DNA Damage and Repair in Human Reproductive Cells



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Genome Organization and Protection in Reproductive Cells
2.1. Gamete Genome Organization: Sperm Protamination
2.2. Genome Domain Protection to Spontaneous Mutations
2.3. Genetic Flaws Affecting Gamete Functionality and Fertility
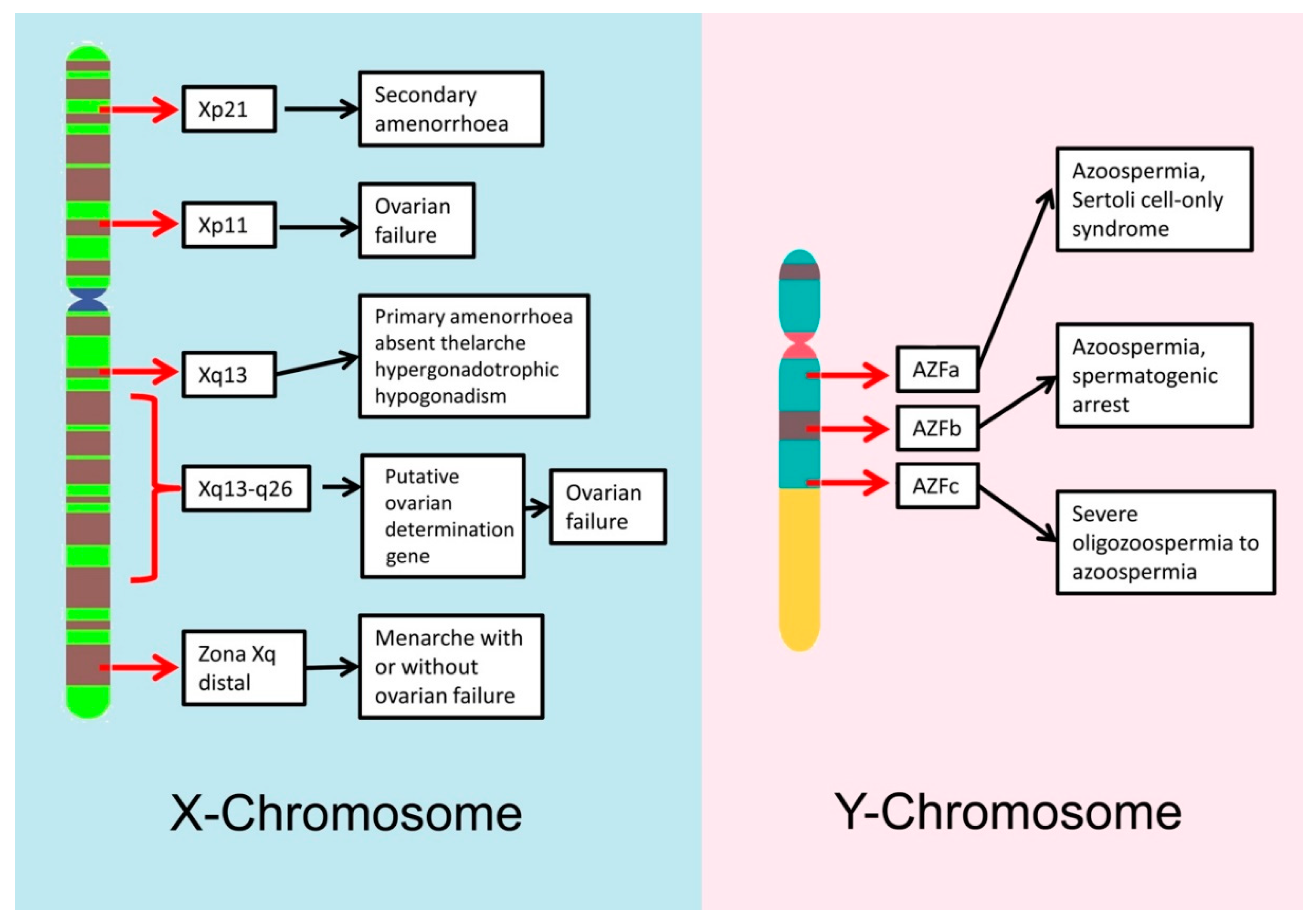
2.3.1. Genetics and Male Infertility
2.3.2. Genetics and Female Infertility
3. DNA Damage in Reproductive Cells
3.1. Origin of DNA Damage in Reproductive Cells
3.2. Defective Protamination and DNA Damage
3.3. Abortive Apoptosis and DNA Damage
3.4. Oxidative Stress and DNA Damage
3.4.1. Oxidative Stress in the Spermatozoa
3.4.2. Oxidative Stress in the Oocyte
3.5. Single-Stranded Breaks versus Double-Stranded Breaks
3.6. Susceptibility to De Novo Mutations
4. DNA Repair in the Reproductive Cells
4.1. DNA Repair Mechanisms during Gametogenesis
4.2. DNA Repair Mechanisms in Male Germ Cells and Spermatozoa
4.3. DNA Repair Mechanisms in the Oocyte
4.4. DNA Repair Mechanisms in the Zygote
5. Conclusions
Funding
Conflicts of Interest
Abbreviations
| Alt-EJ | Alternative NHEJ pathway |
| ART | Assisted reproduction technology |
| AZF | Azoospermia factor |
| BER | Base excision repair |
| CCRs | Complexchromosomerearrangements |
| CNVs | Copynumbervariants |
| DNMs | De novo mutations |
| DNA | Deoxyribonucleic acid |
| DSBs | Double stranded breaks |
| HR | Homologous recombination |
| ICSI | Intracytoplasmic sperm injection |
| LINES | Long interspersed nuclear elements |
| LTR | Long terminal repeats |
| MMR | Mismatch repair |
| NHEJ | Non homologous end joining |
| NER | Nucleotide excision repair |
| PGCs | Primordial germcells |
| PRM | Protamine |
| ROS | Reactive oxygen species |
| SINE | Short interspersed nuclear elements |
| SNVs | Single nucleotide variants |
| SSBs | Single stranded breaks |
| TEL-DNA | Telomeric DNA |
| TNP | Transition nuclear proteins |
References
- Ioannou, D.; Miller, D.; Griffin, D.K.; Tempest, H.G. Impact of Sperm DNA Chromatin in the Clinic. J. Assist. Reprod. Genet. 2016, 33, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Oliva, R.; Dixon, G.H. Vertebrate Protamine Genes and the Histone-to-Protamine Replacement Reaction. Prog. Nucleic Acid Res. Mol. Biol. 1991, 40, 25–94. [Google Scholar] [PubMed]
- Oliva, R. Protamines and Male Infertility. Hum. Reprod. Update 2006, 12, 417–435. [Google Scholar] [CrossRef] [PubMed]
- Balhorn, R. Sperm Chromatin: An Overview. In Sperm Chromatin; Springer: New York, NY, USA, 2011; pp. 3–18. [Google Scholar]
- Kvist, U.; Björndahl, L. Structure of Chromatin in Spermatozoa. Adv. Exp. Med. Biol. 2014, 791, 1–11. [Google Scholar]
- Gusse, M.; Sautière, P.; Bélaiche, D.; Martinage, A.; Roux, C.; Dadoune, J.P.; Chevaillier, P. Purification and Characterization of Nuclear Basic Proteins of Human Sperm. Biochim. Biophys. Acta 1986, 884, 124–134. [Google Scholar] [CrossRef]
- Jodar, M.; Oliva, R. Protamine Alterations in Human Spermatozoa. Adv. Exp. Med. Biol. 2014, 791, 83–102. [Google Scholar] [PubMed]
- Balhorn, R. The Protamine Family of Sperm Nuclear Proteins. Genome Biol. 2007, 8, 227. [Google Scholar] [CrossRef]
- Martins, R.P.; Ostermeier, G.C.; Krawetz, S.A. Nuclear Matrix Interactions at the Human Protamine Domain. J. Biol. Chem. 2004, 279, 51862. [Google Scholar] [CrossRef]
- Chirat, F.; Arkhis, A.; Martinage, A.; Jaquinod, M.; Chevaillier, P.; Sautiere, P. Phosphorylation of Human Sperm Protamines HP1 and HP2: Identification of Phosphorylation Sites. Biochim. Biophys. Acta 1993, 1203, 109–114. [Google Scholar] [CrossRef]
- Koonin, E.V.; Wolf, Y.I. Constraints and Plasticity in Genome and Molecular-Phenome Evolution. Nat. Rev. Genet. 2010, 11, 487–498. [Google Scholar] [CrossRef]
- López-Flores, I.; Garrido-Ramos, M. The Repetitive DNA Content of Eukaryotic Genomes. Genome Dyn. 2012, 7, 1–28. [Google Scholar] [PubMed]
- Biscotti, M.; Olmo, E.; Heslop-Harrison, J. Repetitive DNA in Eukaryotic Genomes. Chromosome Res. 2015, 23, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Gundín, F.; Rivero, M.T.; Gosálvez, J.; Fernández, J.L. Radiation-Induced DNA Breaks in Different Human Satellite DNA Sequence Areas, Analyzed by DNA Breakage Detection-Fluorescence in situ Hybridization. Radiat. Res. 2002, 157, 711–720. [Google Scholar] [CrossRef]
- Fernández, J.L.; Gosálvez, J.; Goyanes, V. High Frequency of Mutagen-Induced Chromatid Exchanges at Interstitial Telomere-Like DNA Sequence Blocks of Chinese Hamster Cells. Chromosome Res. 1995, 3, 281–284. [Google Scholar] [CrossRef] [PubMed]
