Anabolic Therapies in Osteoporosis and Bone Regeneration


{kind=link}
Abstract
:1. Introduction
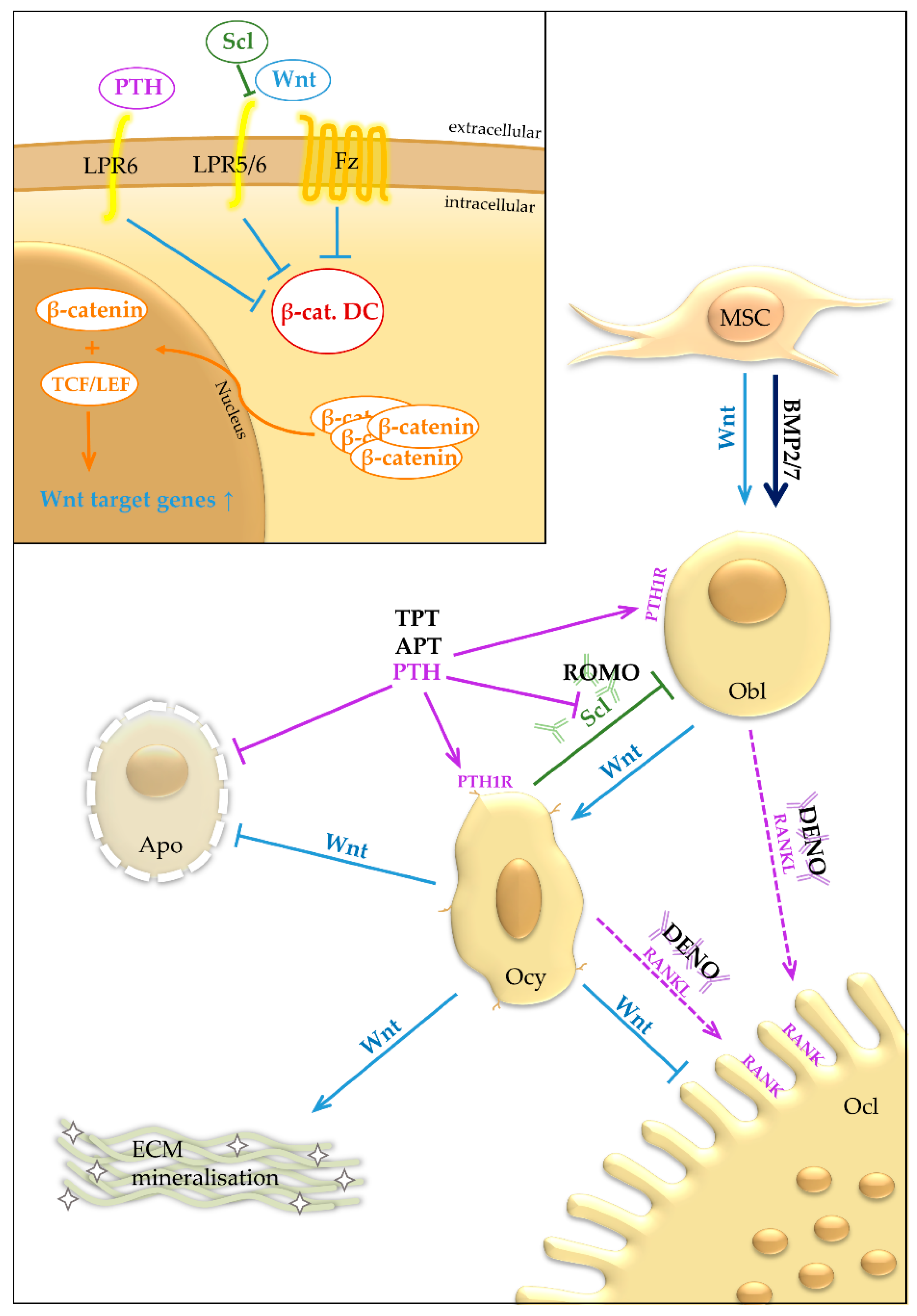
2. Bone Turnover—Osteoporosis
2.1. Osteoporosis—Antiresorptive Therapy
2.2. Osteoporosis—Anabolic Therapy
2.2.1. PTH—Teriparatide and Abaloparatide
2.2.2. Sclerostin-Neutralizing Antibody—Romosozumab
2.2.3. Future Perspectives
3. Fracture Healing—Impaired Bone Regeneration
3.1. Impaired Fracture Healing—Antiresorptive Therapy
3.2. Impaired Fracture Healing—Anabolic Therapy
3.2.1. PTH
3.2.2. Bone Morphogenetic Proteins
3.2.3. Sclerostin-Neutralizing Antibodies
3.2.4. Future Perspectives
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACTIVE | Abaloparatide Comparator Trial In Vertebral Endpoints Trial |
| ARCH | Active-Controlled Fracture Study in Postmenopausal Women with Osteoporosis at High Risk |
| BMD | Bone mineral density |
| BMI | Body mass index |
| BMP | Bone morphogenetic protein |
| cAMP | cyclic Adenosine monophosphate |
| ERK1/2 | Extracellular-signal Regulated Kinase 1 and 2 |
| FDA | United States Food and Drug Administration |
| FRAME | Fracture Study in Postmenopausal Women with Osteoporosis |
| FRAX | Fracture Risk Assessment Score |
| GIOP | Glucocorticoid-induced osteoporosis |
| iPTH | Intermittent PTH |
| LRP5/6 | Low-density lipoprotein receptor-related protein 5 or 6 |
| LRP6 | Low-density lipoprotein receptor-related protein 6 |
| miRNA | Micro RNA |
| NBD | NEMO binding domain peptide |
| NEMO | NF-kappa-B essential modulator |
| NF-κB | Nuclear factor “kappa-light-chain-enhancer” of activated B-cells |
| OPG | osteoprotegerin |
| PTH | Parathyroid hormone |
| PTH1-34 | Teriparatide, peptide Fragment of PTH |
| PTH1R | parathyroid hormone 1 receptor |
| PTHrP | parathyroid hormone-related protein |
| RANK | Receptor Activator of NF-κB |
| RANKL | Receptor Activator of NF-κB Ligand |
| rhBMP | Recombinant BMP |
| Runx2 | Runt-related transcription factor 2 |
| Scl | Sclerostin |
| STRUCTURE | An Open-label Study to Evaluate the Effect of Treatment With Romosozumab or Teriparatide in Postmenopausal Women |
| TCF/LEF | T cell factor/lymphoid enhancer factor |
| Wnt | Wingless-related integration site/Wnt signalling pathway |
| WT | Wild type |
References
- Teitelbaum, S.L. Bone resorption by osteoclasts. Science 2000, 289, 1504–1508. [Google Scholar] [CrossRef] [PubMed]
- Howship, J. Microscopic Observations on the Structure of Bone. Med. Chir. Trans. 1816, 7, 382–592.11. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Zhang, Y.; Miao, G.; Zhang, Y.; Liu, Y.; Huang, Y. Runx1 regulates osteogenic differentiation of BMSCs by inhibiting adipogenesis through Wnt/beta-catenin pathway. Arch. Oral Biol. 2018, 97, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Bahney, C.S.; Zondervan, R.L.; Allison, P.; Theologis, A.; Ashley, J.; Ahn, J.; Miclau, T.; Marcucio, R.; Hankenson, K.D. The Cellular Biology of Fracture Healing. J. Orthop. Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Abdelgawad, M.E.; Delaisse, J.M.; Hinge, M.; Jensen, P.R.; Alnaimi, R.W.; Rolighed, L.; Engelholm, L.H.; Marcussen, N.; Andersen, T.L. Early reversal cells in adult human bone remodeling: OSTEOBLASTIC nature, catabolic functions and interactions with osteoclasts. Histochem. Cell Biol. 2016, 145, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wang, Z.; Duan, N.; Zhu, G.; Schwarz, E.M.; Xie, C. Osteoblast-osteoclast interactions. Connect. Tissue Res. 2018, 59, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Koide, M.; Kobayashi, Y. Regulatory mechanisms of sclerostin expression during bone remodeling. J. Bone Miner. Metab. 2018. [Google Scholar] [CrossRef]
- Lorentzon, M.; Cummings, S.R. Osteoporosis: THE evolution of a diagnosis. J. Intern. Med. 2015, 277, 650–661. [Google Scholar] [CrossRef]
