The Role of APOE and TREM2 in Alzheimer′s Disease—Current Understanding and Perspectives


Abstract
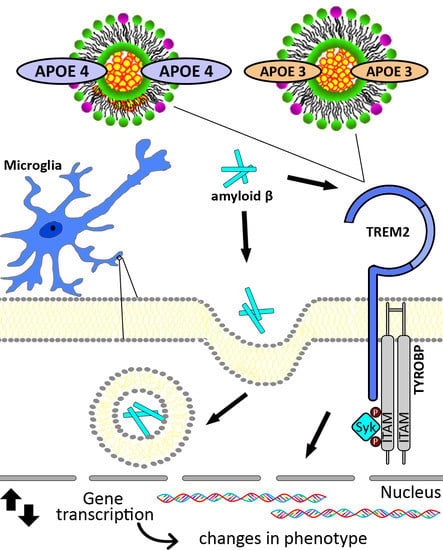
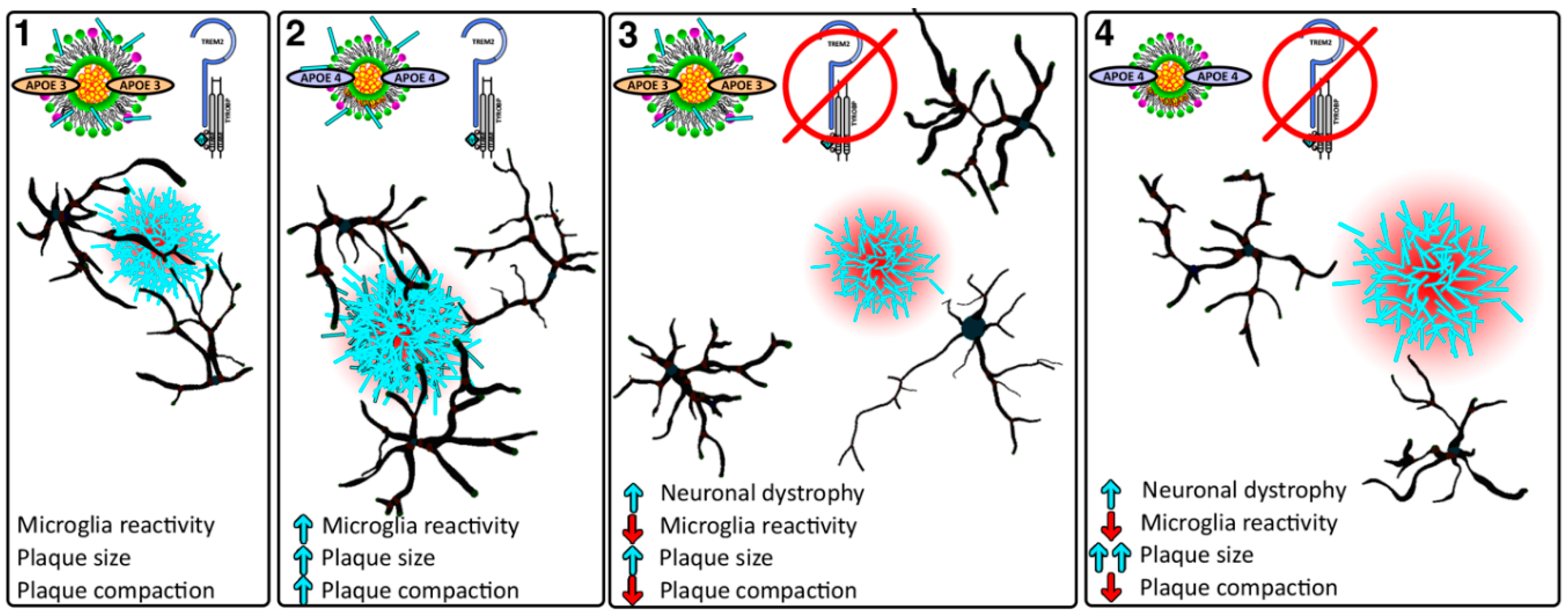
:
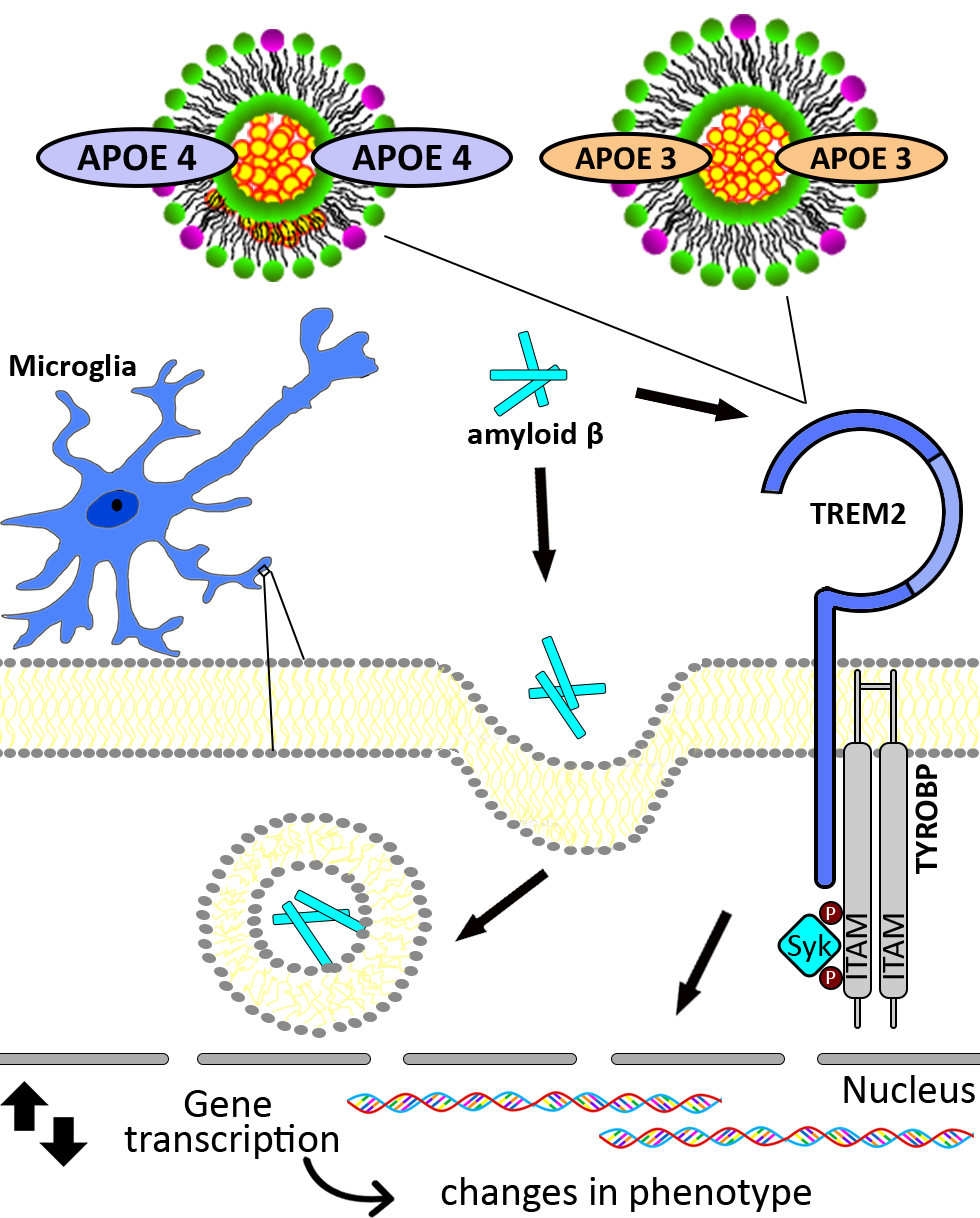{kind=link}
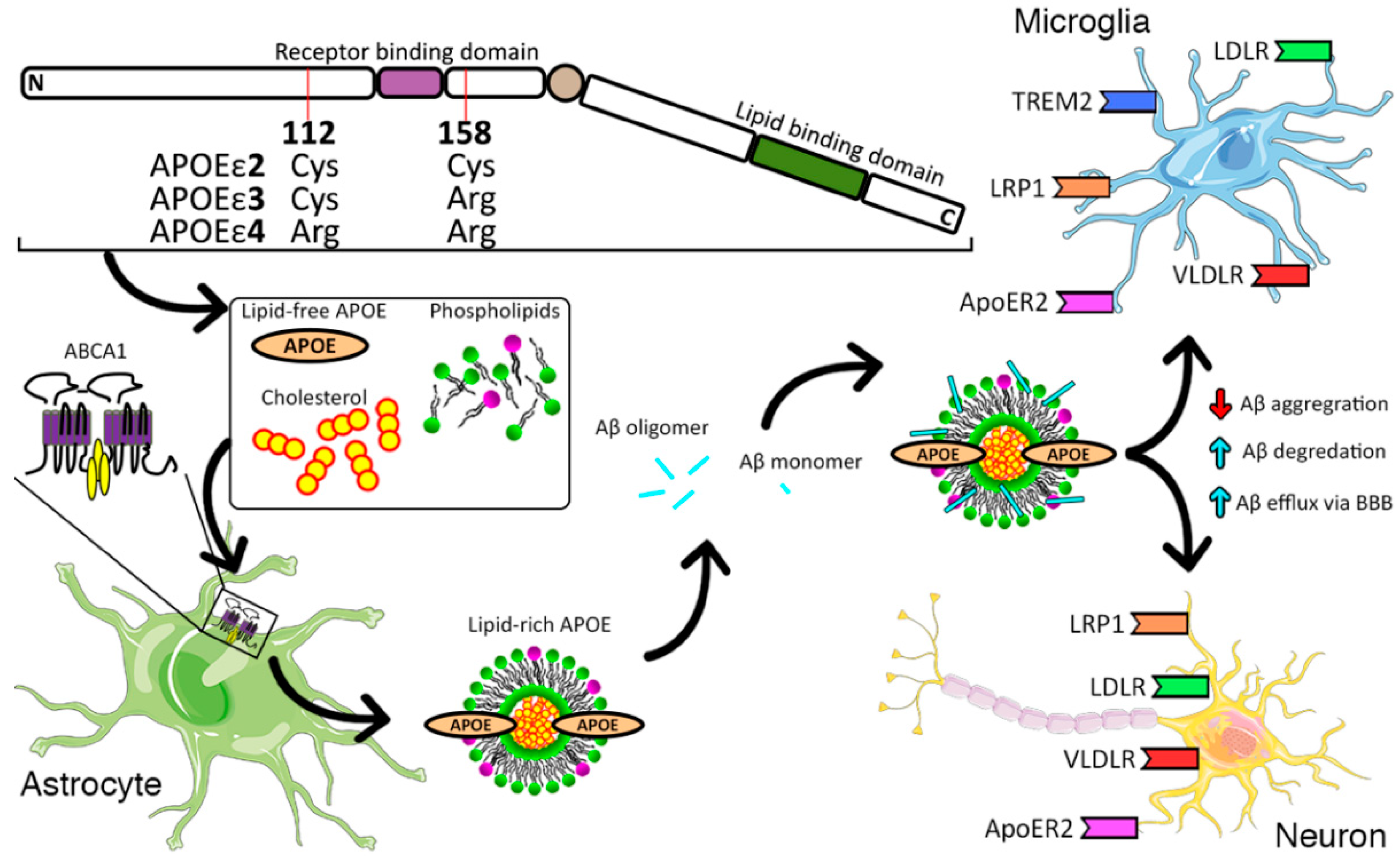{kind=link}
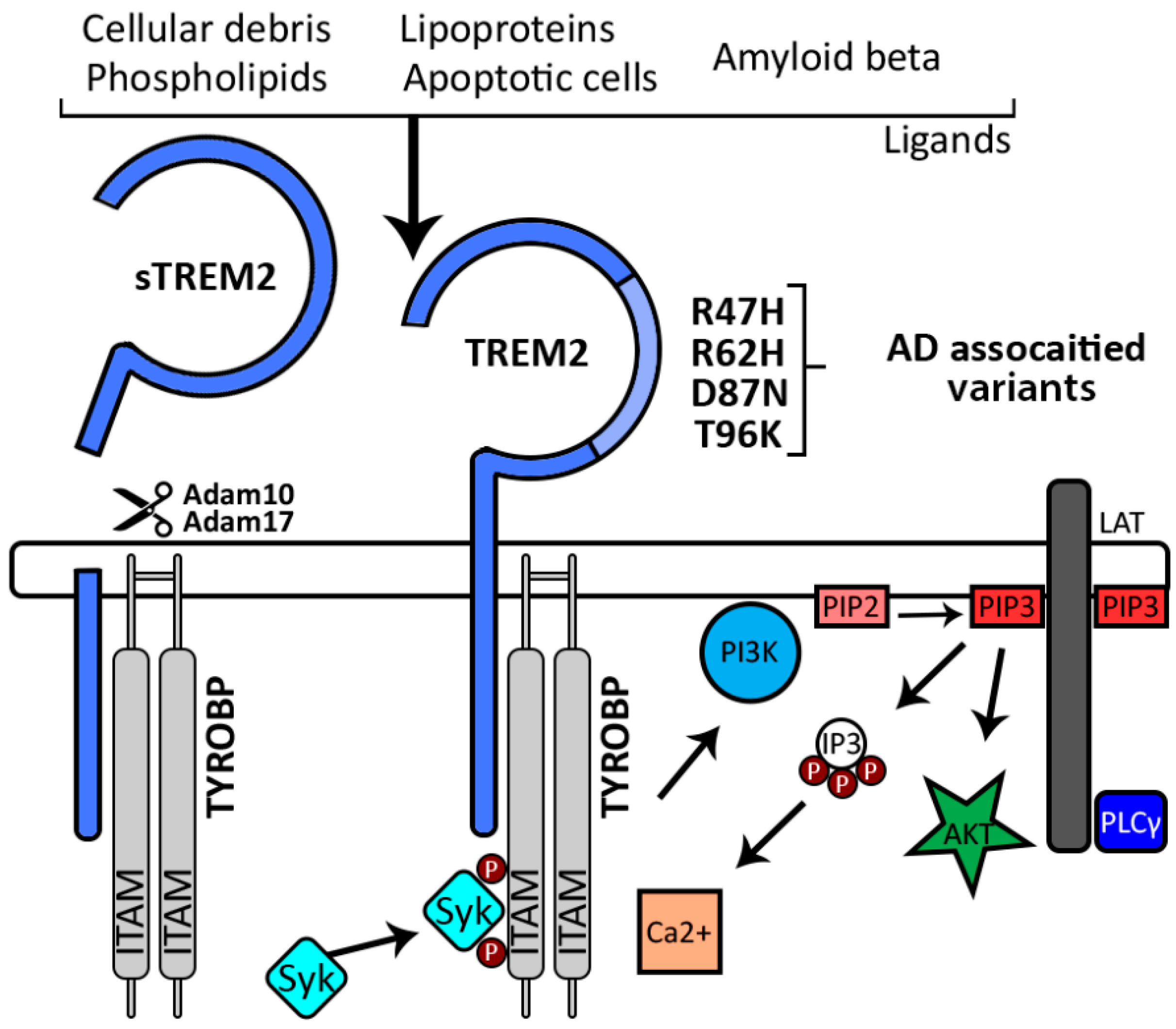{kind=link}
{kind=link}
1. Introduction
2. APOE
2.1. APOE Structure and Isoforms
2.2. APOE Receptors
2.3. APOE Function in the CNS
3. TREM2
3.1. TREM2 Structure and Expression
3.2. TREM2 Function
3.3. TREM2 Variants and Neurodegeneration
4. APOE, TREM2, and AD
4.1. APOE and AD
4.2. TREM2 and Alzheimer’s Disease
4.3. APOE, TREM2, and AD
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| APOE | Apolipoprotein E |
| LOAD | Late Onset AD |
| TREM2 | Triggering receptor expressed on myeloid cells 2 |
| Aβ | β-amyloid peptide |
| EOAD | Early-onset AD |
| APP | Amyloid precursor protein |
| PSEN1 | Presenilin 1 |
| PSEN2 | Presenilin 2 |
| GWAS | Genome-wide association studies |
| TYROBP | Tyro protein tyrosine kinase binding protein |
| R47H | Arginine to histidine at position 47 |
| LDL | Low-density lipoprotein |
| HDL | High-density lipoproteins |
| APOJ | Clusterin |
| ABCA1 | Adenosine triphosphate-binding cassette transporters A1 |
| ABCG1 | Adenosine triphosphate-binding cassette transporters G1 |
| Cys | Cysteine |
| Arg | Arginine |
| LDLR | Low-density lipoprotein receptor |
| LRP1 | LDLR-related receptor 1 |
| VLDLR | Very-low-density lipoprotein receptor |
| APOER2 | APOE receptor 2 |
| SFKs | Src family kinases |
| AKT | Protein kinase B |
| BBB | Blood–brain barrier |
| sTREM2 | Soluble TREM2 |
| CFS | Cerebral spinal fluid |
| NHD | Nasu–Hakola disease |
| FTD | Frontotemporal dementia |
| ALS | Amyotrophic lateral sclerosis |
| LPS | Lipopolysaccharide |
| ITAMs | Immunoreceptor tyrosine-based activation motifs |
| SYK | Spleen tyrosine kinase |
| PI3K | Phosphoinositide 3-kinase |
| PLCγ | Phospholipase Cγ |
| PIP3 | Phosphatidylinositol-3,4,5-trisphosphate |
| IP3 | Inositol trisphosphate |
| ASO | Anti-sense oligonucleotides |
| ADSP | Alzheimer’s Disease Sequencing Project |
References
- Crous-Bou, M.; Minguillon, C.; Gramunt, N.; Molinuevo, J.L. Alzheimer’s disease prevention: From risk factors to early intervention. Alzheimers Res. Ther. 2017, 9, 71. [Google Scholar] [CrossRef] [PubMed]
- Tanzi, R.E. The genetics of Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006296. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, R.J.; Gustafson, D.R.; Hardy, J. The genetic architecture of Alzheimer’s disease: Beyond APP, PSENs and APOE. Neurobiol. Aging 2012, 33, 437–456. [Google Scholar] [CrossRef]
- Campion, D.; Dumanchin, C.; Hannequin, D.; Dubois, B.; Belliard, S.; Puel, M.; Thomas-Anterion, C.; Michon, A.; Martin, C.; Charbonnier, F.; et al. Early-onset autosomal dominant Alzheimer disease: Prevalence, genetic heterogeneity, and mutation spectrum. Am. J. Hum. Genet. 1999, 65, 664–670. [Google Scholar] [CrossRef] [PubMed]
- Pimenova, A.A.; Raj, T.; Goate, A.M. Untangling Genetic Risk for Alzheimer’s Disease. Biol. Psychiatry 2018, 83, 300–310. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, T.; Stefansson, H.; Steinberg, S.; Jonsdottir, I.; Jonsson, P.V.; Snaedal, J.; Bjornsson, S.; Huttenlocher, J.; Levey, A.I.; Lah, J.J.; et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Karch, C.M.; Cruchaga, C.; Goate, A.M. Alzheimer’s disease genetics: From the bench to the clinic. Neuron 2014, 83, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.; Younkin, S.; et al. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef]
- Shi, Y.; Holtzman, D.M. Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight. Nat. Rev. Immunol. 2018. [Google Scholar] [CrossRef]
- Bu, G. Apolipoprotein E and its receptors in Alzheimer’s disease: Pathways, pathogenesis and therapy. Nat. Rev. Neurosci. 2009, 10, 333–344. [Google Scholar] [CrossRef]
- Liu, C.C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Castellano, J.M.; Kim, J.; Stewart, F.R.; Jiang, H.; DeMattos, R.B.; Patterson, B.W.; Fagan, A.M.; Morris, J.C.; Mawuenyega, K.G.; Cruchaga, C.; et al. Human apoE isoforms differentially regulate brain amyloid-beta peptide clearance. Sci. Transl. Med. 2011, 3, 89ra57. [Google Scholar] [CrossRef] [PubMed]
- Mahoney-Sanchez, L.; Belaidi, A.A.; Bush, A.I.; Ayton, S. The Complex Role of Apolipoprotein E in Alzheimer’s Disease: An Overview and Update. J. Mol. Neurosci. 2016, 60, 325–335. [Google Scholar] [CrossRef]
- Ulland, T.K.; Song, W.M.; Huang, S.C.; Ulrich, J.D.; Sergushichev, A.; Beatty, W.L.; Loboda, A.A.; Zhou, Y.; Cairns, N.J.; Kambal, A.; et al. TREM2 Maintains Microglial Metabolic Fitness in Alzheimer’s Disease. Cell 2017, 170, 649–663.e613. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cella, M.; Mallinson, K.; Ulrich, J.D.; Young, K.L.; Robinette, M.L.; Gilfillan, S.; Krishnan, G.M.; Sudhakar, S.; Zinselmeyer, B.H.; et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell 2015, 160, 1061–1071. [Google Scholar] [CrossRef] [PubMed]
- Poliani, P.L.; Wang, Y.; Fontana, E.; Robinette, M.L.; Yamanishi, Y.; Gilfillan, S.; Colonna, M. TREM2 sustains microglial expansion during aging and response to demyelination. J. Clin. Investig. 2015, 125, 2161–2170. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Rochford, C.D.; Neumann, H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J. Exp. Med. 2005, 201, 647–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, C.L.; Koike, M.; Spusta, S.C.; Niemi, E.C.; Yenari, M.; Nakamura, M.C.; Seaman, W.E. A role for TREM2 ligands in the phagocytosis of apoptotic neuronal cells by microglia. J. Neurochem. 2009, 109, 1144–1156. [Google Scholar] [CrossRef] [Green Version]
- Kleinberger, G.; Yamanishi, Y.; Suarez-Calvet, M.; Czirr, E.; Lohmann, E.; Cuyvers, E.; Struyfs, H.; Pettkus, N.; Wenninger-Weinzierl, A.; Mazaheri, F.; et al. TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci. Transl. Med. 2014, 6, 243ra86. [Google Scholar] [CrossRef] [PubMed]