- Reig-Viader, R.; Garcia-Caldés, M.; Ruiz-Herrera, A. Telomere Homeostasis in Mammalian Germ Cells: A Review. Chromosoma 2016, 125, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Blasco, M.A.; Trimarchi, J.R.; Keefe, D.L. An Essential Role for Functional Telomeres in Mouse Germ Cells during Fertilization and Early Development. Dev. Biol. 2002, 249, 74–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, L.E.; Mitchell, S.E.; O’Neill, R.J. Pericentric and Centromeric Transcription: A Perfect Balance Required. Chromosome Res. 2012, 20, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Yang, Q.; Shi, S.; Luo, X.; Sun, Y. Semen Preparation Methods and Sperm Telomere Length: Density Gradient Centrifugation versus the Swim up Procedure. Sci. Rep. 2016, 6, 39051. [Google Scholar] [CrossRef]
- Yang, Q.; Zhang, N.; Zhao, F.; Zhao, W.; Dai, S.; Liu, J.; Bukhari, I.; Xin, H.; Niu, W.; Sun, Y. Processing of Semen by Density Gradient Centrifugation Selects Spermatozoa with Longer Telomeres for Assisted Reproduction Techniques. Reprod. BioMed. Online 2015, 31, 44–50. [Google Scholar] [CrossRef]
- Georgiou, I.; Syrrou, M.; Pardalidis, N.; Karakitsios, K.; Mantzavinos, T.; Giotitsas, N. Genetic and Epigenetic Risks of Intracytoplasmic Sperm Injection Method. Asian J. Androl. 2006, 8, 643–673. [Google Scholar] [CrossRef]
- Foresta, C.; Ferlin, A.; Gianaroli, L.; Dallapiccola, B. Guidelines for the Appropriate use of Genetic Tests in Infertile Couples. Eur. J. Hum. Gen. 2002, 10, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Tahmasbpour, E.; Balasubramanian, D.; Agarwal, A. A Multi-Faceted Approach to Understanding Male Infertility: Gene Mutations, Molecular Defects and Assisted Reproductive Techniques (ART). J. Assist. Reprod. Genet. 2014, 31, 1115–1137. [Google Scholar] [CrossRef]
- Foresta, C.; Ferlin, A. Role of INSL3 and LGR8 in Cryptorchidism and Testicular Functions. Reprod. BioMed. Online 2004, 9, 294–298. [Google Scholar] [CrossRef]
- Greco, E.; Scarselli, F.; Minasi, M.G.; Casciani, V.; Zavaglia, D.; Dente, D.; Tesarik, J.; Franco, G. Birth of 16 Healthy Children After ICSI in Cases of Nonmosaic Klinefelter Syndrome. Hum. Reprod. 2013, 28, 1155–1160. [Google Scholar] [CrossRef]
- Ferlin, A.; Garolla, A.; Foresta, C. Chromosome Abnormalities in Sperm of Individuals with Constitutional Sex Chromosomal Abnormalities. Cytogenet. Genome Res. 2005, 111, 310–316. [Google Scholar] [CrossRef]
- Mau-Holzmann, U.A. Somatic Chromosomal Abnormalities in Infertile Men and Women. Cytogenet. Genome Res. 2005, 111, 317–336. [Google Scholar] [CrossRef] [PubMed]
- Krausz, F.; Riera-Escamilla, A. Testing for Genetic Contributions to Infertility: Potential Clinical Impact. Expert Rev. Mol. Diagn. 2018, 18, 331–346. [Google Scholar] [CrossRef] [PubMed]
- De Braekeleer, M.; Dao, T.N. Cytogenetic Studies in Male Infertility: A Review. Hum. Reprod. 1991, 6, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Hassold, T.; Hall, H.; Hunt, P. The Origin of Human Aneuploidy: Where we have been, Where we are going. Hum. Mol. Genet. 2007. [Google Scholar] [CrossRef]
- Madan, K. What is a Complex Chromosome Rearrangement? Am. J. Med. Genet. Part A 2013, 161, 1181–1184. [Google Scholar] [CrossRef]
- Escudero, T.; Estop, A.; Fischer, J.; Munne, S. Preimplantation Genetic Diagnosis for Complex Chromosome Rearrangements. Am. J. Med. Genet. Part A 2008, 146, 1662–1669. [Google Scholar] [CrossRef] [PubMed]
- Madan, K. Balanced Complex Chromosome Rearrangements: Reproductive Aspects. A Review. Am. J. Med. Genet. Part A 2012, 158, 947–963. [Google Scholar] [CrossRef]
- Kloosterman, W.P.; Guryev, V.; van Roosmalen, M.; Duran, K.J.; de Bruijn, E.; Bakker, S.C.; Letteboer, T.; van Nesselrooij, B.; Hochstenbach, R.; Poot, M.; et al. Chromothripsis as a mechanism driving complex de novo structural rearrangements in the germline. Hum. Mol. Genet. 2011, 12, 1916–1924. [Google Scholar] [CrossRef] [PubMed]
- Fukami, M.; Shima, H.; Suzuki, E.; Ogata, T.; Matsubara, K.; Kamimaki, T. Catastrophic cellular events leading to complex chromosomal rearrangements in the germline. Clin. Genet. 2017, 91, 653–660. [Google Scholar] [CrossRef]
- Colaco, S.; Modi, D. Genetics of the Human Y Chromosome and Its Association with Male Infertility. Reprod. Biol. Endocrinol. 2018, 16, 14. [Google Scholar] [CrossRef]