- Kanis, J.A.; Johansson, H.; Harvey, N.C.; McCloskey, E.V. A brief history of FRAX. Arch. Osteoporos. 2018, 13, 118. [Google Scholar] [CrossRef]
- Siris, E.S.; Adler, R.; Bilezikian, J.; Bolognese, M.; Dawson-Hughes, B.; Favus, M.J.; Harris, S.T.; Jan de Beur, S.M.; Khosla, S.; Lane, N.E.; et al. The clinical diagnosis of osteoporosis: A position statement from the National Bone Health Alliance Working Group. Osteoporos. Int. 2014, 25, 1439–1443. [Google Scholar] [CrossRef]
- Papaioannou, A.; Kennedy, C. Diagnostic criteria for osteoporosis should be expanded. Lancet Diabetes Endocrinol. 2015, 3, 234–236. [Google Scholar] [CrossRef] [Green Version]
- Cosman, F.; de Beur, S.J.; LeBoff, M.S.; Lewiecki, E.M.; Tanner, B.; Randall, S.; Lindsay, R. Clinician’s Guide to Prevention and Treatment of Osteoporosis. Osteoporos. Int. 2014, 25, 2359–2381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canalis, E.; Mazziotti, G.; Giustina, A.; Bilezikian, J.P. Glucocorticoid-induced osteoporosis: PATHOPHYSIOLOGY and therapy. Osteoporos. Int. 2007, 18, 1319–1328. [Google Scholar] [CrossRef] [PubMed]
- Warriner, A.H.; Patkar, N.M.; Curtis, J.R.; Delzell, E.; Gary, L.; Kilgore, M.; Saag, K. Which fractures are most attributable to osteoporosis? J. Clin. Epidemiol. 2011, 64, 46–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deloumeau, A.; Molto, A.; Roux, C.; Briot, K. Determinants of short term fracture risk in patients with a recent history of low-trauma non-vertebral fracture. Bone 2017, 105, 287–291. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, S.L.; Abrahamsen, B.; Napoli, N.; Akesson, K.; Chandran, M.; Eastell, R.; El-Hajj Fuleihan, G.; Josse, R.; Kendler, D.L.; Kraenzlin, M.; et al. Diagnosis and management of bone fragility in diabetes: AN emerging challenge. Osteoporos. Int. 2018, 29, 2585–2596. [Google Scholar] [CrossRef] [PubMed]
- Kanis, J.A.; Johnell, O.; Oden, A.; Johansson, H.; McCloskey, E. FRAX and the assessment of fracture probability in men and women from the UK. Osteoporos. Int. 2008, 19, 385–397. [Google Scholar] [CrossRef]
- Crandall, C.J.; Larson, J.; LaCroix, A.; Cauley, J.A.; LeBoff, M.S.; Li, W.; LeBlanc, E.S.; Edwards, B.J.; Manson, J.E.; Ensrud, K. Predicting Fracture Risk in Younger Postmenopausal Women: Comparison of the Garvan and FRAX Risk Calculators in the Women’s Health Initiative Study. J. Gen. Intern Med. 2018. [Google Scholar] [CrossRef]
- Akinleye, S.D.; Garofolo, G.; Culbertson, M.D.; Homel, P.; Erez, O. The Role of BMI in Hip Fracture Surgery. Geriatr. Orthop. Surg. Rehabil. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Abrahamsen, B.; van Staa, T.; Ariely, R.; Olson, M.; Cooper, C. Excess mortality following hip fracture: A systematic epidemiological review. Osteoporos. Int. 2009, 20, 1633–1650. [Google Scholar] [CrossRef]
- Cummings, S.R.; Black, D.M.; Rubin, S.M. Lifetime risks of hip, Colles’, or vertebral fracture and coronary heart disease among white postmenopausal women. Arch. Intern Med. 1989, 149, 2445–2448. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.Z.; Rames, R.D.; Miller, A.N. Clinical Management of Osteoporotic Fractures. Curr. Osteoporos. Rep. 2018, 16, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Sims, N.A.; Ng, K.W. Implications of osteoblast-osteoclast interactions in the management of osteoporosis by antiresorptive agents denosumab and odanacatib. Curr. Osteoporos. Rep. 2014, 12, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Larsen, M.S.; Schmal, H. The enigma of atypical femoral fractures: A summary of current knowledge. EFORT Open Rev. 2018, 3, 494–500. [Google Scholar] [CrossRef]
