Population Genetics Revealed a New Locus That Underwent Positive Selection in Barley


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
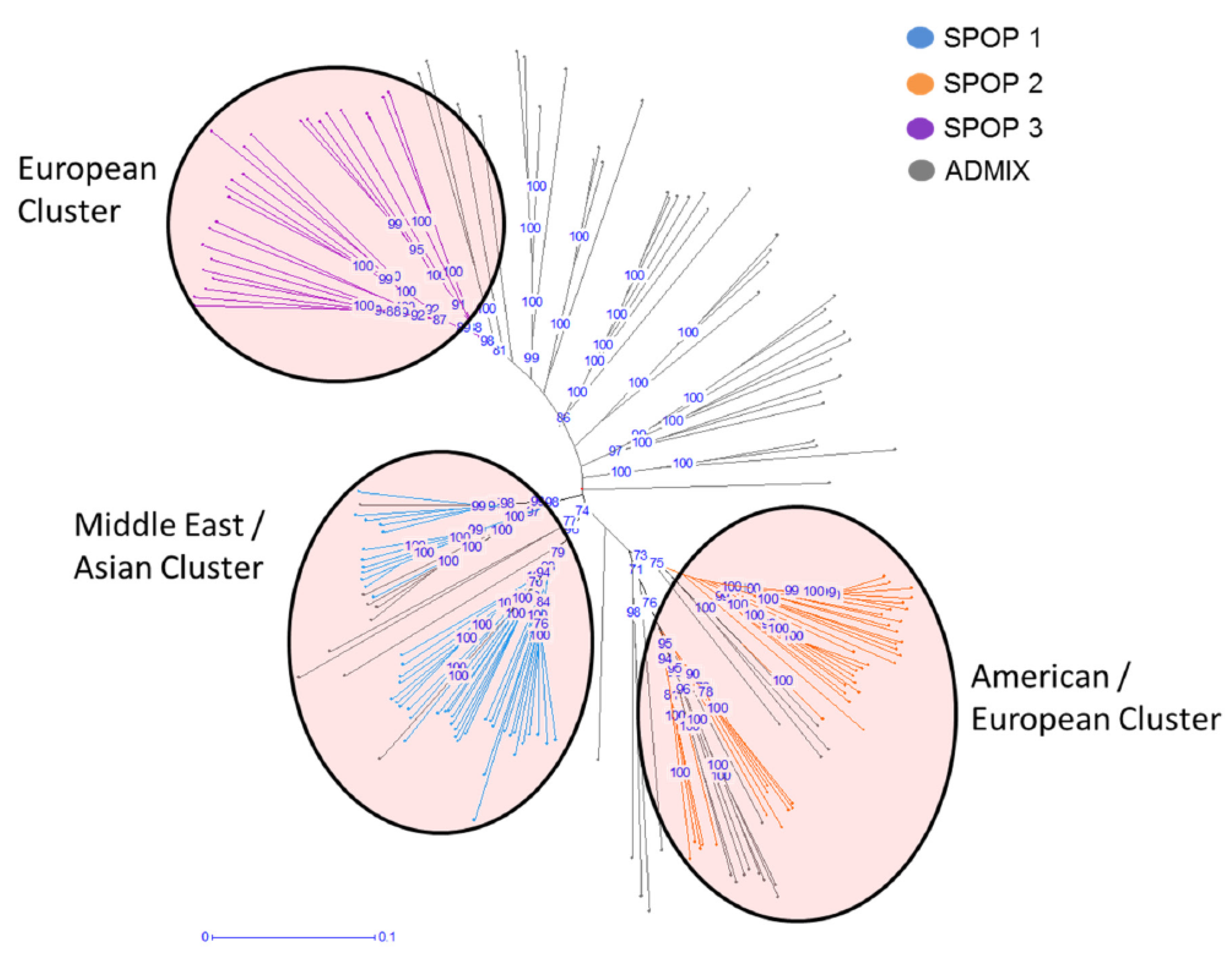
2.1. Population Structure Analysis
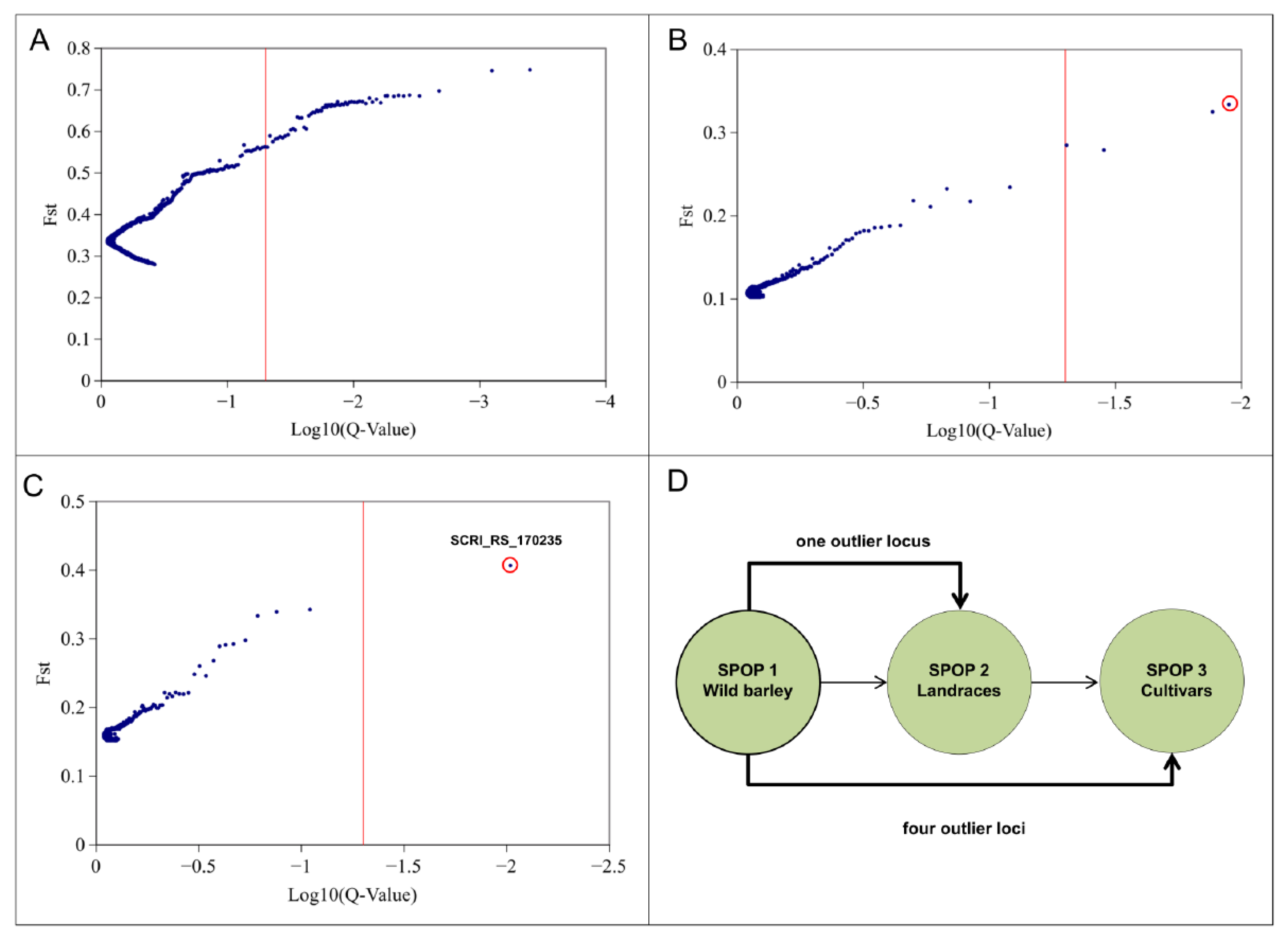
2.2. Genomic Scan for Outlier Loci Detection
2.3. Population Genetic Analysis
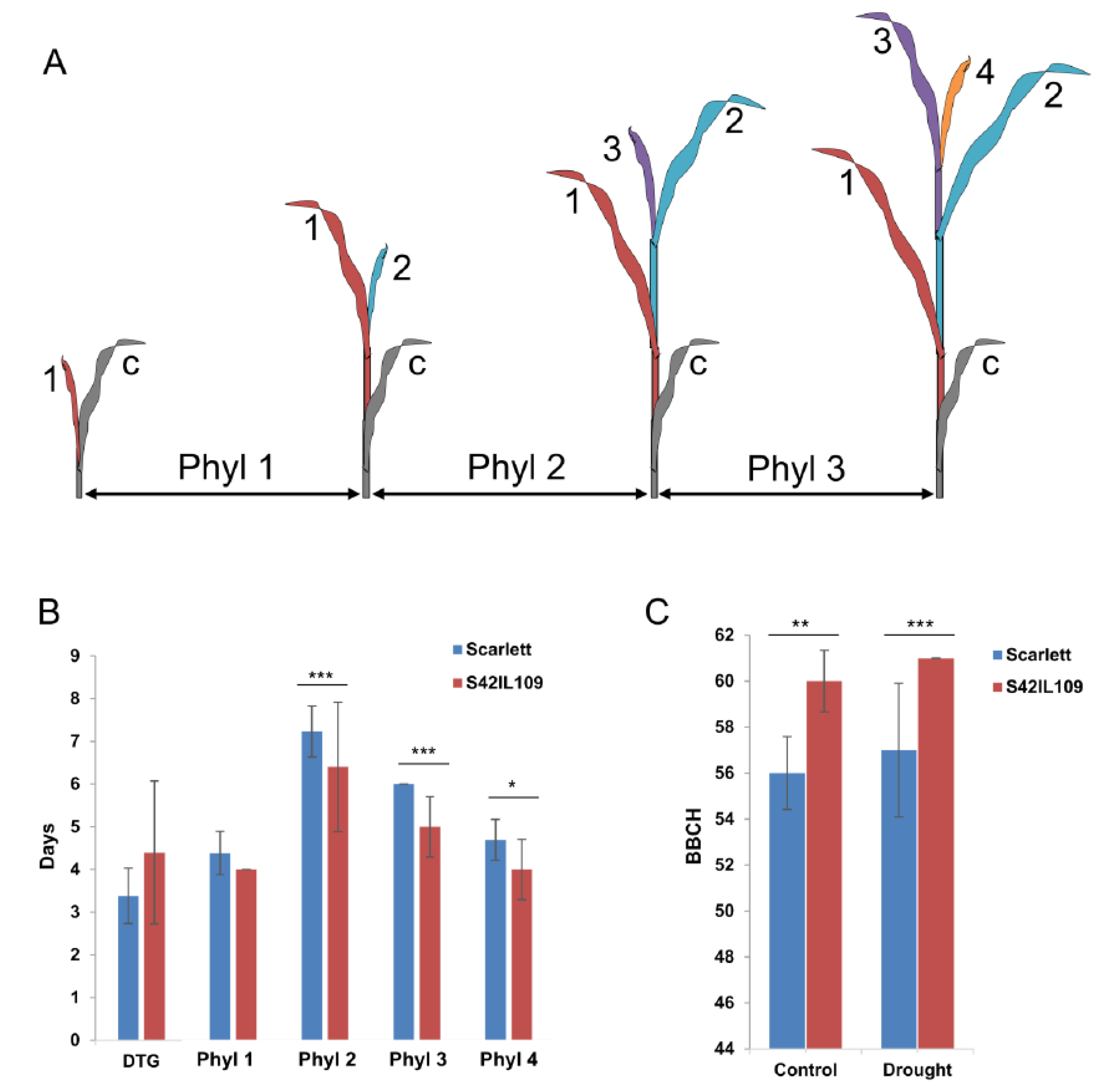
2.4. Phenotypic Evaluation of Outlier Locus Alleles Carrying Genotypes
3. Discussion
4. Materials and Methods
4.1. Plant Material
Genotyping
4.2. Population Structure Analysis
4.3. Loci Outlier Analysis
4.4. Restriction Fragment Polymorphism Analysis
4.5. Phenotypic Comparison of Barley Plant Development
4.6. Greenhouse Experiment
4.7. Climate Chamber Experiment
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schulte, D.; Close, T.J.; Graner, A.; Langridge, P.; Matsumoto, T.; Muehlbauer, G.; Sato, K.; Schulman, A.H.; Waugh, R.; Wise, R.P.; et al. The International Barley Sequencing Consortium—At the Threshold of Efficient Access to the Barley Genome. Plant Physiol. 2009, 149, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Salamini, F.; Ozkan, H.; Brandolini, A.; Schäfer-Pregl, R.; Martin, W. Genetics and geography of wild cereal domestication in the near east. Nat. Rev. Genet. 2002, 3, 429–441. [Google Scholar] [CrossRef] [PubMed]
- Vallage, V.; Hari, S. Domestication of plants in the old World—The origin and spread of cultivated plants in West Asia, Europe, and the Nile Valley. Genet. Resour. Crop. Evol. 1979, 123, 256–275. [Google Scholar]
- Harlan, J.R.; Zohary, D. Distribution of wild wheats and barley. Science 1966, 153, 1074–1080. [Google Scholar] [CrossRef] [PubMed]