- Mazaheri, F.; Snaidero, N.; Kleinberger, G.; Madore, C.; Daria, A.; Werner, G.; Krasemann, S.; Capell, A.; Trumbach, D.; Wurst, W.; et al. TREM2 deficiency impairs chemotaxis and microglial responses to neuronal injury. EMBO Rep. 2017, 18, 1186–1198. [Google Scholar] [CrossRef]
- Zheng, H.; Jia, L.; Liu, C.C.; Rong, Z.; Zhong, L.; Yang, L.; Chen, X.F.; Fryer, J.D.; Wang, X.; Zhang, Y.W.; et al. TREM2 Promotes Microglial Survival by Activating Wnt/beta-Catenin Pathway. J. Neurosci. 2017, 37, 1772–1784. [Google Scholar] [CrossRef]
- Atagi, Y.; Liu, C.C.; Painter, M.M.; Chen, X.F.; Verbeeck, C.; Zheng, H.; Li, X.; Rademakers, R.; Kang, S.S.; Xu, H.; et al. Apolipoprotein E Is a Ligand for Triggering Receptor Expressed on Myeloid Cells 2 (TREM2). J. Biol. Chem. 2015, 290, 26043–26050. [Google Scholar] [CrossRef] [Green Version]
- Bailey, C.C.; DeVaux, L.B.; Farzan, M. The Triggering Receptor Expressed on Myeloid Cells 2 Binds Apolipoprotein E. J. Biol. Chem. 2015, 290, 26033–26042. [Google Scholar] [CrossRef] [PubMed]
- Yeh, F.L.; Wang, Y.; Tom, I.; Gonzalez, L.C.; Sheng, M. TREM2 Binds to Apolipoproteins, Including APOE and CLU/APOJ, and Thereby Facilitates Uptake of Amyloid-Beta by Microglia. Neuron 2016, 91, 328–340. [Google Scholar] [CrossRef]
- Koldamova, R.; Fitz, N.F.; Lefterov, I. ATP-binding cassette transporter A1: From metabolism to neurodegeneration. Neurobiol. Dis. 2014, 72 Pt A, 13–21. [Google Scholar] [CrossRef] [Green Version]
- Koldamova, R.; Fitz, N.F.; Lefterov, I. The role of ATP-binding cassette transporter A1 in Alzheimer’s disease and neurodegeneration. Biochim. Biophys. Acta 2010, 1801, 824–830. [Google Scholar] [CrossRef] [PubMed]
- Nagata, K.O.; Nakada, C.; Kasai, R.S.; Kusumi, A.; Ueda, K. ABCA1 dimer-monomer interconversion during HDL generation revealed by single-molecule imaging. Proc. Natl. Acad. Sci. USA 2013, 110, 5034–5039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Liu, Q. Cholesterol metabolism and homeostasis in the brain. Protein Cell 2015, 6, 254–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lane-Donovan, C.; Wong, W.M.; Durakoglugil, M.S.; Wasser, C.R.; Jiang, S.; Xian, X.; Herz, J. Genetic Restoration of Plasma ApoE Improves Cognition and Partially Restores Synaptic Defects in ApoE-Deficient Mice. J. Neurosci. 2016, 36, 10141–10150. [Google Scholar] [CrossRef] [Green Version]
- Weisgraber, K.H.; Rall, S.C., Jr.; Mahley, R.W. Human E apoprotein heterogeneity. Cysteine-arginine interchanges in the amino acid sequence of the apo-E isoforms. J. Biol. Chem. 1981, 256, 9077–9083. [Google Scholar]
- Mahley, R.W.; Weisgraber, K.H.; Huang, Y. Apolipoprotein E: Structure determines function, from atherosclerosis to Alzheimer’s disease to AIDS. J. Lipid Res. 2009, 50, S183–S188. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.; Wardell, M.R.; Weisgraber, K.H.; Mahley, R.W.; Agard, D.A. Three-dimensional structure of the LDL receptor-binding domain of human apolipoprotein E. Science 1991, 252, 1817–1822. [Google Scholar] [CrossRef] [PubMed]
- Weisgraber, K.H.; Shinto, L.H. Identification of the disulfide-linked homodimer of apolipoprotein E3 in plasma. Impact on receptor binding activity. J. Biol. Chem. 1991, 266, 12029–12034. [Google Scholar] [PubMed]
- Weisgraber, K.H. Apolipoprotein E distribution among human plasma lipoproteins: Role of the cysteine-arginine interchange at residue 112. J. Lipid Res. 1990, 31, 1503–1511. [Google Scholar] [PubMed]
- Dong, L.M.; Weisgraber, K.H. Human apolipoprotein E4 domain interaction. Arginine 61 and glutamic acid 255 interact to direct the preference for very low density lipoproteins. J. Biol. Chem. 1996, 271, 19053–19057. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, M.; Tachibana, M.; Kanekiyo, T.; Bu, G. Role of LRP1 in the pathogenesis of Alzheimer’s disease: Evidence from clinical and preclinical studies. J. Lipid Res. 2017, 58, 1267–1281. [Google Scholar] [CrossRef]
- Kanekiyo, T.; Bu, G. The low-density lipoprotein receptor-related protein 1 and amyloid-beta clearance in Alzheimer’s disease. Front. Aging Neurosci. 2014, 6, 93. [Google Scholar] [CrossRef]
- Lane-Donovan, C.; Philips, G.T.; Herz, J. More than cholesterol transporters: Lipoprotein receptors in CNS function and neurodegeneration. Neuron 2014, 83, 771–787. [Google Scholar] [CrossRef]
- Jeon, H.; Blacklow, S.C. Structure and physiologic function of the low-density lipoprotein receptor. Annu. Rev. Biochem. 2005, 74, 535–562. [Google Scholar] [CrossRef]
- Herz, J.; Clouthier, D.E.; Hammer, R.E. LDL receptor-related protein internalizes and degrades uPA-PAI-1 complexes and is essential for embryo implantation. Cell 1992, 71, 411–421. [Google Scholar] [CrossRef]
- May, P.; Rohlmann, A.; Bock, H.H.; Zurhove, K.; Marth, J.D.; Schomburg, E.D.; Noebels, J.L.; Beffert, U.; Sweatt, J.D.; Weeber, E.J.; et al. Neuronal LRP1 functionally associates with postsynaptic proteins and is required for normal motor function in mice. Mol. Cell. Biol. 2004, 24, 8872–8883. [Google Scholar] [CrossRef] [PubMed]
- Fryer, J.D.; Demattos, R.B.; McCormick, L.M.; O’Dell, M.A.; Spinner, M.L.; Bales, K.R.; Paul, S.M.; Sullivan, P.M.; Parsadanian, M.; Bu, G.; et al. The low density lipoprotein receptor regulates the level of central nervous system human and murine apolipoprotein E but does not modify amyloid plaque pathology in PDAPP mice. J. Biol. Chem. 2005, 280, 25754–25759. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zerbinatti, C.V.; Zhang, J.; Hoe, H.S.; Wang, B.; Cole, S.L.; Herz, J.; Muglia, L.; Bu, G. Amyloid precursor protein regulates brain apolipoprotein E and cholesterol metabolism through lipoprotein receptor LRP1. Neuron 2007, 56, 66–78. [Google Scholar] [CrossRef] [PubMed]
- Trommsdorff, M.; Gotthardt, M.; Hiesberger, T.; Shelton, J.; Stockinger, W.; Nimpf, J.; Hammer, R.E.; Richardson, J.A.; Herz, J. Reeler/Disabled-like disruption of neuronal migration in knockout mice lacking the VLDL receptor and ApoE receptor 2. Cell 1999, 97, 689–701. [Google Scholar] [CrossRef]
- Zhang, G.; Assadi, A.H.; McNeil, R.S.; Beffert, U.; Wynshaw-Boris, A.; Herz, J.; Clark, G.D.; D’Arcangelo, G. The Pafah1b complex interacts with the reelin receptor VLDLR. PLoS ONE 2007, 2, e252. [Google Scholar] [CrossRef] [PubMed]