- Lim, S.J.; Yeo, I.; Yoon, P.W.; Yoo, J.J.; Rhyu, K.H.; Han, S.B.; Lee, W.S.; Song, J.H.; Min, B.W.; Park, Y.S. Incidence, risk factors, and fracture healing of atypical femoral fractures: A multicenter case-control study. Osteoporos. Int. 2018, 29, 2427–2435. [Google Scholar] [CrossRef] [PubMed]
- Wojda, S.J.; Donahue, S.W. Parathyroid hormone for bone regeneration. J. Orthop. Res. 2018, 36, 2586–2594. [Google Scholar] [CrossRef]
- Langdahl, B.L.; Silverman, S.; Fujiwara, S.; Saag, K.; Napoli, N.; Soen, S.; Enomoto, H.; Melby, T.E.; Disch, D.P.; Marin, F.; Krege, J.H. Real-world effectiveness of teriparatide on fracture reduction in patients with osteoporosis and comorbidities or risk factors for fractures: Integrated analysis of 4 prospective observational studies. Bone 2018, 116, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Swarthout, J.T.; D’Alonzo, R.C.; Selvamurugan, N.; Partridge, N.C. Parathyroid hormone-dependent signaling pathways regulating genes in bone cells. Gene 2002, 282, 1–17. [Google Scholar] [CrossRef]
- Cheng, Z.Y.; Ye, T.; Ling, Q.Y.; Wu, T.; Wu, G.Y.; Zong, G.J. Parathyroid hormone promotes osteoblastic differentiation of endothelial cells via the extracellular signal-regulated protein kinase 1/2 and nuclear factor-kappaB signaling pathways. Exp. Ther. Med. 2018, 15, 1754–1760. [Google Scholar] [CrossRef]
- Kakar, S.; Einhorn, T.A.; Vora, S.; Miara, L.J.; Hon, G.; Wigner, N.A.; Toben, D.; Jacobsen, K.A.; Al-Sebaei, M.O.; Song, M.; Trackman, P.C.; et al. Enhanced chondrogenesis and Wnt signaling in PTH-treated fractures. J. Bone Miner. Res. 2007, 22, 1903–1912. [Google Scholar] [CrossRef]
- Krishnan, V.; Bryant, H.U.; Macdougald, O.A. Regulation of bone mass by Wnt signaling. J. Clin. Investig. 2006, 116, 1202–1209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tobimatsu, T.; Kaji, H.; Sowa, H.; Naito, J.; Canaff, L.; Hendy, G.N.; Sugimoto, T.; Chihara, K. Parathyroid hormone increases beta-catenin levels through Smad3 in mouse osteoblastic cells. Endocrinology 2006, 147, 2583–2590. [Google Scholar] [CrossRef] [PubMed]
- Wan, M.; Yang, C.; Li, J.; Wu, X.; Yuan, H.; Ma, H.; He, X.; Nie, S.; Chang, C.; Cao, X. Parathyroid hormone signaling through low-density lipoprotein-related protein 6. Genes Dev. 2008, 22, 2968–2979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keller, H.; Kneissel, M. SOST is a target gene for PTH in bone. Bone 2005, 37, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, Y.; Kang, H.; Liu, W.; Liu, P.; Zhang, J.; Harris, S.E.; Wu, D. Sclerostin binds to LRP5/6 and antagonizes canonical Wnt signaling. J. Biol. Chem. 2005, 280, 19883–19887. [Google Scholar] [CrossRef] [PubMed]
- Drake, M.T.; Srinivasan, B.; Modder, U.I.; Peterson, J.M.; McCready, L.K.; Riggs, B.L.; Dwyer, D.; Stolina, M.; Kostenuik, P.; Khosla, S. Effects of parathyroid hormone treatment on circulating sclerostin levels in postmenopausal women. J. Clin. Endocrinol. Metab. 2010, 95, 5056–5062. [Google Scholar] [CrossRef] [PubMed]
- Bellido, T.; Ali, A.A.; Gubrij, I.; Plotkin, L.I.; Fu, Q.; O’Brien, C.A.; Manolagas, S.C.; Jilka, R.L. Chronic elevation of parathyroid hormone in mice reduces expression of sclerostin by osteocytes: A novel mechanism for hormonal control of osteoblastogenesis. Endocrinology 2005, 146, 4577–4583. [Google Scholar] [CrossRef]
- Kramer, I.; Loots, G.G.; Studer, A.; Keller, H.; Kneissel, M. Parathyroid hormone (PTH)-induced bone gain is blunted in SOST overexpressing and deficient mice. J. Bone Miner. Res. 2010, 25, 178–189. [Google Scholar] [CrossRef]
- Robling, A.G.; Kedlaya, R.; Ellis, S.N.; Childress, P.J.; Bidwell, J.P.; Bellido, T.; Turner, C.H. Anabolic and catabolic regimens of human parathyroid hormone 1-34 elicit bone- and envelope-specific attenuation of skeletal effects in Sost-deficient mice. Endocrinology 2011, 152, 2963–2975. [Google Scholar] [CrossRef]