- Krausz, C.; Hoefsloot, L.; Simoni, M.; Tüttelmann, F. EAA/EMQN Best Practice Guidelines for Molecular Diagnosis of Y-chromosomal Microdeletions: State-of-the-art 2013. Andrology 2014, 2, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Krausz, C. Y Chromosome and Male Infertility. Andrologia 2005, 37, 219–223. [Google Scholar] [CrossRef]
- Foresta, C.; Moro, E.; Ferlin, A. Y Chromosome Microdeletions and Alterations of Spermatogenesis 1. Endocr. Rev. 2001, 22, 226–239. [Google Scholar] [CrossRef] [PubMed]
- Vogt, P.H. Azoospermia Factor (AZF) in Yq11: Towards a Molecular Understanding of its Function for Human Male Fertility and Spermatogenesis. Reprod. BioMed. Online 2005, 10, 81–93. [Google Scholar] [CrossRef]
- Ferlin, A.; Raicu, F.; Gatta, V.; Zuccarello, D.; Palka, G.; Foresta, C. Male Infertility: Role of Genetic Background. Reprod. BioMed. Online 2007, 14, 734–745. [Google Scholar] [CrossRef]
- O’Flynn O’Brien, K.L.; Varghese, A.C.; Agarwal, A. The Genetic Causes of Male Factor Infertility: A Review. Fertil. Steril. 2010, 93, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Stuppia, L.; Antonucci, I.; Binni, F.; Brandi, A.; Grifone, N.; Colosimo, A.; De Santo, M.; Gatta, V.; Gelli, G.; Guida, V.; et al. Screening of Mutations in the CFTR Gene in 1195 Couples Entering Assisted Reproduction Technique Programs. Eur. J. Hum. Genet. 2005, 13, 959–964. [Google Scholar] [CrossRef] [PubMed]
- Tamburino, L.; Guglielmino, A.; Venti, E.; Chamayou, S. Molecular Analysis of Mutations and Polymorphisms in the CFTR Gene in Male Infertility. Reprod. BioMed. Online 2008, 17, 27–35. [Google Scholar] [CrossRef]
- Ferlin, A.; Vinanzi, C.; Garolla, A.; Selice, R.; Zuccarello, D.; Cazzadore, C.; Foresta, C. Male Infertility and Androgen Receptor Gene Mutations: Clinical Features and Identification of Seven Novel Mutations. Clin. Endocrinol. 2006, 65, 606–610. [Google Scholar] [CrossRef] [PubMed]
- Bogatcheva, N.; Agoulnik, A. INSL3/LGR8 Role in Testicular Descent and Cryptorchidism. Reprod. BioMed. Online 2005, 10, 49–54. [Google Scholar] [CrossRef]
- Tarani, L.; Lampariello, S.; Raguso, G.; Colloridi, F.; Pucarelli, I.; Pasquino, A.M.; Bruni, L.A. Pregnancy in Patients with Turner’s Syndrome: Six New Cases and Review of Literature. Gynecol. Endocrinol. 1998, 12, 83–87. [Google Scholar] [CrossRef]
- Barber JCKMcKinlay Gardner, R.J.; Sutherland, G.R.; Shaffer, L.G. Chromosome Abnormalities and Genetic Counselling. Hum. Genet. 2012, 131, 1393. [Google Scholar]
- Waters, J.; Campbell, P.; Crocker, A.; Campbell, C. Phenotypic Effects of Balanced X-Autosome Translocations in Females: A Retrospective Survey of 104 Cases Reported from UK Laboratories. Hum. Genet. 2001, 108, 318–327. [Google Scholar] [CrossRef]
- Mortlock, D.P.; Innis, J.W. Mutation of HOXA13 in Hand-Foot-Genital Syndrome. Nat. Genet. 1997, 15, 179–180. [Google Scholar] [CrossRef]
- Forges, T.; Monnier-Barbarino, P. Premature Ovarian Failure in Galactosaemia: Pathophysiology and Clinical Management. Pathol. Biol. 2003, 51, 47–56. [Google Scholar] [CrossRef]
- Layman, L.C. Human Gene Mutations Causing Infertility. J. Med. Genet. 2002, 39, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Marcon, L.; Boissonneault, G. Transient DNA Strand Breaks during Mouse and Human Spermiogenesis: New Insights in Stage Specificity and Link to Chromatin Remodeling. Biol. Reprod. 2004, 70, 910–918. [Google Scholar] [CrossRef] [PubMed]
- Sakkas, D.; Moffatt, O.; Manicardi, G.C.; Mariethoz, E.; Tarozzi, N.; Bizzaro, D. Nature of DNA Damage in Ejaculated Human Spermatozoa and the Possible Involvement of Apoptosis. Biol. Reprod. 2002, 66, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.; Saleh, R.; Bedaiwy, M. Role of Reactive Oxygen Species in the Pathophysiology of Human Reproduction. Fertil. Steril. 2003, 79, 829–843. [Google Scholar] [CrossRef]
- Castilla, J.A.; Zamora, S.; Gonzalvo, M.C.; Luna del Castillo, J.D.; Roldan-Nofuentes, J.A.; Clavero, A.; Björndahl, L.; Martínez, L. Sperm Chromatin Structure Assay and Classical Semen Parameters: Systematic Review. Reprod. BioMed. Online 2010, 20, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Gosálvez, J.; García-Ochoa, C.; Ruíz-Jorro, M.; Martínez-Moya, M.; Sánchez-Martín, P.; Caballero, P. ¿A Qué Velocidad “muere” El ADN Del Espermatozoide Tras Descongelar Muestras Seminales Procedentes De Donantes? Rev. Int. Androl. 2013, 11, 85–93. [Google Scholar]
- Simon, L.; Zini, A.; Dyachenko, A.; Ciampi, A.; Carrell, D. A Systematic Review and Meta-Analysis to Determine the Effect of Sperm DNA Damage on in vitro Fertilization and Intracytoplasmic Sperm Injection Outcome. Asian J. Androl. 2017, 19, 80–90. [Google Scholar] [PubMed]