- Hiesberger, T.; Trommsdorff, M.; Howell, B.W.; Goffinet, A.; Mumby, M.C.; Cooper, J.A.; Herz, J. Direct binding of Reelin to VLDL receptor and ApoE receptor 2 induces tyrosine phosphorylation of disabled-1 and modulates tau phosphorylation. Neuron 1999, 24, 481–489. [Google Scholar] [CrossRef]
- Kowal, R.C.; Herz, J.; Weisgraber, K.H.; Mahley, R.W.; Brown, M.S.; Goldstein, J.L. Opposing effects of apolipoproteins E and C on lipoprotein binding to low density lipoprotein receptor-related protein. J. Biol. Chem. 1990, 265, 10771–10779. [Google Scholar]
- Ruiz, J.; Kouiavskaia, D.; Migliorini, M.; Robinson, S.; Saenko, E.L.; Gorlatova, N.; Li, D.; Lawrence, D.; Hyman, B.T.; Weisgraber, K.H.; et al. The apoE isoform binding properties of the VLDL receptor reveal marked differences from LRP and the LDL receptor. J. Lipid Res. 2005, 46, 1721–1731. [Google Scholar] [CrossRef] [Green Version]
- Bjorkhem, I.; Meaney, S. Brain cholesterol: Long secret life behind a barrier. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 806–815. [Google Scholar] [CrossRef]
- Mauch, D.H.; Nagler, K.; Schumacher, S.; Goritz, C.; Muller, E.C.; Otto, A.; Pfrieger, F.W. CNS synaptogenesis promoted by glia-derived cholesterol. Science 2001, 294, 1354–1357. [Google Scholar] [CrossRef]
- Fitz, N.F.; Tapias, V.; Cronican, A.A.; Castranio, E.L.; Saleem, M.; Carter, A.Y.; Lefterova, M.; Lefterov, I.; Koldamova, R. Opposing effects of Apoe/Apoa1 double deletion on amyloid-beta pathology and cognitive performance in APP mice. Brain 2015, 138, 3699–3715. [Google Scholar] [CrossRef] [PubMed]
- Koffie, R.M.; Hyman, B.T.; Spires-Jones, T.L. Alzheimer’s disease: Synapses gone cold. Mol. Neurodegener. 2011, 6, 63. [Google Scholar] [CrossRef] [PubMed]
- Sheng, M.; Sabatini, B.L.; Sudhof, T.C. Synapses and Alzheimer’s disease. Cold Spring Harb. Perspect. Biol. 2012, 4, a005777. [Google Scholar] [CrossRef] [PubMed]
- Azarnia Tehran, D.; Kuijpers, M.; Haucke, V. Presynaptic endocytic factors in autophagy and neurodegeneration. Curr. Opin. Neurobiol. 2018, 48, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A. Amyloid precursor protein and endosomal-lysosomal dysfunction in Alzheimer’s disease: Inseparable partners in a multifactorial disease. FASEB J. 2017, 31, 2729–2743. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Gong, Y.; Gan, W.; Beach, T.; Holtzman, D.M.; Wisniewski, T. Apolipoprotein E isoform-specific regulation of dendritic spine morphology in apolipoprotein E transgenic mice and Alzheimer’s disease patients. Neuroscience 2003, 122, 305–315. [Google Scholar] [CrossRef]
- Love, S.; Siew, L.K.; Dawbarn, D.; Wilcock, G.K.; Ben-Shlomo, Y.; Allen, S.J. Premorbid effects of APOE on synaptic proteins in human temporal neocortex. Neurobiol. Aging 2006, 27, 797–803. [Google Scholar] [CrossRef]
- Zhu, Y.; Nwabuisi-Heath, E.; Dumanis, S.B.; Tai, L.M.; Yu, C.; Rebeck, G.W.; LaDu, M.J. APOE genotype alters glial activation and loss of synaptic markers in mice. Glia 2012, 60, 559–569. [Google Scholar] [CrossRef] [Green Version]
- Yong, S.M.; Lim, M.L.; Low, C.M.; Wong, B.S. Reduced neuronal signaling in the ageing apolipoprotein-E4 targeted replacement female mice. Sci. Rep. 2014, 4, 6580. [Google Scholar] [CrossRef] [Green Version]
- Mounier, A.; Georgiev, D.; Nam, K.N.; Fitz, N.F.; Castranio, E.; Wolfe, C.; Cronican, A.A.; Schug, J.; Lefterov, I.; Koldamova, R. Bexarotene-Activated Retinoid X Receptors Regulate Neuronal Differentiation and Dendritic Complexity. J. Neurosci. 2015, 35, 11862–11876. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Durakoglugil, M.S.; Xian, X.; Herz, J. ApoE4 reduces glutamate receptor function and synaptic plasticity by selectively impairing ApoE receptor recycling. Proc. Natl. Acad. Sci. USA 2010, 107, 12011–12016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumanis, S.B.; Tesoriero, J.A.; Babus, L.W.; Nguyen, M.T.; Trotter, J.H.; Ladu, M.J.; Weeber, E.J.; Turner, R.S.; Xu, B.; Rebeck, G.W.; et al. ApoE4 decreases spine density and dendritic complexity in cortical neurons in vivo. J. Neurosci. 2009, 29, 15317–15322. [Google Scholar] [CrossRef] [PubMed]
- Wisdom, N.M.; Callahan, J.L.; Hawkins, K.A. The effects of apolipoprotein E on non-impaired cognitive functioning: A meta-analysis. Neurobiol. Aging 2011, 32, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Small, B.J.; Rosnick, C.B.; Fratiglioni, L.; Backman, L. Apolipoprotein E and cognitive performance: A meta-analysis. Psychol. Aging 2004, 19, 592–600. [Google Scholar] [CrossRef] [PubMed]
- O’Donoghue, M.C.; Murphy, S.E.; Zamboni, G.; Nobre, A.C.; Mackay, C.E. APOE genotype and cognition in healthy individuals at risk of Alzheimer’s disease: A review. Cortex 2018, 104, 103–123. [Google Scholar] [CrossRef] [PubMed]
- Colonna, M.; Wang, Y. TREM2 variants: New keys to decipher Alzheimer disease pathogenesis. Nat. Rev. Neurosci. 2016, 17, 201–207. [Google Scholar] [CrossRef]
- Paloneva, J.; Manninen, T.; Christman, G.; Hovanes, K.; Mandelin, J.; Adolfsson, R.; Bianchin, M.; Bird, T.; Miranda, R.; Salmaggi, A.; et al. Mutations in two genes encoding different subunits of a receptor signaling complex result in an identical disease phenotype. Am. J. Hum. Genet. 2002, 71, 656–662. [Google Scholar] [CrossRef]
- Guerreiro, R.J.; Lohmann, E.; Bras, J.M.; Gibbs, J.R.; Rohrer, J.D.; Gurunlian, N.; Dursun, B.; Bilgic, B.; Hanagasi, H.; Gurvit, H.; et al. Using exome sequencing to reveal mutations in TREM2 presenting as a frontotemporal dementia-like syndrome without bone involvement. JAMA Neurol. 2013, 70, 78–84. [Google Scholar] [CrossRef]
- Klesney-Tait, J.; Turnbull, I.R.; Colonna, M. The TREM receptor family and signal integration. Nat. Immunol. 2006, 7, 1266–1273. [Google Scholar] [CrossRef]
- Painter, M.M.; Atagi, Y.; Liu, C.C.; Rademakers, R.; Xu, H.; Fryer, J.D.; Bu, G. TREM2 in CNS homeostasis and neurodegenerative disease. Mol. Neurodegener. 2015, 10, 43. [Google Scholar] [CrossRef]