- Cheloha, R.W.; Gellman, S.H.; Vilardaga, J.P.; Gardella, T.J. PTH receptor-1 signalling-mechanistic insights and therapeutic prospects. Nat. Rev. Endocrinol. 2015, 11, 712–724. [Google Scholar] [CrossRef]
- Pazianas, M. Anabolic effects of PTH and the ‘anabolic window’. Trends Endocrinol. Metab. 2015, 26, 111–113. [Google Scholar] [CrossRef] [PubMed]
- Chandler, H.; Lanske, B.; Varela, A.; Guillot, M.; Boyer, M.; Brown, J.; Pierce, A.; Ominsky, M.; Mitlak, B.; Baron, R.; Kostenuik, P.; Hattersley, G. Abaloparatide, a novel osteoanabolic PTHrP analog, increases cortical and trabecular bone mass and architecture in orchiectomized rats by increasing bone formation without increasing bone resorption. Bone 2018, 120, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Miller, P.D.; Hattersley, G.; Riis, B.J.; Williams, G.C.; Lau, E.; Russo, L.A.; Alexandersen, P.; Zerbini, C.A.; Hu, M.Y.; Harris, A.G.; et al. Effect of Abaloparatide vs Placebo on New Vertebral Fractures in Postmenopausal Women with Osteoporosis: A Randomized Clinical Trial. JAMA 2016, 316, 722–733. [Google Scholar] [CrossRef] [PubMed]
- Reginster, J.Y.; Hattersley, G.; Williams, G.C.; Hu, M.Y.; Fitzpatrick, L.A.; Lewiecki, E.M. Abaloparatide is an Effective Treatment Option for Postmenopausal Osteoporosis: Review of the Number Needed to Treat Compared with Teriparatide. Calcif. Tissue Int. 2018, 103, 540–545. [Google Scholar] [CrossRef] [PubMed]
- Miller, P.D.; Hattersley, G.; Lau, E.; Fitzpatrick, L.A.; Harris, A.G.; Williams, G.C.; Hu, M.Y.; Riis, B.J.; Russo, L.; Christiansen, C. Bone mineral density response rates are greater in patients treated with abaloparatide compared with those treated with placebo or teriparatide: Results from the ACTIVE phase 3 trial. Bone 2018, 120, 137–140. [Google Scholar] [CrossRef] [PubMed]
- Boyce, E.G.; Mai, Y.; Pham, C. Abaloparatide: Review of a Next-Generation Parathyroid Hormone Agonist. Ann. Pharmacother. 2018, 52, 462–472. [Google Scholar] [CrossRef] [PubMed]
- Van Lierop, A.H.; Appelman-Dijkstra, N.M.; Papapoulos, S.E. Sclerostin deficiency in humans. Bone 2017, 96, 51–62. [Google Scholar] [CrossRef]
- Li, X.; Ominsky, M.S.; Niu, Q.T.; Sun, N.; Daugherty, B.; D’Agostin, D.; Kurahara, C.; Gao, Y.; Cao, J.; Gong, J.; et al. Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J. Bone Miner. Res. 2008, 23, 860–869. [Google Scholar] [CrossRef]
- Shi, C.; Li, J.; Wang, W.; Cao, W.; Cao, X.; Wan, M. Antagonists of LRP6 regulate PTH-induced cAMP generation. Ann. N. Y. Acad. Sci. 2011, 1237, 39–46. [Google Scholar] [CrossRef]
- Wijenayaka, A.R.; Kogawa, M.; Lim, H.P.; Bonewald, L.F.; Findlay, D.M.; Atkins, G.J. Sclerostin stimulates osteocyte support of osteoclast activity by a RANKL-dependent pathway. PLoS ONE 2011, 6, e25900. [Google Scholar] [CrossRef]
- Alaee, F.; Virk, M.S.; Tang, H.; Sugiyama, O.; Adams, D.J.; Stolina, M.; Dwyer, D.; Ominsky, M.S.; Ke, H.Z.; Lieberman, J.R. Evaluation of the effects of systemic treatment with a sclerostin neutralizing antibody on bone repair in a rat femoral defect model. J. Orthop. Res. 2014, 32, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Ominsky, M.S.; Brown, D.L.; Van, G.; Cordover, D.; Pacheco, E.; Frazier, E.; Cherepow, L.; Higgins-Garn, M.; Aguirre, J.I.; Wronski, T.J.; et al. Differential temporal effects of sclerostin antibody and parathyroid hormone on cancellous and cortical bone and quantitative differences in effects on the osteoblast lineage in young intact rats. Bone 2015, 81, 380–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cosman, F.; Crittenden, D.B.; Ferrari, S.; Khan, A.; Lane, N.E.; Lippuner, K.; Matsumoto, T.; Milmont, C.E.; Libanati, C.; Grauer, A. FRAME Study: The Foundation Effect of Building Bone With 1 Year of Romosozumab Leads to Continued Lower Fracture Risk After Transition to Denosumab. J. Bone Miner. Res. 2018, 33, 1219–1226. [Google Scholar] [CrossRef] [PubMed]