- Badr, A.; Müller, K.; Schäfer-Pregl, R.; El Rabey, H.; Effgen, S.; Ibrahim, H.H.; Pozzi, C.; Rohde, W.; Salamini, F. On the origin and domestication history of Barley (Hordeum vulgare). Mol. Biol. Evol. 2000, 17, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Stebbins, G.L.; Yagil, E. Morphogenetic Effects of Hooded Gene in Barley. 1. Course of Development in Hooded and Awned Genotypes. Genetics 1966, 54, 727. [Google Scholar] [PubMed]
- Badr, A.; El-Shazly, H. Molecular approaches to origin, ancestry and domestication history of crop plants: Barley and clover as examples. J. Genet. Eng. Biotechnol. 2012, 10, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Von Bothmer, R.; Jacobsen, N.; Baden, C.; Jørgensen, R.B.; Linde-Laursen, I. An Ecogeographical Study of the Genus Hordeum, 2nd ed.; IPGRI: Rome, Italy, 1995. [Google Scholar]
- Wang, Y.; Ren, X.; Sun, D.; Sun, G. Origin of worldwide cultivated barley revealed by NAM-1 gene and grain protein content. Front. Plant Sci. 2015, 6, 803. [Google Scholar] [CrossRef] [PubMed]
- Dai, F.; Nevo, E.; Wu, D.; Comadran, J.; Zhou, M.; Qiu, L.; Chen, Z.; Beiles, A.; Chen, G.; Zhang, G. Tibet is one of the centers of domestication of cultivated barley. Proc. Natl. Acad. Sci. USA 2012, 109, 16969–16973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrell, P.L.; Clegg, M.T. Genetic evidence for a second domestication of barley (Hordeum vulgare) east of the Fertile Crescent. Proc. Natl. Acad. Sci. USA 2007, 104, 3289–3294. [Google Scholar] [CrossRef] [PubMed]