- Wunderlich, P.; Glebov, K.; Kemmerling, N.; Tien, N.T.; Neumann, H.; Walter, J. Sequential proteolytic processing of the triggering receptor expressed on myeloid cells-2 (TREM2) protein by ectodomain shedding and gamma-secretase-dependent intramembranous cleavage. J. Biol. Chem. 2013, 288, 33027–33036. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Cao, B.; Zhao, Y.; Huang, H.; McIntyre, R.S.; Rosenblat, J.D.; Zhou, H. Soluble TREM2 changes during the clinical course of Alzheimer’s disease: A meta-analysis. Neurosci. Lett. 2018. [Google Scholar] [CrossRef] [PubMed]
- Daws, M.R.; Sullam, P.M.; Niemi, E.C.; Chen, T.T.; Tchao, N.K.; Seaman, W.E. Pattern recognition by TREM-2: Binding of anionic ligands. J. Immunol. 2003, 171, 594–599. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wu, X.; Li, X.; Jiang, L.L.; Gui, X.; Liu, Y.; Sun, Y.; Zhu, B.; Pina-Crespo, J.C.; Zhang, M.; et al. TREM2 Is a Receptor for beta-Amyloid that Mediates Microglial Function. Neuron 2018, 97, 1023–1031.e1027. [Google Scholar] [CrossRef] [PubMed]
- Peng, Q.; Malhotra, S.; Torchia, J.A.; Kerr, W.G.; Coggeshall, K.M.; Humphrey, M.B. TREM2- and DAP12-dependent activation of PI3K requires DAP10 and is inhibited by SHIP1. Sci. Signal. 2010, 3, ra38. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Chen, X.F.; Wang, T.; Wang, Z.; Liao, C.; Wang, Z.; Huang, R.; Wang, D.; Li, X.; Wu, L.; et al. Soluble TREM2 induces inflammatory responses and enhances microglial survival. J. Exp. Med. 2017, 214, 597–607. [Google Scholar] [CrossRef] [Green Version]
- Wu, K.; Byers, D.E.; Jin, X.; Agapov, E.; Alexander-Brett, J.; Patel, A.C.; Cella, M.; Gilfilan, S.; Colonna, M.; Kober, D.L.; et al. TREM-2 promotes macrophage survival and lung disease after respiratory viral infection. J. Exp. Med. 2015, 212, 681–697. [Google Scholar] [CrossRef] [Green Version]
- Xiang, X.; Werner, G.; Bohrmann, B.; Liesz, A.; Mazaheri, F.; Capell, A.; Feederle, R.; Knuesel, I.; Kleinberger, G.; Haass, C. TREM2 deficiency reduces the efficacy of immunotherapeutic amyloid clearance. EMBO Mol. Med. 2016, 8, 992–1004. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.; Liu, C.C.; Atagi, Y.; Chen, X.F.; Jia, L.; Yang, L.; He, W.; Zhang, X.; Kang, S.S.; Rosenberry, T.L.; et al. Opposing roles of the triggering receptor expressed on myeloid cells 2 and triggering receptor expressed on myeloid cells-like transcript 2 in microglia activation. Neurobiol. Aging 2016, 42, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Saber, M.; Kokiko-Cochran, O.; Puntambekar, S.S.; Lathia, J.D.; Lamb, B.T. Triggering Receptor Expressed on Myeloid Cells 2 Deficiency Alters Acute Macrophage Distribution and Improves Recovery after Traumatic Brain Injury. J. Neurotrauma 2017, 34, 423–435. [Google Scholar] [CrossRef]
- Carbajosa, G.; Malki, K.; Lawless, N.; Wang, H.; Ryder, J.W.; Wozniak, E.; Wood, K.; Mein, C.A.; Dobson, R.J.B.; Collier, D.A.; et al. Loss of Trem2 in microglia leads to widespread disruption of cell coexpression networks in mouse brain. Neurobiol. Aging 2018, 69, 151–166. [Google Scholar] [CrossRef] [PubMed]
- Chitu, V.; Stanley, E.R. Colony-stimulating factor-1 in immunity and inflammation. Curr. Opin. Immunol. 2006, 18, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Mitrasinovic, O.M.; Vincent, V.A.; Simsek, D.; Murphy, G.M., Jr. Macrophage colony stimulating factor promotes phagocytosis by murine microglia. Neurosci. Lett. 2003, 344, 185–188. [Google Scholar] [CrossRef]
- Otero, K.; Turnbull, I.R.; Poliani, P.L.; Vermi, W.; Cerutti, E.; Aoshi, T.; Tassi, I.; Takai, T.; Stanley, S.L.; Miller, M.; et al. Macrophage colony-stimulating factor induces the proliferation and survival of macrophages via a pathway involving DAP12 and beta-catenin. Nat. Immunol. 2009, 10, 734–743. [Google Scholar] [CrossRef] [PubMed]
- Cuyvers, E.; Bettens, K.; Philtjens, S.; Van Langenhove, T.; Gijselinck, I.; van der Zee, J.; Engelborghs, S.; Vandenbulcke, M.; Van Dongen, J.; Geerts, N.; et al. Investigating the role of rare heterozygous TREM2 variants in Alzheimer’s disease and frontotemporal dementia. Neurobiol. Aging 2014, 35, 726.e11–726.e19. [Google Scholar] [CrossRef] [PubMed]
- Klunemann, H.H.; Ridha, B.H.; Magy, L.; Wherrett, J.R.; Hemelsoet, D.M.; Keen, R.W.; De Bleecker, J.L.; Rossor, M.N.; Marienhagen, J.; Klein, H.E.; et al. The genetic causes of basal ganglia calcification, dementia, and bone cysts: DAP12 and TREM2. Neurology 2005, 64, 1502–1507. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, A.; Kakita, A.; Yoshida, K.; Konno, T.; Ikeuchi, T.; Hayashi, S.; Matsuo, H.; Shioda, K. Variable expression of microglial DAP12 and TREM2 genes in Nasu-Hakola disease. Neurogenetics 2015, 16, 265–276. [Google Scholar] [CrossRef]
- Satoh, J.; Tabunoki, H.; Ishida, T.; Yagishita, S.; Jinnai, K.; Futamura, N.; Kobayashi, M.; Toyoshima, I.; Yoshioka, T.; Enomoto, K.; et al. Immunohistochemical characterization of microglia in Nasu-Hakola disease brains. Neuropathol. Off. J. Jpn. Soc. Neuropathol. 2011, 31, 363–375. [Google Scholar] [CrossRef]
- Giraldo, M.; Lopera, F.; Siniard, A.L.; Corneveaux, J.J.; Schrauwen, I.; Carvajal, J.; Munoz, C.; Ramirez-Restrepo, M.; Gaiteri, C.; Myers, A.J.; et al. Variants in triggering receptor expressed on myeloid cells 2 are associated with both behavioral variant frontotemporal lobar degeneration and Alzheimer’s disease. Neurobiol. Aging 2013, 34, 2077.e11-8. [Google Scholar] [CrossRef]
- Le Ber, I.; De Septenville, A.; Guerreiro, R.; Bras, J.; Camuzat, A.; Caroppo, P.; Lattante, S.; Couarch, P.; Kabashi, E.; Bouya-Ahmed, K.; et al. Homozygous TREM2 mutation in a family with atypical frontotemporal dementia. Neurobiol. Aging 2014, 35, 2419.e2423–2419.e2425. [Google Scholar] [CrossRef]
- Paloneva, J.; Mandelin, J.; Kiialainen, A.; Bohling, T.; Prudlo, J.; Hakola, P.; Haltia, M.; Konttinen, Y.T.; Peltonen, L. DAP12/TREM2 deficiency results in impaired osteoclast differentiation and osteoporotic features. J. Exp. Med. 2003, 198, 669–675. [Google Scholar] [CrossRef] [PubMed]
- Soragna, D.; Papi, L.; Ratti, M.T.; Sestini, R.; Tupler, R.; Montalbetti, L. An Italian family affected by Nasu-Hakola disease with a novel genetic mutation in the TREM2 gene. J. Neurol. Neurosurg. Psychiatry 2003, 74, 825–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Numasawa, Y.; Yamaura, C.; Ishihara, S.; Shintani, S.; Yamazaki, M.; Tabunoki, H.; Satoh, J.I. Nasu-Hakola disease with a splicing mutation of TREM2 in a Japanese family. Eur. J. Neurol. 2011, 18, 1179–1183. [Google Scholar] [CrossRef] [PubMed]