- Zenzes, M. Smoking and Reproduction: Gene Damage to Human Gametes and Embryos. Hum. Reprod. Update 2000, 6, 122–131. [Google Scholar] [CrossRef]
- Agarwal, A.; Said, T.; Bedaiwy, M.; Banerjee, J.; Alvarez, J. Oxidative Stress in an Assisted Reproductive Techniques Setting. Fertil. Steril. 2006, 86, 503–512. [Google Scholar] [CrossRef]
- Kopeika, J.; Thornhill, A.; Khalaf, Y. The Effect of Cryopreservation on the Genome of Gametes and Embryos: Principles of Cryobiology and Critical Appraisal of the Evidence. Hum. Reprod. Update 2015, 21, 209–227. [Google Scholar] [CrossRef]
- Carrell, D.T.; Emery, B.R.; Hammoud, S. Altered Protamine Expression and Diminished Spermatogenesis: What is the Link? Hum. Reprod. Update 2007, 13, 313–327. [Google Scholar] [CrossRef] [PubMed]
- Aoki, V.W.; Moskovtsev, S.I.; Willis, J.; Liu, L.; Mullen, J.B.M.; Carrell, D.T. DNA Integrity is Compromised in Protamine-Deficient Human Sperm. J. Androl. 2005, 26, 741–748. [Google Scholar] [CrossRef] [PubMed]
- Aoki, V.W.; Liu, L.; Jones, K.P.; Hatasaka, H.H.; Gibson, M.; Peterson, C.M.; Carrell, D.T. Sperm Protamine 1/Protamine 2 Ratios are Related to in vitro Fertilization Pregnancy Rates and Predictive of Fertilization Ability. Fertil. Steril. 2006, 86, 1408–1415. [Google Scholar] [CrossRef] [PubMed]
- De Mateo, S.; Gazquez, C.; Guimera, M.; Balasch, J.; Meistrich, M.L.; Luis Ballesca, J.; Oliva, R. Protamine 2 Precursors (Pre-P2), Protamine 1 to Protamine 2 Ratio (P1/P2), and Assisted Reproduction Outcome. Fertil. Steril. 2009, 91, 715–722. [Google Scholar] [CrossRef] [PubMed]
- Torregrosa, N.; Dominguez-Fandos, D.; Camejo, M.I.; Shirley, C.R.; Meistrich, M.L.; Ballesca, J.L.; Oliva, R. Protamine 2 Precursors, Protamine 1/Protamine 2 Ratio, DNA Integrity and Other Sperm Parameters in Infertile Patients. Hum. Reprod. 2006, 21, 2084–2089. [Google Scholar] [CrossRef] [PubMed]
- Rajender, S.; Avery, K.; Agarwal, A. Epigenetics, Spermatogenesis and Male Infertility. Mutat. Res. 2011, 727, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Sakkas, D.; Seli, E.; Manicardi, G.C.; Nijs, M.; Ombelet, W.; Bizzaro, D. The presence of abnormal spermatozoa in the ejaculate: Did apoptosis fail? Hum. Fertil. 2004, 7, 99–103. [Google Scholar] [CrossRef]
- Sakkas, D.; Mariethoz, E.; St John, J.C. Abnormal Sperm Parameters in Humans Are Indicative of an Abortive Apoptotic Mechanism Linked to the Fas-Mediated Pathway. Exp. Cell Res. 1999, 251, 350–355. [Google Scholar] [CrossRef]
- Lavranos, G.; Balla, M.; Tzortzopoulou, A.; Syriou, V.; Angelopoulou, R. Investigating ROS Sources in Male Infertility: A Common End for Numerous Pathways. Reprod. Toxicol. 2012, 34, 298–307. [Google Scholar] [CrossRef]
- Ohno, M.; Sakumi, K.; Fukumura, R.; Furuichi, M.; Iwasaki, Y.; Hokama, M.; Ikemura, T.; Tsuzuki, T.; Gondo, Y.; Nakabeppu, Y. 8-oxoguanine causes spontaneous de novo germline mutations in mice. Sci. Rep. 2014, 4, 4689. [Google Scholar] [CrossRef]
- Agarwal, A.; Durairajanayagam, D.; du Plessis, S.S. Utility of Antioxidants during Assisted Reproductive Techniques: An Evidence Based Review. Reprod. Biol. Endocrinol. 2014, 12, 112. [Google Scholar] [CrossRef] [PubMed]
- Aitken, R.; Smith, T.; Jobling, M.; Baker, M.; de Lulliis, N. Oxidative Stress and Male Reproductive Health. Asian J. Androl. 2014, 16, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Kothari, S.; Thompson, A.; Agarwal, A.; du Plessis, S.S. Free Radicals: Their Beneficial and Detrimental Effects on Sperm Function. Indian J. Exp. Biol. 2010, 48, 425–435. [Google Scholar] [PubMed]
- Kemal Duru, N.; Morshedi, M.; Oehninger, S. Effects of Hydrogen Peroxide on DNA and Plasma Membrane Integrity of Human Spermatozoa. Fertil. Steril. 2000, 74, 1200–1207. [Google Scholar] [CrossRef]
- Agarwal, A.; Virk, G.; Ong, C.; du Plessis, S.S. Effect of Oxidative Stress on Male Reproduction. World J. Men’s Health 2014, 32, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Aitken, R.; Gibb, Z.; Baker, M.; Drevet, J.; Gharagozloo, P. Causes and Consequences of Oxidative Stress in Spermatozoa. Reprod. Fertil. Dev. 2016, 28, 1–10. [Google Scholar] [CrossRef]
- Du Plessis, S.S.; Makker, K.; Desai, N.R.; Agarwal, A. Impact of Oxidative Stress on IVF. Expert Rev. Obstet. Gynecol. 2008, 3, 539–554. [Google Scholar] [CrossRef]
- O’Flaherty, C. The Enzymatic Antioxidant System of Human Spermatozoa. Adv. Androl. 2014, 2014, 1–15. [Google Scholar] [Green Version]