- Sang, T.; Li, J. Molecular genetic basis of the domestication syndrome in cereals. In Cereal Genomics II; Pushpendra, K.G., Rajeev, K.V., Eds.; Springer: Dordrecht, The Netherlands, 2013; Volume 9789400764, pp. 319–340. [Google Scholar]
- Li, Y.-H.; Reif, J.C.; Jackson, S.A.; Ma, Y.-S.; Chang, R.-Z.; Qiu, L.-J. Detecting SNPs underlying domestication-related traits in soybean. BMC Plant Biol. 2014, 14, 251. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhou, A.; Sang, T. Rice Domestication by Reducing Shattering. Science 2006, 311, 1936–1939. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, D.A.; Balázs, E.; Heslop-Harrison, J.S. From crop domestication to super-domestication. Ann. Bot. 2007, 100, 893–901. [Google Scholar] [CrossRef]
- Avni, R.; Nave, M.; Barad, O.; Baruch, K.; Twardziok, S.O.; Gundlach, H.; Hale, I.; Mascher, M.; Spannagl, M.; Wiebe, K.; et al. Wild emmer genome architecture and diversity elucidate wheat evolution and domestication. Science 2017, 357, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Azhaguvel, P.; Komatsuda, T. A Phylogenetic Analysis Based on Nucleotide Sequence of a Marker Linked to the Brittle Rachis Locus Indicates a Diphyletic Origin of Barley. Ann. Bot. 2007, 100, 1009–1015. [Google Scholar] [CrossRef] [Green Version]
- Komatsuda, T.; Pourkheirandish, M.; He, C.; Azhaguvel, P.; Kanamori, H.; Perovic, D.; Stein, N.; Graner, A.; Wicker, T.; Tagiri, A.; et al. Six-rowed barley originated from a mutation in a homeodomain-leucine zipper I-class homeobox gene. Proc. Natl. Acad. Sci. USA 2007, 104, 1424–1429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akpinar, B.A.; Biyiklioglu, S.; Alptekin, B.; Havránková, M.; Vrána, J.; Doležel, J.; Distelfeld, A.; Hernandez, P.; Budak, H. Chromosome-based survey sequencing reveals the genome organization of wild wheat progenitor Triticum dicoccoides. Plant Biotechnol. J. 2018. [Google Scholar] [CrossRef]