- Chouery, E.; Delague, V.; Bergougnoux, A.; Koussa, S.; Serre, J.L.; Megarbane, A. Mutations in TREM2 lead to pure early-onset dementia without bone cysts. Hum. Mutat. 2008, 29, E194–E204. [Google Scholar] [CrossRef] [PubMed]
- Thelen, M.; Razquin, C.; Hernandez, I.; Gorostidi, A.; Sanchez-Valle, R.; Ortega-Cubero, S.; Wolfsgruber, S.; Drichel, D.; Fliessbach, K.; Duenkel, T.; et al. Investigation of the role of rare TREM2 variants in frontotemporal dementia subtypes. Neurobiol. Aging 2014, 35, 2657.e2613–2657.e2619. [Google Scholar] [CrossRef] [PubMed]
- Rayaprolu, S.; Mullen, B.; Baker, M.; Lynch, T.; Finger, E.; Seeley, W.W.; Hatanpaa, K.J.; Lomen-Hoerth, C.; Kertesz, A.; Bigio, E.H.; et al. TREM2 in neurodegeneration: Evidence for association of the p.R47H variant with frontotemporal dementia and Parkinson’s disease. Mol. Neurodegener. 2013, 8, 19. [Google Scholar] [CrossRef] [PubMed]
- Cady, J.; Koval, E.D.; Benitez, B.A.; Zaidman, C.; Jockel-Balsarotti, J.; Allred, P.; Baloh, R.H.; Ravits, J.; Simpson, E.; Appel, S.H.; et al. TREM2 variant p.R47H as a risk factor for sporadic amyotrophic lateral sclerosis. JAMA Neurol. 2014, 71, 449–453. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Chen, Y.; Wei, Q.; Guo, X.; Cao, B.; Ou, R.; Zhao, B.; Shang, H.F. Assessment of TREM2 rs75932628 association with amyotrophic lateral sclerosis in a Chinese population. J. Neurol. Sci. 2015, 355, 193–195. [Google Scholar] [CrossRef]
- Walton, R.L.; Soto-Ortolaza, A.I.; Murray, M.E.; Lorenzo-Betancor, O.; Ogaki, K.; Heckman, M.G.; Rayaprolu, S.; Rademakers, R.; Ertekin-Taner, N.; Uitti, R.J.; et al. TREM2 p.R47H substitution is not associated with dementia with Lewy bodies. Neurol. Genet. 2016, 2, e85. [Google Scholar] [CrossRef]
- Carrasquillo, M.M.; Barber, I.; Lincoln, S.J.; Murray, M.E.; Camsari, G.B.; Khan, Q.; Nguyen, T.; Ma, L.; Bisceglio, G.D.; Crook, J.E.; et al. Evaluating pathogenic dementia variants in posterior cortical atrophy. Neurobiol. Aging 2016, 37, 38–44. [Google Scholar] [CrossRef] [Green Version]
- Slattery, C.F.; Beck, J.A.; Harper, L.; Adamson, G.; Abdi, Z.; Uphill, J.; Campbell, T.; Druyeh, R.; Mahoney, C.J.; Rohrer, J.D.; et al. R47H TREM2 variant increases risk of typical early-onset Alzheimer’s disease but not of prion or frontotemporal dementia. Alzheimers Dement. 2014, 10, 602–608.e604. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, X.; Guo, X.; Song, W.; Cao, B.; Wei, Q.; Ou, R.; Zhao, B.; Shang, H.F. Assessment of TREM2 rs75932628 association with Parkinson’s disease and multiple system atrophy in a Chinese population. Neurol. Sci. 2015, 36, 1903–1906. [Google Scholar] [CrossRef] [PubMed]
- Cruchaga, C.; Kauwe, J.S.; Harari, O.; Jin, S.C.; Cai, Y.; Karch, C.M.; Benitez, B.A.; Jeng, A.T.; Skorupa, T.; Carrell, D.; et al. GWAS of cerebrospinal fluid tau levels identifies risk variants for Alzheimer’s disease. Neuron 2013, 78, 256–268. [Google Scholar] [CrossRef] [PubMed]
- Hooli, B.V.; Lill, C.M.; Mullin, K.; Qiao, D.; Lange, C.; Bertram, L.; Tanzi, R.E. PLD3 gene variants and Alzheimer’s disease. Nature 2015, 520, E7–E8. [Google Scholar] [CrossRef] [PubMed]
- Lill, C.M.; Rengmark, A.; Pihlstrom, L.; Fogh, I.; Shatunov, A.; Sleiman, P.M.; Wang, L.S.; Liu, T.; Lassen, C.F.; Meissner, E.; et al. The role of TREM2 R47H as a risk factor for Alzheimer’s disease, frontotemporal lobar degeneration, amyotrophic lateral sclerosis, and Parkinson’s disease. Alzheimers Dement. 2015, 11, 1407–1416. [Google Scholar] [CrossRef] [PubMed]
- Sims, R.; van der Lee, S.J.; Naj, A.C.; Bellenguez, C.; Badarinarayan, N.; Jakobsdottir, J.; Kunkle, B.W.; Boland, A.; Raybould, R.; Bis, J.C.; et al. Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat. Genet. 2017, 49, 1373–1384. [Google Scholar] [CrossRef]
- Jin, S.C.; Carrasquillo, M.M.; Benitez, B.A.; Skorupa, T.; Carrell, D.; Patel, D.; Lincoln, S.; Krishnan, S.; Kachadoorian, M.; Reitz, C.; et al. TREM2 is associated with increased risk for Alzheimer’s disease in African Americans. Mol. Neurodegener. 2015, 10, 19. [Google Scholar] [CrossRef]
- Yu, J.T.; Jiang, T.; Wang, Y.L.; Wang, H.F.; Zhang, W.; Hu, N.; Tan, L.; Sun, L.; Tan, M.S.; Zhu, X.C.; et al. Triggering receptor expressed on myeloid cells 2 variant is rare in late-onset Alzheimer’s disease in Han Chinese individuals. Neurobiol. Aging 2014, 35, 937.e931–937.e933. [Google Scholar] [CrossRef]
- Benitez, B.A.; Jin, S.C.; Guerreiro, R.; Graham, R.; Lord, J.; Harold, D.; Sims, R.; Lambert, J.C.; Gibbs, J.R.; Bras, J.; et al. Missense variant in TREML2 protects against Alzheimer’s disease. Neurobiol. Aging 2014, 35, 1510.e1519–1510.e1526. [Google Scholar] [CrossRef]
- Kober, D.L.; Alexander-Brett, J.M.; Karch, C.M.; Cruchaga, C.; Colonna, M.; Holtzman, M.J.; Brett, T.J. Neurodegenerative disease mutations in TREM2 reveal a functional surface and distinct loss-of-function mechanisms. eLife 2016, 5, e20391. [Google Scholar] [CrossRef]
- Kelker, M.S.; Debler, E.W.; Wilson, I.A. Crystal structure of mouse triggering receptor expressed on myeloid cells 1 (TREM-1) at 1.76 A. J. Mol. Biol. 2004, 344, 1175–1181. [Google Scholar] [CrossRef] [PubMed]
- Kelker, M.S.; Foss, T.R.; Peti, W.; Teyton, L.; Kelly, J.W.; Wuthrich, K.; Wilson, I.A. Crystal structure of human triggering receptor expressed on myeloid cells 1 (TREM-1) at 1.47 A. J. Mol. Biol. 2004, 342, 1237–1248. [Google Scholar] [CrossRef] [PubMed]
- Strittmatter, W.J.; Saunders, A.M.; Schmechel, D.; Pericak-Vance, M.; Enghild, J.; Salvesen, G.S.; Roses, A.D. Apolipoprotein E: High-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 1977–1981. [Google Scholar] [CrossRef] [PubMed]
- Koldamova, R.P.; Lefterov, I.M.; Lefterova, M.I.; Lazo, J.S. Apolipoprotein A-I directly interacts with amyloid precursor protein and inhibits A beta aggregation and toxicity. Biochemistry 2001, 40, 3553–3560. [Google Scholar] [CrossRef]
- Manelli, A.M.; Stine, W.B.; Van Eldik, L.J.; LaDu, M.J. ApoE and Abeta1-42 interactions: Effects of isoform and conformation on structure and function. J. Mol. Neurosci. 2004, 23, 235–246. [Google Scholar] [CrossRef]
