Sciences. 2018; 19(8):2151. https://doi.org/10.3390/ijms19082151
Chicago/Turabian StyleVisentin, Cristina, Susanna Navarro, Gianvito Grasso, Maria Elena Regonesi, Marco Agostino Deriu, Paolo Tortora, and Salvador Ventura. 2018. "Protein Environment: A Crucial Triggering Factor in Josephin Domain Aggregation: The Role of 2,2,2-Trifluoroethanol" International Journal of Molecular Sciences 19, no. 8: 2151. https://doi.org/10.3390/ijms19082151
APA StyleVisentin, C., Navarro, S., Grasso, G., Regonesi, M. E., Deriu, M. A., Tortora, P., & Ventura, S. (2018). Protein Environment: A Crucial Triggering Factor in Josephin Domain Aggregation: The Role of 2,2,2-Trifluoroethanol. International Journal of Molecular Sciences, 19(8), 2151. https://doi.org/10.3390/ijms19082151