- Burden, H.P.; Holmes, C.H.; Persad, R.; Whittington, K. Prostasomes—Their Effects on Human Male Reproduction and Fertility. Hum. Reprod. Update 2006, 12, 283–292. [Google Scholar] [CrossRef]
- Krisher, R.L. In vivo and in vitro Environmental Effects on Mammalian Oocyte Quality. Annu. Rev. Anim. Biosci. 2013, 1, 393–417. [Google Scholar] [CrossRef]
- Agarwal, A.; Aziz, N.; Rizk, B. Studies on Women’s Health; Humana Press: Totowa, NJ, USA, 2013. [Google Scholar]
- Espey, L.L. Current Status of the Hypothesis that Mammalian Ovulation is Comparable to an Inflammatory Reaction. Biol. Reprod. 1994, 50, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Richards, J.S.; Russell, D.L.; Ochsner, S.; Espey, L.L. Ovulation: New Dimensions and New Regulators of the Inflammatory-Like Response. Annu. Rev. Physiol. 2002, 64, 69–92. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.; Gupta, S.; Sharma, R. Oxidative Stress and its Implications in Female Infertility—A Clinician’s Perspective. Reprod. BioMed. Online 2005, 11, 641–650. [Google Scholar] [CrossRef]
- Carbone, M.; Tatone, C.; Delle Monache, S.; Marci, R.; Caserta, D.; Colonna, R.; Amicarelli, F. Antioxidant Enzymatic Defences in Human Follicular Fluid: Characterization and Age-dependent Changes. Mol. Hum. Reprod. 2003, 9, 639–643. [Google Scholar] [CrossRef] [PubMed]
- Marco, M.; Emanuela, M.; Simona, C.; Luisa, P.; Alberto, R.; Paolo, R. Follicular Fluid Content and Oocyte Quality: From Single Biochemical Markers to Metabolomics. Reprod. Biol. Endocrinol. 2009, 7, 40. [Google Scholar]
- Guerin, P.; El Mouatassim, S.; Menezo, Y. Oxidative Stress and Protection Against Reactive Oxygen Species in the Pre-Implantation Embryo and its Surroundings. Hum. Reprod. Update 2001, 7, 175–189. [Google Scholar] [CrossRef] [PubMed]
- Jozwik, M.; Wolczynski, S.; Jozwik, M.; Szamatowicz, M. Oxidative Stress Markers in Preovulatory Follicular Fluid in Humans. Mol. Hum. Reprod. 1999, 5, 409–413. [Google Scholar] [CrossRef]
- Behrman, H.R.; Kodaman, P.H.; Preston, S.L.; Gao, S. Oxidative Stress and the Ovary. J. Soc. Gynecol. Investig. 2001, 8, 40–42. [Google Scholar]
- Szczepańska, M.; Koźlik, J.; Skrzypczak, J.; Mikołajczyk, M. Oxidative Stress may be a Piece in the Endometriosis Puzzle. Fertil. Steril. 2003, 79, 1288–1293. [Google Scholar] [CrossRef]
- McKelvey-Martin, V.; Green, M.; Schmezer, P.; Pool-Zobe, L.L.; De Meo, M.P.; Collins, A. The Single Cell Gel Electrophoresis Assay (Comet Assay): A European Review. Mutat. Res. 1993, 288, 47–63. [Google Scholar] [CrossRef]
- Fairbairn, D.W.; Olive, P.L.; O’Neill, K.L. The Comet Assay: A Comprehensive Review. Mutat. Res. 1995, 339, 37–59. [Google Scholar] [CrossRef]
- Cortés-Gutiérrez, E.I. Two-Tailed Comet Assay (2T-Comet): Simultaneous Detection of DNA Single and Double Strand Breaks. Methods Mol. Biol. 2017, 1560, 285–293. [Google Scholar]
- Laberge, R.; Boissonneault, G. On the Nature and Origin of DNA Strand Breaks in Elongating Spermatids. Biol. Reprod. 2005, 73, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Leduc, F.; Nkoma, G.B.; Boissonneault, G. Spermiogenesis and DNA Repair: A Possible Etiology of Human Infertility and Genetic Disorders. Syst. Biol. Reprod. Med. 2008, 54, 3–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fatehi, A.N.; Bevers, M.M.; Schoevers, E.; Roelen, B.A.J.; Colenbrander, B.; Gadella, B.M. DNA Damage in Bovine Sperm does Not Block Fertilization and Early Embryonic Development but Induces Apoptosis after the First Cleavages. J. Androl. 2006, 27, 176–188. [Google Scholar] [CrossRef]
- Gawecka, J.E.; Marh, J.; Ortega, M.; Yamauchi, Y.; Ward, M.A.; Ward, W.S. Mouse Zygotes Respond to Severe Sperm DNA Damage by Delaying Paternal DNA Replication and Embryonic Development. PLoS ONE 2013, 8, e56385. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, F.; Essers, J.; Kanaar, R.; Wyrobek, A. Disruption of Maternal DNA Repair Increases Sperm-Derived Chromosomal Aberrations. Proc. Natl. Acad. Sci. USA 2007, 104, 17725–17729. [Google Scholar] [CrossRef]
- Lynch, M. Rate, Molecular Spectrum, and Consequences of Human Mutation. Proc. Natl. Acad. Sci. USA 2010, 107, 961–968. [Google Scholar] [CrossRef]
- Ségurel, L.; Wyman, M.; Przeworski, M. Determinants of Mutation Rate Variation in the Human Germline. Annu. Rev. Genom. Hum. Genet. 2014, 15, 47–70. [Google Scholar] [CrossRef]
- Veltman, J.A.; Brunner, H.G. De novo Mutations in Human Genetic Disease. Nat. Rev. Genet. 2012, 13, 565–575. [Google Scholar] [CrossRef]