- Ghiso, J.; Matsubara, E.; Koudinov, A.; Choi-Miura, N.H.; Tomita, M.; Wisniewski, T.; Frangione, B. The cerebrospinal-fluid soluble form of Alzheimer’s amyloid beta is complexed to SP-40,40 (apolipoprotein J), an inhibitor of the complement membrane-attack complex. Biochem. J. 1993, 293 Pt 1, 27–30. [Google Scholar] [CrossRef]
- Wisniewski, T.; Golabek, A.; Matsubara, E.; Ghiso, J.; Frangione, B. Apolipoprotein E: Binding to soluble Alzheimer’s beta-amyloid. Biochem. Biophys. Res. Commun. 1993, 192, 359–365. [Google Scholar] [CrossRef]
- LaDu, M.J.; Falduto, M.T.; Manelli, A.M.; Reardon, C.A.; Getz, G.S.; Frail, D.E. Isoform-specific binding of apolipoprotein E to beta-amyloid. J. Biol. Chem. 1994, 269, 23403–23406. [Google Scholar]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef]
- Schmechel, D.E.; Saunders, A.M.; Strittmatter, W.J.; Crain, B.J.; Hulette, C.M.; Joo, S.H.; Pericak-Vance, M.A.; Goldgaber, D.; Roses, A.D. Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 9649–9653. [Google Scholar] [CrossRef]
- Holtzman, D.M.; Morris, J.C.; Goate, A.M. Alzheimer’s disease: The challenge of the second century. Sci. Transl. Med. 2011, 3, 77sr71. [Google Scholar] [CrossRef] [PubMed]
- Cosentino, S.; Scarmeas, N.; Helzner, E.; Glymour, M.M.; Brandt, J.; Albert, M.; Blacker, D.; Stern, Y. APOE epsilon 4 allele predicts faster cognitive decline in mild Alzheimer disease. Neurology 2008, 70, 1842–1849. [Google Scholar] [CrossRef] [PubMed]
- Agosta, F.; Vossel, K.A.; Miller, B.L.; Migliaccio, R.; Bonasera, S.J.; Filippi, M.; Boxer, A.L.; Karydas, A.; Possin, K.L.; Gorno-Tempini, M.L. Apolipoprotein E epsilon4 is associated with disease-specific effects on brain atrophy in Alzheimer’s disease and frontotemporal dementia. Proc. Natl. Acad. Sci. USA 2009, 106, 2018–2022. [Google Scholar] [CrossRef] [PubMed]
- Kanekiyo, T.; Xu, H.; Bu, G. ApoE and Abeta in Alzheimer’s disease: Accidental encounters or partners? Neuron 2014, 81, 740–754. [Google Scholar] [CrossRef] [PubMed]
- Bales, K.R.; Verina, T.; Dodel, R.C.; Du, Y.; Altstiel, L.; Bender, M.; Hyslop, P.; Johnstone, E.M.; Little, S.P.; Cummins, D.J.; et al. Lack of apolipoprotein E dramatically reduces amyloid beta-peptide deposition. Nat.Genet. 1997, 17, 263–264. [Google Scholar] [CrossRef] [PubMed]
- Holtzman, D.M.; Bales, K.R.; Wu, S.; Bhat, P.; Parsadanian, M.; Fagan, A.M.; Chang, L.K.; Sun, Y.; Paul, S.M. Expression of human apolipoprotein E reduces amyloid-beta deposition in a mouse model of Alzheimer’s disease. J. Clin. Investig. 1999, 103, R15–R21. [Google Scholar] [CrossRef] [PubMed]
- Holtzman, D.M.; Bales, K.R.; Tenkova, T.; Fagan, A.M.; Parsadanian, M.; Sartorius, L.J.; Mackey, B.; Olney, J.; McKeel, D.; Wozniak, D.; et al. Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2000, 97, 2892–2897. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, J.D.; Ulland, T.K.; Mahan, T.E.; Nystrom, S.; Nilsson, K.P.; Song, W.M.; Zhou, Y.; Reinartz, M.; Choi, S.; Jiang, H.; et al. ApoE facilitates the microglial response to amyloid plaque pathology. J. Exp. Med. 2018, 215, 1047–1058. [Google Scholar] [CrossRef]
- Kim, J.; Jiang, H.; Park, S.; Eltorai, A.E.; Stewart, F.R.; Yoon, H.; Basak, J.M.; Finn, M.B.; Holtzman, D.M. Haploinsufficiency of human APOE reduces amyloid deposition in a mouse model of amyloid-beta amyloidosis. J. Neurosci. 2011, 31, 18007–18012. [Google Scholar] [CrossRef]
- Fitz, N.F.; Cronican, A.A.; Saleem, M.; Fauq, A.H.; Chapman, R.; Lefterov, I.; Koldamova, R. Abca1 deficiency affects Alzheimer’s disease-like phenotype in human ApoE4 but not in ApoE3-targeted replacement mice. J. Neurosci. 2012, 32, 13125–13136. [Google Scholar] [CrossRef]
- Bales, K.R.; Liu, F.; Wu, S.; Lin, S.; Koger, D.; DeLong, C.; Hansen, J.C.; Sullivan, P.M.; Paul, S.M. Human APOE isoform-dependent effects on brain beta-amyloid levels in PDAPP transgenic mice. J. Neurosci. 2009, 29, 6771–6779. [Google Scholar] [CrossRef]
- Liu, C.C.; Zhao, N.; Fu, Y.; Wang, N.; Linares, C.; Tsai, C.W.; Bu, G. ApoE4 Accelerates Early Seeding of Amyloid Pathology. Neuron 2017, 96, 1024–1032.e1023. [Google Scholar] [CrossRef] [PubMed]
- Huynh, T.V.; Liao, F.; Francis, C.M.; Robinson, G.O.; Serrano, J.R.; Jiang, H.; Roh, J.; Finn, M.B.; Sullivan, P.M.; Esparza, T.J.; et al. Age-Dependent Effects of apoE Reduction Using Antisense Oligonucleotides in a Model of beta-amyloidosis. Neuron 2017, 96, 1013–1023.e1014. [Google Scholar] [CrossRef] [PubMed]
- Liao, F.; Li, A.; Xiong, M.; Bien-Ly, N.; Jiang, H.; Zhang, Y.; Finn, M.B.; Hoyle, R.; Keyser, J.; Lefton, K.B.; et al. Targeting of nonlipidated, aggregated apoE with antibodies inhibits amyloid accumulation. J. Clin. Investig. 2018, 128, 2144–2155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clayton, K.A.; Van Enoo, A.A.; Ikezu, T. Alzheimer’s Disease: The Role of Microglia in Brain Homeostasis and Proteopathy. Front. Neurosci. 2017, 11, 680. [Google Scholar] [CrossRef]
- Acosta, C.; Anderson, H.D.; Anderson, C.M. Astrocyte dysfunction in Alzheimer disease. J. Neurosci. Res. 2017, 95, 2430–2447. [Google Scholar] [CrossRef]
- Chakraborty, A.; de Wit, N.M.; van der Flier, W.M.; de Vries, H.E. The blood brain barrier in Alzheimer’s disease. Vasc. Pharmacol. 2016. [Google Scholar] [CrossRef]
- Bakker, E.N.; Bacskai, B.J.; Arbel-Ornath, M.; Aldea, R.; Bedussi, B.; Morris, A.W.; Weller, R.O.; Carare, R.O. Lymphatic Clearance of the Brain: Perivascular, Paravascular and Significance for Neurodegenerative Diseases. Cell. Mol. Neurobiol. 2016, 36, 181–194. [Google Scholar] [CrossRef] [Green Version]
- El Khoury, J.; Luster, A.D. Mechanisms of microglia accumulation in Alzheimer’s disease: Therapeutic implications. Trends Pharmacol. Sci. 2008, 29, 626–632. [Google Scholar] [CrossRef]
- Koistinaho, M.; Lin, S.; Wu, X.; Esterman, M.; Koger, D.; Hanson, J.; Higgs, R.; Liu, F.; Malkani, S.; Bales, K.R.; et al. Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-beta peptides. Nat. Med. 2004, 10, 719–726. [Google Scholar] [CrossRef]