- Graeff, C.; Campbell, G.M.; Pena, J.; Borggrefe, J.; Padhi, D.; Kaufman, A.; Chang, S.; Libanati, C.; Gluer, C.C. Administration of romosozumab improves vertebral trabecular and cortical bone as assessed with quantitative computed tomography and finite element analysis. Bone 2015, 81, 364–369. [Google Scholar] [CrossRef] [PubMed]
- Saag, K.G.; Petersen, J.; Brandi, M.L.; Karaplis, A.C.; Lorentzon, M.; Thomas, T.; Maddox, J.; Fan, M.; Meisner, P.D.; Grauer, A. Romosozumab or Alendronate for Fracture Prevention in Women with Osteoporosis. N. Engl. J. Med. 2017, 377, 1417–1427. [Google Scholar] [CrossRef] [PubMed]
- Weske, S.; Vaidya, M.; Reese, A.; von Wnuck Lipinski, K.; Keul, P.; Bayer, J.K.; Fischer, J.W.; Flogel, U.; Nelsen, J.; Epple, M.; et al. Targeting sphingosine-1-phosphate lyase as an anabolic therapy for bone loss. Nat. Med. 2018, 24, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Yallowitz, A.; Qin, A.; Wu, Z.; Shin, D.Y.; Kim, J.M.; Debnath, S.; Ji, G.; Bostrom, M.P.; Yang, X.; Zhang, C.; et al. Targeting skeletal endothelium to ameliorate bone loss. Nat. Med. 2018, 24, 823–833. [Google Scholar] [CrossRef]
- Baltzer, A.W.A.; Whalen, J.D.; Wooley, P.; Latterman, C.; Truchan, L.M.; Robbins, P.D.; Evans, C.H. Gene therapy for osteoporosis: EVALUATION in a murine ovariectomy model. Gene Ther. 2001, 8, 1770–1776. [Google Scholar] [CrossRef]
- Zhang, W.; De La Vega, R.E.; Coenen, M.J.; Muller, S.A.; Peniche Silva, C.J.; Aneja, M.K.; Plank, C.; van Griensven, M.; Evans, C.H.; Balmayor, E.R. An Improved, Chemically Modified RNA Encoding BMP-2 Enhances Osteogenesis In Vitro and In Vivo. Tissue Eng. Part A 2018. [Google Scholar] [CrossRef]
- Bolon, B.; Carter, C.; Daris, M.; Morony, S.; Capparelli, C.; Hsieh, A.; Mao, M.; Kostenuik, P.; Dunstan, C.R.; Lacey, D.L.; Sheng, J.Z. Adenoviral delivery of osteoprotegerin ameliorates bone resorption in a mouse ovariectomy model of osteoporosis. Mol. Ther. 2001, 3, 197–205. [Google Scholar] [CrossRef]
- Engstrand, T.; Daluiski, A.; Bahamonde, M.E.; Melhus, H.; Lyons, K.M. Transient production of bone morphogenetic protein 2 by allogeneic transplanted transduced cells induces bone formation. Hum. Gene Ther. 2000, 11, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Kawai, M.; Bessho, K.; Maruyama, H.; Miyazaki, J.; Yamamoto, T. Human BMP-2 gene transfer using transcutaneous in vivo electroporation induced both intramembranous and endochondral ossification. Anat. Rec. A Discov. Mol. Cell. Evol. Biol. 2005, 287, 1264–1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kostenuik, P.J.; Bolon, B.; Morony, S.; Daris, M.; Geng, Z.; Carter, C.; Sheng, J. Gene therapy with human recombinant osteoprotegerin reverses established osteopenia in ovariectomized mice. Bone 2004, 34, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Tang, J.; Ji, J.; Gu, J. The expression of functional human parathyroid hormone in a gene therapy model for osteoporosis. Cell Tissue Res. 2004, 317, 57–63. [Google Scholar] [CrossRef]
- Luk, K.D.; Chen, Y.; Cheung, K.M.; Kung, H.F.; Lu, W.W.; Leong, J.C. Adeno-associated virus-mediated bone morphogenetic protein-4 gene therapy for in vivo bone formation. Biochem. Biophys. Res. Commun. 2003, 308, 636–645. [Google Scholar] [CrossRef]
- Ulrich-Vinther, M.; Schwarz, E.M.; Pedersen, F.S.; Soballe, K.; Andreassen, T.T. Gene therapy with human osteoprotegerin decreases callus remodeling with limited effects on biomechanical properties. Bone 2005, 37, 751–758. [Google Scholar] [CrossRef]