- Iossifov, I.; O’Roak, B.J.; Sanders, S.J.; Ronemus, M.; Krumm, N.; Levy, D.; Stessman, H.A.; Witherspoon, K.T.; Vives, L.; Patterson, K.E.; et al. The Contribution of De novo Coding Mutations to Autism Spectrum Disorder. Nature 2014, 515, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Lisenka, E.L.M.V.; Ligt, J.D.; Gilissen, C.; Janssen, I.; Steehouwer, M.; Vries, P.D.; Bart, V.L.; Arts, P.; Wieskamp, N.; Marisol, D.R.; et al. A De novo Paradigm for Mental Retardation. Nat. Genet. 2010, 42, 1109–1112. [Google Scholar]
- Homsy, J.; Zaidi, S.; Shen, Y.; Ware, J.; Samocha, K.; Karczewski, K.; Depalma, S.; Mckean, D.; Wakimoto, H.; Gorham, J.; et al. De novo Mutations in Congenital Heart Disease with Neurodevelopmental and Other Congenital Anomalies. Science 2015, 350, 1262–1266. [Google Scholar] [CrossRef] [PubMed]
- Pamphlett, R.; Morahan, J.M.; Yu, B. Using Case-Parent Trios to Look for Rare De novo Genetic Variants in Adult-Onset Neurodegenerative Diseases. J. Neurosci. Methods 2011, 197, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Geschwind, D.; Flint, J. Genetics and Genomics of Psychiatric Disease. Science 2015, 349, 1489–1494. [Google Scholar] [CrossRef]
- Goriely, A. Decoding Germline de novo Point Mutations. Nat. Genet. 2016, 48, 823–824. [Google Scholar] [CrossRef]
- Rahbari, R.; Wuster, A.; Sarah, J.L.; Robert, J.H.; Ludmil, B.A.; Turki, S.A.; Dominiczak, A.; Morris, A.; Porteous, D.; Smith, B.; et al. Timing, Rates and Spectra of Human Germline Mutation. Nat. Genet. 2016, 48, 126–133. [Google Scholar] [CrossRef]
- Gao, Z.; Wyman, M.; Sella, G.; Przeworski, M. Interpreting the Dependence of Mutation Rates on Age and Time. PLoS Biol. 2016, 14, e1002355. [Google Scholar] [CrossRef]
- Kong, A.; Frigge, M.L.; Masson, G.; Besenbacher, S.; Sulem, P.; Magnusson, G.; Gudjonsson, S.A.; Sigurdsson, A.; Jonasdottir, A.; Jonasdottir, A.; et al. Rate of De novo Mutations and the Importance of Father’s Age to Disease Risk. Nature 2012, 488, 471–475. [Google Scholar] [CrossRef]
- Jakob, M.G.; Wendy, S.W.W.; Pinelli, M.; Farrah, T.; Bodian, D.; Anna, B.S.; Glusman, G.; Lisenka, E.L.M.V.; Hoischen, A.; Jared, C.R.; et al. Parent-of-Origin-Specific Signatures of De novo Mutations. Nat. Genet. 2016, 48, 935–939. [Google Scholar]
- Acuna-Hidalgo, R.; Veltman, J.A.; Hoischen, A. New Insights into the Generation and Role of De novo Mutations in Health and Disease. Genome Biol. 2016, 17, 241. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, C.M.; Lupski, J.R. Mechanisms underlying structural variant formation in genomic disorders. Nat. Rev. Genet. 2016, 17, 224–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, I.M.; Shaw, C.A.; Stankiewicz, P.; Lupski, J.R. Somatic mosaicism: Implications for disease and transmission genetics. Trends Genet. 2015, 31, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Thomas, N.S.; Durkie, M.; Van Zyl, B.; Sanford, R.; Potts, G.; Youings, S.; Dennis, N.; Jacobs, P. Parental and chromosomal origin of unbalanced de novo structural chromosome abnormalities in man. Hum. Genet. 2006, 119, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Crow, F.J. The origins, patterns and implications of human spontaneous mutation. Nat. Rev. Genet. 2000, 1, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Nouspikel, T.; Hanawalt, P. DNA Repair in Terminally Differentiated Cells. DNA Repair 2002, 1, 59–75. [Google Scholar] [CrossRef]
- Skinner, A.M.; Turker, M.S. Oxidative Mutagenesis, Mismatch Repair, and Aging. Sci. Aging Knowl. Environ. 2005, 2005. [Google Scholar] [CrossRef]
- Karran, P. Microsatellite Instability and DNA Mismatch Repair in Human Cancer. Semin. Cancer Biol. 1996, 7, 15–24. [Google Scholar] [CrossRef]
- Wilson, D.M.; Bohr, V.A. The Mechanics of Base Excision Repair, and its Relationship to Aging and Disease. DNA Repair 2007, 6, 544–559. [Google Scholar] [CrossRef]
- Almeida, K.H.; Sobol, R.W. A Unified View of Base Excision Repair: Lesion-Dependent Protein Complexes Regulated by Post-Translational Modification. DNA Repair 2007, 6, 695–711. [Google Scholar] [CrossRef]
- Ceccaldi, R.; Rondinelli, B.; D’Andrea, A. Repair Pathway Choices and Consequences at the Double-Strand Break. Trends Cell Biol. 2016, 26, 52–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chengqi, Y.; Chuan, H. DNA repair by reversal of DNA damage. In Additional Perspectives on DNA Repair, Mutagenesis, and Other Responses to DNA Damage; Cold Spring Harbor Perspectives in Biology, Friedberg, E.C., Elledge, S.J., Lehmann, A.R., Lindahl, T., Muzi-Falconi, M., Eds.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2013. [Google Scholar]