- Koldamova, R.; Staufenbiel, M.; Lefterov, I. Lack of ABCA1 considerably decreases brain ApoE level and increases amyloid deposition in APP23 mice. J. Biol. Chem. 2005, 280, 43224–43235. [Google Scholar] [CrossRef] [PubMed]
- Wahrle, S.E.; Jiang, H.; Parsadanian, M.; Hartman, R.E.; Bales, K.R.; Paul, S.M.; Holtzman, D.M. Deletion of Abca1 increases Abeta deposition in the PDAPP transgenic mouse model of Alzheimer disease. J. Biol. Chem. 2005, 280, 43236–43242. [Google Scholar] [CrossRef] [PubMed]
- Hirsch-Reinshagen, V.; Maia, L.F.; Burgess, B.L.; Blain, J.F.; Naus, K.E.; McIsaac, S.A.; Parkinson, P.F.; Chan, J.Y.; Tansley, G.H.; Hayden, M.R.; et al. The Absence of ABCA1 Decreases Soluble ApoE Levels but Does Not Diminish Amyloid Deposition in Two Murine Models of Alzheimer Disease. J. Biol. Chem. 2005, 280, 43243–43256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahrle, S.E.; Jiang, H.; Parsadanian, M.; Kim, J.; Li, A.; Knoten, A.; Jain, S.; Hirsch-Reinshagen, V.; Wellington, C.L.; Bales, K.R.; et al. Overexpression of ABCA1 reduces amyloid deposition in the PDAPP mouse model of Alzheimer disease. J. Clin. Investig. 2008, 118, 671–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strittmatter, W.J.; Saunders, A.M.; Goedert, M.; Weisgraber, K.H.; Dong, L.M.; Jakes, R.; Huang, D.Y.; Pericak-Vance, M.; Schmechel, D.; Roses, A.D. Isoform-specific interactions of apolipoprotein E with microtubule-associated protein tau: Implications for Alzheimer disease. Proc. Natl. Acad. Sci. USA 1994, 91, 11183–11186. [Google Scholar] [CrossRef] [PubMed]
- Fleming, L.M.; Weisgraber, K.H.; Strittmatter, W.J.; Troncoso, J.C.; Johnson, G.V. Differential binding of apolipoprotein E isoforms to tau and other cytoskeletal proteins. Exp. Neurol. 1996, 138, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Yamada, K.; Liddelow, S.A.; Smith, S.T.; Zhao, L.; Luo, W.; Tsai, R.M.; Spina, S.; Grinberg, L.T.; Rojas, J.C.; et al. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 2017, 549, 523–527. [Google Scholar] [CrossRef] [Green Version]
- Yamazaki, Y.; Painter, M.M.; Bu, G.; Kanekiyo, T. Apolipoprotein E as a Therapeutic Target in Alzheimer’s Disease: A Review of Basic Research and Clinical Evidence. CNS Drugs 2016, 30, 773–789. [Google Scholar] [CrossRef] [Green Version]
- Koldamova, R.P.; Lefterov, I.M.; Staufenbiel, M.; Wolfe, D.; Huang, S.; Glorioso, J.C.; Walter, M.; Roth, M.G.; Lazo, J.S. The Liver X Receptor Ligand T0901317 Decreases Amyloid {beta} Production in Vitro and in a Mouse Model of Alzheimer’s Disease. J. Biol. Chem. 2005, 280, 4079–4088. [Google Scholar] [CrossRef]
- Jiang, Q.; Lee, C.Y.; Mandrekar, S.; Wilkinson, B.; Cramer, P.; Zelcer, N.; Mann, K.; Lamb, B.; Willson, T.M.; Collins, J.L.; et al. ApoE promotes the proteolytic degradation of Abeta. Neuron 2008, 58, 681–693. [Google Scholar] [CrossRef]
- Vanmierlo, T.; Rutten, K.; Dederen, J.; Bloks, V.W.; van Vark-van der Zee, L.C.; Kuipers, F.; Kiliaan, A.; Blokland, A.; Sijbrands, E.J.; Steinbusch, H.; et al. Liver X receptor activation restores memory in aged AD mice without reducing amyloid. Neurobiol. Aging 2011, 32, 1262–1272. [Google Scholar] [CrossRef] [PubMed]
- Skerrett, R.; Malm, T.; Landreth, G. Nuclear receptors in neurodegenerative diseases. Neurobiol. Dis. 2014, 72 Pt A, 104–116. [Google Scholar] [CrossRef] [Green Version]
- Mandrekar-Colucci, S.; Karlo, J.C.; Landreth, G.E. Mechanisms underlying the rapid peroxisome proliferator-activated receptor-gamma-mediated amyloid clearance and reversal of cognitive deficits in a murine model of Alzheimer’s disease. J. Neurosci. 2012, 32, 10117–10128. [Google Scholar] [CrossRef]
- Cramer, P.E.; Cirrito, J.R.; Wesson, D.W.; Lee, C.Y.; Karlo, J.C.; Zinn, A.E.; Casali, B.T.; Restivo, J.L.; Goebel, W.D.; James, M.J.; et al. ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models. Science 2012, 335, 1503–1506. [Google Scholar] [CrossRef] [PubMed]
- Fitz, N.F.; Cronican, A.A.; Lefterov, I.; Koldamova, R. Comment on “ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models”. Science 2013, 340, 924–992c. [Google Scholar] [CrossRef] [PubMed]
- Boehm-Cagan, A.; Michaelson, D.M. Reversal of apoE4-driven brain pathology and behavioral deficits by bexarotene. J. Neurosci. 2014, 34, 7293–7301. [Google Scholar] [CrossRef] [PubMed]
- Price, A.R.; Xu, G.; Siemienski, Z.B.; Smithson, L.A.; Borchelt, D.R.; Golde, T.E.; Felsenstein, K.M. Comment on “ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models”. Science 2013, 340, 924-d. [Google Scholar] [CrossRef]
- Tesseur, I.; Lo, A.C.; Roberfroid, A.; Dietvorst, S.; Van Broeck, B.; Borgers, M.; Gijsen, H.; Moechars, D.; Mercken, M.; Kemp, J.; et al. Comment on “ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models”. Science 2013, 340, 924-e. [Google Scholar] [CrossRef]
- Veeraraghavalu, K.; Zhang, C.; Miller, S.; Hefendehl, J.K.; Rajapaksha, T.W.; Ulrich, J.; Jucker, M.; Holtzman, D.M.; Tanzi, R.E.; Vassar, R.; et al. Comment on “ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models”. Science 2013, 340, 924-f. [Google Scholar] [CrossRef]
- Kim, J.; Eltorai, A.E.; Jiang, H.; Liao, F.; Verghese, P.B.; Kim, J.; Stewart, F.R.; Basak, J.M.; Holtzman, D.M. Anti-apoE immunotherapy inhibits amyloid accumulation in a transgenic mouse model of Abeta amyloidosis. J. Exp. Med. 2012, 209, 2149–2156. [Google Scholar] [CrossRef]
- Chen, H.K.; Ji, Z.S.; Dodson, S.E.; Miranda, R.D.; Rosenblum, C.I.; Reynolds, I.J.; Freedman, S.B.; Weisgraber, K.H.; Huang, Y.; Mahley, R.W. Apolipoprotein E4 domain interaction mediates detrimental effects on mitochondria and is a potential therapeutic target for Alzheimer disease. J. Biol. Chem. 2011, 286, 5215–5221. [Google Scholar] [CrossRef]
- Brodbeck, J.; McGuire, J.; Liu, Z.; Meyer-Franke, A.; Balestra, M.E.; Jeong, D.E.; Pleiss, M.; McComas, C.; Hess, F.; Witter, D.; et al. Structure-dependent impairment of intracellular apolipoprotein E4 trafficking and its detrimental effects are rescued by small-molecule structure correctors. J. Biol. Chem. 2011, 286, 17217–17226. [Google Scholar] [CrossRef] [PubMed]