- Yue, B.; Lu, B.; Dai, K.R.; Zhang, X.L.; Yu, C.F.; Lou, J.R.; Tang, T.T. BMP2 gene therapy on the repair of bone defects of aged rats. Calcif. Tissue Int. 2005, 77, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Zheng, S.; Zheng, J. The emerging role of microRNAs in bone remodeling and its therapeutic implications for osteoporosis. Biosci. Rep. 2018, 38, BSR20180453. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Bleek, K.; Kwee, B.J.; Mooney, D.J.; Duda, G.N. Boon and Bane of Inflammation in Bone Tissue Regeneration and Its Link with Angiogenesis. Tissue Eng. Part B Rev. 2015, 21, 354–364. [Google Scholar] [CrossRef] [Green Version]
- Winkler, T.; Sass, F.A.; Duda, G.N.; Schmidt-Bleek, K. A review of biomaterials in bone defect healing, remaining shortcomings and future opportunities for bone tissue engineering: The unsolved challenge. Bone Jt. Res. 2018, 7, 232–243. [Google Scholar] [CrossRef]
- Hak, D.J.; Fitzpatrick, D.; Bishop, J.A.; Marsh, J.L.; Tilp, S.; Schnettler, R.; Simpson, H.; Alt, V. Delayed union and nonunions: EPIDEMIOLOGY, clinical issues, and financial aspects. Injury 2014, 45 (Suppl. 2), S3–S7. [Google Scholar] [CrossRef] [PubMed]
- Wells, G.A.; Cranney, A.; Peterson, J.; Boucher, M.; Shea, B.; Robinson, V.; Coyle, D.; Tugwell, P. Risedronate for the primary and secondary prevention of osteoporotic fractures in postmenopausal women. Cochrane Database Syst. Rev. 2008. [Google Scholar] [CrossRef] [PubMed]
- Wells, G.A.; Cranney, A.; Peterson, J.; Boucher, M.; Shea, B.; Robinson, V.; Coyle, D.; Tugwell, P. Etidronate for the primary and secondary prevention of osteoporotic fractures in postmenopausal women. Cochrane Database Syst. Rev. 2008. [Google Scholar] [CrossRef] [PubMed]
- Wells, G.A.; Cranney, A.; Peterson, J.; Boucher, M.; Shea, B.; Robinson, V.; Coyle, D.; Tugwell, P. Alendronate for the primary and secondary prevention of osteoporotic fractures in postmenopausal women. Cochrane Database Syst. Rev. 2008, CD001155. [Google Scholar] [CrossRef] [PubMed]
- Goodship, A.E.; Walker, P.C.; McNally, D.; Chambers, T.; Green, J.R. Use of a bisphosphonate (pamidronate) to modulate fracture repair in ovine bone. Ann. Oncol. 1994, 5 (Suppl. 7), S53–S55. [Google Scholar] [PubMed]
- Peter, C.P.; Cook, W.O.; Nunamaker, D.M.; Provost, M.T.; Seedor, J.G.; Rodan, G.A. Effect of alendronate on fracture healing and bone remodeling in dogs. J. Orthop. Res. 1996, 14, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, S.S.; Jaatinen, J.; Pelttari, A.; Lappalainen, R.; Monkkonen, J.; Venesmaa, P.K.; Kroger, H.P. Effect of locally administered zoledronic acid on injury-induced intramembranous bone regeneration and osseointegration of a titanium implant in rats. J. Orthop. Sci. 2009, 14, 431–436. [Google Scholar] [CrossRef]
- Skripitz, R.; Johansson, H.R.; Ulrich, S.D.; Werner, A.; Aspenberg, P. Effect of alendronate and intermittent parathyroid hormone on implant fixation in ovariectomized rats. J. Orthop. Sci. 2009, 14, 138–143. [Google Scholar] [CrossRef] [Green Version]
- Gerstenfeld, L.C.; Sacks, D.J.; Pelis, M.; Mason, Z.D.; Graves, D.T.; Barrero, M.; Ominsky, M.S.; Kostenuik, P.J.; Morgan, E.F.; Einhorn, T.A. Comparison of effects of the bisphosphonate alendronate versus the RANKL inhibitor denosumab on murine fracture healing. J. Bone Miner. Res. 2009, 24, 196–208. [Google Scholar] [CrossRef]
- Saito, T.; Sterbenz, J.M.; Malay, S.; Zhong, L.; MacEachern, M.P.; Chung, K.C. Effectiveness of anti-osteoporotic drugs to prevent secondary fragility fractures: SYSTEMATIC review and meta-analysis. Osteoporos. Int. 2017, 28, 3289–3300. [Google Scholar] [CrossRef]
- Adami, S.; Libanati, C.; Boonen, S.; Cummings, S.R.; Ho, P.R.; Wang, A.; Siris, E.; Lane, J.; Adachi, J.D.; Bhandari, M.; et al. Denosumab treatment in postmenopausal women with osteoporosis does not interfere with fracture-healing: RESULTS from the FREEDOM trial. J. Bone Jt. Surg. Am. 2012, 94, 2113–2119. [Google Scholar] [CrossRef] [PubMed]