- Baarends, W.; van der Laan, R.; Grootegoed, J. DNA Repair Mechanisms and Gametogenesis. Reproduction 2001, 121, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, E.A.; van der Vaart, A.; Barten, A.; Kal, H.B.; Chen, J.; Lou, Z.; Minter-Dykhouse, K.; Bartkova, J.; Bartek, J.; de Boer, P.; et al. Differences in DNA double strand breaks repair in male germ cell types: Lessons learned from a differential expression of Mdc1 and 53BP1. DNA Repair 2007, 6, 1243–1254. [Google Scholar] [CrossRef] [PubMed]
- Iyama, T.; Wilson, D.M., 3rd. DNA repair mechanisms in dividing and non-dividing cells. DNA Repair 2013, 12, 620–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gouraud, A.; Brazeau, M.; Grégoire, M.; Simard, O.; Massonneau, J.; Arguin, M.; Boissonneault, G. “Breaking News” from Spermatids. Basic Clin. Androl. 2013, 23, 11. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, E.; Scherthan, H.; de Rooij, D. DNA Double Strand Break Response and Limited Repair Capacity in Mouse Elongated Spermatids. Int. J. Mol. Sci. 2015, 16, 29923–29935. [Google Scholar] [CrossRef] [Green Version]
- Aitken, R.J.; De Iuliis, G.N. On the Possible Origins of DNA Damage in Human Spermatozoa. Mol. Hum. Reprod. 2010, 16, 3–13. [Google Scholar] [CrossRef]
- Ward, W.S.; Coffey, D.S. DNA Packaging and Organization in Mammalian Spermatozoa: Comparison with Somatic Cells. Biol. Reprod. 1991, 44, 569–574. [Google Scholar] [CrossRef]
- Smith, T.B.; Dun, M.D.; Smith, N.D.; Curry, B.J.; Connaughton, H.S.; Aitken, R.J. The Presence of a Truncated Base Excision Repair Pathway in Human Spermatozoa that is Mediated by OGG1. J. Cell. Sci. 2013, 126, 1488–1497. [Google Scholar] [CrossRef]
- Suganuma, R.; Yanagimachi, R.; Meistrich, M.L. Decline in Fertility of Mouse Sperm with Abnormal Chromatin during Epididymal Passage as Revealed by ICSI. Hum. Reprod. 2005, 20, 3101–3108. [Google Scholar] [CrossRef]
- Hajkova, P.; Erhardt, S.; Lane, N.; Haaf, T.; El-Maarri, O.; Reik, W.; Walter, J.; Surani, M.A. Epigenetic Reprogramming in Mouse Primordial Germ Cells. Mech. Dev. 2002, 117, 15–23. [Google Scholar] [CrossRef]
- Ashwood-Smith, M.J.; Edwards, R.G. Genetics and Human Conception: DNA Repair by Oocytes. Mol. Hum. Reprod. 1996, 2, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.E.; Pollack, S.E.; Pollard, J.W. Genetic Analysis of Chromosome Pairing, Recombination, and Cell Cycle Control during First Meiotic Prophase in Mammals. Endocr. Rev. 2006, 27, 398–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanoux, V.; Pairault, C.; Bakalska, M.; Habert, R.; Livera, G. Caspase-2 Involvement during Ionizing Radiation-Induced Oocyte Death in the Mouse Ovary. Cell Death Differ. 2007, 14, 671–681. [Google Scholar] [CrossRef] [PubMed]
- Mira, A. Why is Meiosis Arrested? J. Theor. Biol. 1998, 194, 275–287. [Google Scholar] [CrossRef] [PubMed]
- Stringer, J.M.; Winship, A.; Liew, S.H.; Hutt, K. The capacity of oocytes for DNA repair. Cell Mol. Life Sci. 2018, 75, 2777–2792. [Google Scholar] [CrossRef] [PubMed]
- Menezo, Y.J.; Russo, G.; Tosti, E.; Mouatassim, S.E.; Benkhalifa, M. Expression Profile of Genes Coding for DNA Repair in Human Oocytes using Pangenomic Microarrays, with a Special Focus on ROS Linked Decays. J. Assist. Reprod. Genet. 2007, 24, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Jaroudi, S.; Kakourou, G.; Cawood, S.; Doshi, A.; Ranieri, D.M.; Serhal, P.; Harper, J.C.; SenGupta, S.B. Expression profiling of DNA repair genes in human oocytes and blastocysts using microarrays. Hum. Reprod. 2009, 24, 2649–2655. [Google Scholar] [CrossRef] [Green Version]
- Zeng, F.; Baldwin, D.A.; Schultz, R.M. Transcript profiling during preimplantation mouse development. Dev. Biol. 2004, 272, 483–496. [Google Scholar] [CrossRef]
- Zheng, P.; Schramm, R.D.; Latham, K.E. Developmental regulation and in vitro culture effects on expression of DNA repair and cell cycle checkpoint control genes in rhesus monkey oocytes and embryos. Biol. Reprod. 2005, 72, 1359–1369. [Google Scholar] [CrossRef]
- Martin, J.H.; Bromfield, E.G.; Aitken, R.J.; Lord, T.; Nixon, B. Double Strand Break DNA Repair occurs via non-Homologous End-Joining in Mouse MII Oocytes. Sci. Rep. 2018, 8, 9685. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, D.; He, D.; Suo, S.; Xia, X.; He, X.; Han, J.-D.; Zheng, P. Transcriptome analyses of rhesus monkey preimplantation embryos reveal a reduced capacity for DNA double-strand break repair in primate oocytes and early embryos. GenomeRes 2005, 27, 567–579. [Google Scholar]