- Pourkheirandish, M.; Komatsuda, T. The importance of barley genetics and domestication in a global perspective. Ann. Bot. 2007, 100, 999–1008. [Google Scholar] [CrossRef]
- Fang, Z.; Gonzales, A.M.; Clegg, M.T.; Smith, K.P.; Muehlbauer, G.J.; Steffenson, B.J.; Morrell, P.L. Two genomic regions contribute disproportionately to geographic differentiation in wild barley. Genes Genomes Genet. 2014, 4, 1193–1203. [Google Scholar] [CrossRef]
- Abebe, T.D.; Naz, A.A.; Léon, J. Landscape genomics reveal signatures of local adaptation in barley (Hordeum vulgare L.). Front. Plant Sci. 2015, 6, 813. [Google Scholar] [CrossRef]
- Kawecki, T.J.; Ebert, D. Conceptual issues in local adaptation. Ecol. Lett. 2004, 7, 1225–1241. [Google Scholar] [CrossRef] [Green Version]
- Bellucci, E.; Bitocchi, E.; Rau, D.; Nanni, L.; Ferradini, N.; Giardini, A.; Rodriguez, M.; Attene, G.; Papa, R. Population structure of barley landrace populations and gene-flow with modern varieties. PLoS ONE 2013, 8, e83891. [Google Scholar] [CrossRef]
- Huebner, S.; Bdolach, E.; Ein-Gedy, S.; Schmid, K.J.; Korol, A.; Fridman, E. Phenotypic landscapes: Phenological patterns in wild and cultivated barley. J. Evol. Biol. 2013, 26, 163–174. [Google Scholar] [CrossRef]
- Huebner, S.; Günther, T.; Flavell, A.; Fridman, E.; Graner, A.; Korol, A.; Schmid, K.J. Islands and streams: Clusters and gene flow in wild barley populations from the Levant. Mol. Ecol. 2012, 21, 1115–1129. [Google Scholar] [CrossRef] [PubMed]