- Andreassen, T.T.; Ejersted, C.; Oxlund, H. Intermittent parathyroid hormone (1-34) treatment increases callus formation and mechanical strength of healing rat fractures. J. Bone Miner. Res. 1999, 14, 960–968. [Google Scholar] [CrossRef] [PubMed]
- Tagil, M.; McDonald, M.M.; Morse, A.; Peacock, L.; Mikulec, K.; Amanat, N.; Godfrey, C.; Little, D.G. Intermittent PTH(1-34) does not increase union rates in open rat femoral fractures and exhibits attenuated anabolic effects compared to closed fractures. Bone 2010, 46, 852–859. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; D’Amelio, P.; Tyagi, A.M.; Vaccaro, C.; Li, J.Y.; Hsu, E.; Buondonno, I.; Sassi, F.; Adams, J.; Weitzmann, M.N.; et al. Regulatory T cells are expanded by Teriparatide treatment in humans and mediate intermittent PTH-induced bone anabolism in mice. EMBO Rep. 2018, 19, 156–171. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, L.; Kikuiri, T.; Akiyama, K.; Chen, C.; Xu, X.; Yang, R.; Chen, W.; Wang, S.; Shi, S. Mesenchymal stem cell-based tissue regeneration is governed by recipient T lymphocytes via IFN-gamma and TNF-alpha. Nat. Med. 2011, 17, 1594–1601. [Google Scholar] [CrossRef] [PubMed]
- Bernhardsson, M.; Aspenberg, P. Abaloparatide versus teriparatide: A head to head comparison of effects on fracture healing in mouse models. Acta Orthop. 2018. [Google Scholar] [CrossRef] [PubMed]
- Subbiah, V.; Madsen, V.S.; Raymond, A.K.; Benjamin, R.S.; Ludwig, J.A. Of mice and men: DIVERGENT risks of teriparatide-induced osteosarcoma. Osteoporos. Int. 2010, 21, 1041–1045. [Google Scholar] [CrossRef] [PubMed]
- Lou, S.; Lv, H.; Li, Z.; Tang, P.; Wang, Y. Parathyroid hormone analogues for fracture healing: PROTOCOL for a systematic review and meta-analysis of randomised controlled trials. BMJ Open 2018, 8, e019291. [Google Scholar] [CrossRef]
- Jacobson, J.A.; Yanoso-Scholl, L.; Reynolds, D.G.; Dadali, T.; Bradica, G.; Bukata, S.; Puzas, E.J.; Zuscik, M.J.; Rosier, R.; O’Keefe, R.J.; et al. Teriparatide therapy and beta-tricalcium phosphate enhance scaffold reconstruction of mouse femoral defects. Tissue Eng. Part A 2011, 17, 389–398. [Google Scholar] [CrossRef]
- Murray, S.S.; Brochmann Murray, E.J.; Wang, J.C.; Duarte, M.E. The history and histology of bone morphogenetic protein. Histol. Histopathol. 2016, 31, 721–732. [Google Scholar]
- Onishi, T.; Ishidou, Y.; Nagamine, T.; Yone, K.; Imamura, T.; Kato, M.; Sampath, T.K.; ten Dijke, P.; Sakou, T. Distinct and overlapping patterns of localization of bone morphogenetic protein (BMP) family members and a BMP type II receptor during fracture healing in rats. Bone 1998, 22, 605–612. [Google Scholar] [CrossRef]
- Canalis, E.; Economides, A.N.; Gazzerro, E. Bone morphogenetic proteins, their antagonists, and the skeleton. Endocr. Rev. 2003, 24, 218–235. [Google Scholar] [CrossRef] [PubMed]
- Pluhar, G.E.; Turner, A.S.; Pierce, A.R.; Toth, C.A.; Wheeler, D.L. A comparison of two biomaterial carriers for osteogenic protein-1 (BMP-7) in an ovine critical defect model. J. Bone Jt. Surg. Br. 2006, 88, 960–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawyer, A.A.; Song, S.J.; Susanto, E.; Chuan, P.; Lam, C.X.; Woodruff, M.A.; Hutmacher, D.W.; Cool, S.M. The stimulation of healing within a rat calvarial defect by mPCL-TCP/collagen scaffolds loaded with rhBMP-2. Biomaterials 2009, 30, 2479–2488. [Google Scholar] [CrossRef] [PubMed]
- Cipitria, A.; Reichert, J.C.; Epari, D.R.; Saifzadeh, S.; Berner, A.; Schell, H.; Mehta, M.; Schuetz, M.A.; Duda, G.N.; Hutmacher, D.W. Polycaprolactone scaffold and reduced rhBMP-7 dose for the regeneration of critical-sized defects in sheep tibiae. Biomaterials 2013, 34, 9960–9968. [Google Scholar] [CrossRef] [PubMed]