- Hamatani, T.; Falco, G.; Carter, M.G.; Akutsu, H.; Stagg, C.A.; Sharov, A.A.; Dudekula, D.B.; VanBuren, V.; Ko, M.S.H. Age-Associated Alteration of Gene Expression Patterns in Mouse Oocytes. Hum. Mol. Genet. 2004, 13, 2263–2278. [Google Scholar] [CrossRef] [PubMed]
- Van der Heijden, G.W.; Dieker, J.W.; Derijck, A.A.; Muller, S.; Berden, J.H.; Braat, D.D.; van der Vlag, J.; de Boer, P. Asymmetry in Histone H3 Variants and Lysine Methylation between Paternal and Maternal Chromatin of the Early Mouse Zygote. Mech. Dev. 2005, 122, 1008–1022. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, Y.; Tobari, I.; Yamagiwa, J.; Utsugi, T.; Okamoto, M.; Nakai, S. Dose-Response Relationship of γ-Ray-Induced Reciprocal Translocations at Low Doses in Spermatogonia of the Crab-Eating Monkey (Macaca fascicularis). Mutat. Res. 1985, 151, 121–127. [Google Scholar] [CrossRef]
- Moore, S.; Braude, P.; Bolton, V. Human Gene Expression First Occurs between the Four- and Eight-Cell Stages of Preimplantation Development. Nature 1988, 332, 459–461. [Google Scholar]
- Genescà, A.; Caballín, M.R.; Miró, R.; Benet, J.; Germà, J.R.; Egozcue, J. Repair of Human Sperm Chromosome Aberrations in the Hamster Egg. Hum. Genet. 1992, 89, 181–186. [Google Scholar] [CrossRef]
- Ahmadi, A.; Ng, S. Fertilizing Ability of DNA-damaged Spermatozoa. J. Exp. Zool. 1999, 284, 696–704. [Google Scholar] [CrossRef]
- Seli, E.; Gardner, D.K.; Schoolcraft, W.B.; Moffatt, O.; Sakkas, D. Extent of Nuclear DNA Damage in Ejaculated Spermatozoa Impacts on Blastocyst Development After in vitro Fertilization. Fertil. Steril. 2004, 82, 378–383. [Google Scholar] [CrossRef]
- Shoukir, Y.; Chardonnens, D.; Campana, A.; Sakkas, D. Blastocyst Development from Supernumerary Embryos After Intracytoplasmic Sperm Injection: A Paternal Influence? Hum. Reprod. 1998, 13, 1632–1637. [Google Scholar] [CrossRef]
- Dumoulin, J.C.M.; Coonen, E.; Bras, M.; Van Wissen, L.C.P.; Ignoul-Vanvuchelen, R.; Bergers-Jansen, J.M.; Derhaag, J.G.; Geraedts, J.P.M.; Evers, J.L.H. Comparison of in-Vitro Development of Embryos Originating from either Conventional in-Vitro Fertilization or Intracytoplasmic Sperm Injection. Hum. Reprod. 2000, 15, 402–409. [Google Scholar] [CrossRef] [PubMed]
- Borini, A.; Tarozzi, N.; Bizzaro, D.; Bonu, M.A.; Fava, L.; Flamigni, C.; Coticchio, G. Sperm DNA Fragmentation: Paternal Effect on Early Post-Implantation Embryo Development in ART. Hum. Reprod. 2006, 21, 2876–2881. [Google Scholar] [CrossRef] [PubMed]
- Lord, T.; Aitken, R.J. Fertilization Stimulates 8-Hydroxy-2′-Deoxyguanosine Repair and Antioxidant Activity to Prevent Mutagenesis in the Embryo. Dev. Biol. 2015, 406, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Essers, J.; van Steeg, H.; de Wit, J.; Swagemakers, S.M.; Vermeij, M.; Hoeijmakers, J.H.; Kanaar, R. Homologous and non-homologous recombination differentially affect DNA damage repair in mice. EMBO J. 2000, 19, 1703–1710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothkamm, K.; Krüger, I.; Thompson, L.H.; Löbrich, M. Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol. Cell Biol. 2003, 23, 5706–5715. [Google Scholar] [CrossRef]
- Derijck, A.; van der Heijden, G.; Giele, M.; Philippens, M.; de Boer, P. DNA Double-Strand Break Repair in Parental Chromatin of Mouse Zygotes, the First Cell Cycle as an Origin of de novo Mutation. Hum. Mol. Genet. 2008, 17, 1922–1937. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Rodríguez, A.; Gosálvez, J.; Agarwal, A.; Roy, R.; Johnston, S. DNA Damage and Repair in Human Reproductive Cells. Int. J. Mol. Sci. 2019, 20, 31. https://doi.org/10.3390/ijms20010031
García-Rodríguez A, Gosálvez J, Agarwal A, Roy R, Johnston S. DNA Damage and Repair in Human Reproductive Cells. International Journal of Molecular Sciences. 2019; 20(1):31. https://doi.org/10.3390/ijms20010031
Chicago/Turabian StyleGarcía-Rodríguez, Anaís, Jaime Gosálvez, Ashok Agarwal, Rosa Roy, and Stephen Johnston. 2019. "DNA Damage and Repair in Human Reproductive Cells" International Journal of Molecular Sciences 20, no. 1: 31. https://doi.org/10.3390/ijms20010031
APA StyleGarcía-Rodríguez, A., Gosálvez, J., Agarwal, A., Roy, R., & Johnston, S. (2019). DNA Damage and Repair in Human Reproductive Cells. International Journal of Molecular Sciences, 20(1), 31. https://doi.org/10.3390/ijms20010031