- Von Bothmer, R.; van Hintum, T.; Knüpffer, H.; Sato, K. Diversity in Barley, 1st ed.; von Bothmer, R., van Hintum, T., Knüpffer, H., Sato, K., Eds.; Elsevier: Amsterdam, The Netherlands, 2003. [Google Scholar]
- Allaby, R.G. Barley domestication: The end of a central dogma? Genome Biol. 2015, 16, 176. [Google Scholar] [CrossRef] [PubMed]
- Zamir, D. Improving plant breeding with exotic genetic libraries. Nat. Rev. Genet. 2001, 2, 983–989. [Google Scholar] [CrossRef]
- Obando, J.; Fernandez-Trujillo, J.P.; Martinez, J.A.; Alarcon, A.L.; Eduardo, I.; Arus, P.; Monforte, A.J. Identification of melon fruit quality quantitative trait loci using near-isogenic lines. J. Am. Soc. Hortic. Sci. 2008, 133, 139–151. [Google Scholar]
- Rousseaux, M.C.; Jones, C.M.; Adams, D.; Chetelat, R.; Bennett, A.; Powell, A. QTL analysis of fruit antioxidants in tomato using Lycopersicon pennellii introgression lines. Theor. Appl. Genet. 2005, 111, 1396–1408. [Google Scholar] [CrossRef]
- Liu, S.; Zhao, S.; Chen, F.; Xia, G. Generation of novel high quality HMW-GS genes in two introgression lines of Triticum aestivum/Agropyron elongatum. BMC Evol. Biol. 2007, 7, 76. [Google Scholar] [CrossRef]
- Wilhelm, W.W.; McMaster, G.S. Importance of the phyllochron in studying development and growth in grasses. Crop. Sci. 1995, 35, 1–3. [Google Scholar] [CrossRef]
- Reinert, S. A Global Barley Diversity Panel Uncovered Novel Drought Inducible QTL and Footprints of Evolution. 2017. Available online: http://hss.ulb.uni-bonn.de/2017/4668/4668.pdf (accessed on 2 January 2019).