- Pankiewicz, J.E.; Guridi, M.; Kim, J.; Asuni, A.A.; Sanchez, S.; Sullivan, P.M.; Holtzman, D.M.; Sadowski, M.J. Blocking the apoE/Abeta interaction ameliorates Abeta-related pathology in APOE epsilon2 and epsilon4 targeted replacement Alzheimer model mice. Acta Neuropathol. Commun. 2014, 2, 75. [Google Scholar] [PubMed]
- Kuszczyk, M.A.; Sanchez, S.; Pankiewicz, J.; Kim, J.; Duszczyk, M.; Guridi, M.; Asuni, A.A.; Sullivan, P.M.; Holtzman, D.M.; Sadowski, M.J. Blocking the interaction between apolipoprotein E and Abeta reduces intraneuronal accumulation of Abeta and inhibits synaptic degeneration. Am. J. Pathol. 2013, 182, 1750–1768. [Google Scholar] [CrossRef]
- Butovsky, O.; Weiner, H.L. Microglial signatures and their role in health and disease. Nat. Rev. Neurosci. 2018. [Google Scholar] [CrossRef] [PubMed]
- Yuan, P.; Condello, C.; Keene, C.D.; Wang, Y.; Bird, T.D.; Paul, S.M.; Luo, W.; Colonna, M.; Baddeley, D.; Grutzendler, J. TREM2 Haplodeficiency in Mice and Humans Impairs the Microglia Barrier Function Leading to Decreased Amyloid Compaction and Severe Axonal Dystrophy. Neuron 2016, 90, 724–739. [Google Scholar] [CrossRef]
- Wang, Y.; Ulland, T.K.; Ulrich, J.D.; Song, W.; Tzaferis, J.A.; Hole, J.T.; Yuan, P.; Mahan, T.E.; Shi, Y.; Gilfillan, S.; et al. TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J. Exp. Med. 2016, 213, 667–675. [Google Scholar] [CrossRef] [Green Version]
- Ulrich, J.D.; Ulland, T.K.; Colonna, M.; Holtzman, D.M. Elucidating the Role of TREM2 in Alzheimer’s Disease. Neuron 2017, 94, 237–248. [Google Scholar] [CrossRef]
- Jay, T.R.; Miller, C.M.; Cheng, P.J.; Graham, L.C.; Bemiller, S.; Broihier, M.L.; Xu, G.; Margevicius, D.; Karlo, J.C.; Sousa, G.L.; et al. TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer’s disease mouse models. J. Exp. Med. 2015, 212, 287–295. [Google Scholar] [CrossRef]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581.e569. [Google Scholar] [CrossRef]
- Jay, T.R.; Hirsch, A.M.; Broihier, M.L.; Miller, C.M.; Neilson, L.E.; Ransohoff, R.M.; Lamb, B.T.; Landreth, G.E. Disease Progression-Dependent Effects of TREM2 Deficiency in a Mouse Model of Alzheimer’s Disease. J. Neurosci. 2017, 37, 637–647. [Google Scholar] [CrossRef] [PubMed]
- Song, W.M.; Joshita, S.; Zhou, Y.; Ulland, T.K.; Gilfillan, S.; Colonna, M. Humanized TREM2 mice reveal microglia-intrinsic and -extrinsic effects of R47H polymorphism. J. Exp. Med. 2018, 215, 745–760. [Google Scholar] [CrossRef] [PubMed]
- Cheng-Hathaway, P.J.; Reed-Geaghan, E.G.; Jay, T.R.; Casali, B.T.; Bemiller, S.M.; Puntambekar, S.S.; von Saucken, V.E.; Williams, R.Y.; Karlo, J.C.; Moutinho, M.; et al. The Trem2 R47H variant confers loss-of-function-like phenotypes in Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 29. [Google Scholar] [CrossRef] [PubMed]
- Heslegrave, A.; Heywood, W.; Paterson, R.; Magdalinou, N.; Svensson, J.; Johansson, P.; Ohrfelt, A.; Blennow, K.; Hardy, J.; Schott, J.; et al. Increased cerebrospinal fluid soluble TREM2 concentration in Alzheimer’s disease. Mol. Neurodegener. 2016, 11, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suarez-Calvet, M.; Kleinberger, G.; Araque Caballero, M.A.; Brendel, M.; Rominger, A.; Alcolea, D.; Fortea, J.; Lleo, A.; Blesa, R.; Gispert, J.D.; et al. sTREM2 cerebrospinal fluid levels are a potential biomarker for microglia activity in early-stage Alzheimer’s disease and associate with neuronal injury markers. EMBO Mol. Med. 2016, 8, 466–476. [Google Scholar] [CrossRef] [PubMed]
- Piccio, L.; Deming, Y.; Del-Aguila, J.L.; Ghezzi, L.; Holtzman, D.M.; Fagan, A.M.; Fenoglio, C.; Galimberti, D.; Borroni, B.; Cruchaga, C. Cerebrospinal fluid soluble TREM2 is higher in Alzheimer disease and associated with mutation status. Acta Neuropathol. 2016, 131, 925–933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rauchmann, B.S.; Schneider-Axmann, T.; Alexopoulos, P.; Perneczky, R.; Alzheimer’s Disease Neuroimaging, I. CSF soluble TREM2 as a measure of immune response along the Alzheimer’s disease continuum. Neurobiol. Aging 2018, 74, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, R.; Orme, T.; Naj, A.C.; Kuzma, A.B.; Schellenberg, G.D.; Bras, J. Is APOE epsilon4 required for Alzheimer’s disease to develop in TREM2 p.R47H variant carriers? Neuropathol. Appl. Neurobiol. 2018. [Google Scholar] [CrossRef]
- Villegas-Llerena, C.; Phillips, A.; Garcia-Reitboeck, P.; Hardy, J.; Pocock, J.M. Microglial genes regulating neuroinflammation in the progression of Alzheimer’s disease. Curr. Opin. Neurobiol. 2016, 36, 74–81. [Google Scholar] [CrossRef]
- Jendresen, C.; Arskog, V.; Daws, M.R.; Nilsson, L.N. The Alzheimer’s disease risk factors apolipoprotein E and TREM2 are linked in a receptor signaling pathway. J. Neuroinflamm. 2017, 14, 59. [Google Scholar] [CrossRef]
- Lee, C.Y.D.; Daggett, A.; Gu, X.; Jiang, L.L.; Langfelder, P.; Li, X.; Wang, N.; Zhao, Y.; Park, C.S.; Cooper, Y.; et al. Elevated TREM2 Gene Dosage Reprograms Microglia Responsivity and Ameliorates Pathological Phenotypes in Alzheimer’s Disease Models. Neuron 2018, 97, 1032–1048.e1035. [Google Scholar] [CrossRef] [PubMed]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e1217. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wolfe, C.M.; Fitz, N.F.; Nam, K.N.; Lefterov, I.; Koldamova, R. The Role of APOE and TREM2 in Alzheimer′s Disease—Current Understanding and Perspectives. Int. J. Mol. Sci. 2019, 20, 81. https://doi.org/10.3390/ijms20010081
Wolfe CM, Fitz NF, Nam KN, Lefterov I, Koldamova R. The Role of APOE and TREM2 in Alzheimer′s Disease—Current Understanding and Perspectives. International Journal of Molecular Sciences. 2019; 20(1):81. https://doi.org/10.3390/ijms20010081
Chicago/Turabian StyleWolfe, Cody M., Nicholas F. Fitz, Kyong Nyon Nam, Iliya Lefterov, and Radosveta Koldamova. 2019. "The Role of APOE and TREM2 in Alzheimer′s Disease—Current Understanding and Perspectives" International Journal of Molecular Sciences 20, no. 1: 81. https://doi.org/10.3390/ijms20010081
APA StyleWolfe, C. M., Fitz, N. F., Nam, K. N., Lefterov, I., & Koldamova, R. (2019). The Role of APOE and TREM2 in Alzheimer′s Disease—Current Understanding and Perspectives. International Journal of Molecular Sciences, 20(1), 81. https://doi.org/10.3390/ijms20010081