- Guzman, J.Z.; Merrill, R.K.; Kim, J.S.; Overley, S.C.; Dowdell, J.E.; Somani, S.; Hecht, A.C.; Cho, S.K.; Qureshi, S.A. Bone morphogenetic protein use in spine surgery in the United States: HOW have we responded to the warnings? Spine J. 2017, 17, 1247–1254. [Google Scholar] [CrossRef]
- Carragee, E.J.; Hurwitz, E.L.; Weiner, B.K. A critical review of recombinant human bone morphogenetic protein-2 trials in spinal surgery: EMERGING safety concerns and lessons learned. Spine J. 2011, 11, 471–491. [Google Scholar] [CrossRef]
- James, A.W.; LaChaud, G.; Shen, J.; Asatrian, G.; Nguyen, V.; Zhang, X.; Ting, K.; Soo, C. A Review of the Clinical Side Effects of Bone Morphogenetic Protein-2. Tissue Eng. Part B Rev. 2016, 22, 284–297. [Google Scholar] [CrossRef] [Green Version]
- Glaeser, J.D.; Salehi, K.; Kanim, L.E.A.; Sheyn, D.; NaPier, Z.; Behrens, P.H.; Garcia, L.; Cuellar, J.M.; Bae, H.W. Anti-Inflammatory Peptide Attenuates Edema and Promotes BMP-2-Induced Bone Formation in Spine Fusion. Tissue Eng. Part A 2018, 24, 1641–1651. [Google Scholar] [CrossRef]
- Tinsley, B.A.; Dukas, A.; Pensak, M.J.; Adams, D.J.; Tang, A.H.; Ominsky, M.S.; Ke, H.Z.; Lieberman, J.R. Systemic Administration of Sclerostin Antibody Enhances Bone Morphogenetic Protein-Induced Femoral Defect Repair in a Rat Model. J. Bone Jt. Surg. Am. 2015, 97, 1852–1859. [Google Scholar] [CrossRef]
- Virk, M.S.; Alaee, F.; Tang, H.; Ominsky, M.S.; Ke, H.Z.; Lieberman, J.R. Systemic administration of sclerostin antibody enhances bone repair in a critical-sized femoral defect in a rat model. J. Bone Jt. Surg. Am. 2013, 95, 694–701. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.S.; Lastfogel, J.; Ackerman, L.L.; Jea, A.; Robling, A.G.; Tholpady, S.S. Loss of mechanosensitive sclerostin may accelerate cranial bone growth and regeneration. J. Neurosurg. 2018, 129, 1085–1091. [Google Scholar] [CrossRef]
- Wang, M.; Park, S.; Nam, Y.; Nielsen, J.; Low, S.A.; Srinivasarao, M.; Low, P.S. Bone-Fracture-Targeted Dasatinib-Oligoaspartic Acid Conjugate Potently Accelerates Fracture Repair. Bioconjug. Chem. 2018, 29, 3800–3809. [Google Scholar] [CrossRef] [PubMed]
- Bara, J.J.; Dresing, I.; Zeiter, S.; Anton, M.; Daculsi, G.; Eglin, D.; Nehrbass, D.; Stadelmann, V.A.; Betts, D.C.; Muller, R.; et al. A doxycycline inducible, adenoviral bone morphogenetic protein-2 gene delivery system to bone. J. Tissue Eng. Regen. Med. 2018, 12, e106–e118. [Google Scholar] [CrossRef] [PubMed]
- Kolk, A.; Tischer, T.; Koch, C.; Vogt, S.; Haller, B.; Smeets, R.; Kreutzer, K.; Plank, C.; Bissinger, O. A novel nonviral gene delivery tool of BMP-2 for the reconstitution of critical-size bone defects in rats. J. Biomed. Mater. Res. A 2016, 104, 2441–2455. [Google Scholar] [CrossRef]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Russow, G.; Jahn, D.; Appelt, J.; Märdian, S.; Tsitsilonis, S.; Keller, J. Anabolic Therapies in Osteoporosis and Bone Regeneration. Int. J. Mol. Sci. 2019, 20, 83. https://doi.org/10.3390/ijms20010083
Russow G, Jahn D, Appelt J, Märdian S, Tsitsilonis S, Keller J. Anabolic Therapies in Osteoporosis and Bone Regeneration. International Journal of Molecular Sciences. 2019; 20(1):83. https://doi.org/10.3390/ijms20010083
Chicago/Turabian StyleRussow, Gabriele, Denise Jahn, Jessika Appelt, Sven Märdian, Serafeim Tsitsilonis, and Johannes Keller. 2019. "Anabolic Therapies in Osteoporosis and Bone Regeneration" International Journal of Molecular Sciences 20, no. 1: 83. https://doi.org/10.3390/ijms20010083
APA StyleRussow, G., Jahn, D., Appelt, J., Märdian, S., Tsitsilonis, S., & Keller, J. (2019). Anabolic Therapies in Osteoporosis and Bone Regeneration. International Journal of Molecular Sciences, 20(1), 83. https://doi.org/10.3390/ijms20010083