- Schmalenbach, I.; Körber, N.; Pillen, K. Selecting a set of wild barley introgression lines and verification of QTL effects for resistance to powdery mildew and leaf rust. Theor. Appl. Genet. 2008, 117, 1093–1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayer, K.F.X.; Waugh, R.; Brown, J.W.S.; Schulman, A.; Langridge, P.; Platzer, M.; Fincher, G.B.; Muehlbauer, G.J.; Sato, K.; Close, T.J.; et al. A physical, genetic and functional sequence assembly of the barley genome. Nature 2012, 491, 711–716. [Google Scholar] [PubMed] [Green Version]
- Miyagawa, T.; Nishida, N.; Ohashi, J.; Kimura, R.; Fujimoto, A.; Kawashima, M.; Koike, A.; Sasaki, T.; Tanii, H.; Otowa, T.; et al. Appropriate data cleaning methods for genome-wide association study. J. Hum. Genet. 2008, 53, 886–893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopelman, N.M.; Mayzel, J.; Jakobsson, M.; Rosenberg, N.A.; Mayrose, I. CLUMPAK: A program for identifying clustering modes and packaging population software inferences across K. Mol. Ecol. Resour. 2015, 15, 1179–1191. [Google Scholar] [CrossRef] [PubMed]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Perrier, X.; Jacquemoud-Collet, J.P. DARwin—Dissimilarity Analysis and Representation for Windows. 2006. Available online: http://darwin.cirad.fr/ (accessed on 6 October 2016).
- Weir, B.S.; Hill, W.G. Estimating F-Statistics. Annu. Rev. Genet. 2002, 36, 721–750. [Google Scholar] [CrossRef] [PubMed]
- Hack, H.; Weber, E.; Feller, C.; Hess, M.; Wicke, H.; van den Boom, T.; Lancashire, P.D.; Buhr, L.; Klose, R.; Stauss, R.; et al. Entwicklungsstadien mono- und dikotyler Pflanzen. Biol. Bundesanst. Forstwirtsch. 1992, 2, 1–165. [Google Scholar]
- Forster, B.P.; Franckowiak, J.D.; Lundqvist, U.; Lyon, J.; Pitkethly, I.; Thomas, W.T.B. The barley phytomer. Ann. Bot. 2007, 100, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Itoh, Y.; Sato, S.; Sano, Y. Developmental changes of phyllochron in near-isogenic lines of rice (Oryza sativa L.) with different growth durations. Euphytica 2001, 119, 271–278. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reinert, S.; Osthoff, A.; Léon, J.; Naz, A.A. Population Genetics Revealed a New Locus That Underwent Positive Selection in Barley. Int. J. Mol. Sci. 2019, 20, 202. https://doi.org/10.3390/ijms20010202
Reinert S, Osthoff A, Léon J, Naz AA. Population Genetics Revealed a New Locus That Underwent Positive Selection in Barley. International Journal of Molecular Sciences. 2019; 20(1):202. https://doi.org/10.3390/ijms20010202
Chicago/Turabian StyleReinert, Stephan, Alina Osthoff, Jens Léon, and Ali Ahmad Naz. 2019. "Population Genetics Revealed a New Locus That Underwent Positive Selection in Barley" International Journal of Molecular Sciences 20, no. 1: 202. https://doi.org/10.3390/ijms20010202
APA StyleReinert, S., Osthoff, A., Léon, J., & Naz, A. A. (2019). Population Genetics Revealed a New Locus That Underwent Positive Selection in Barley. International Journal of Molecular Sciences, 20(1), 202. https://doi.org/10.3390/ijms20010202