p53 at the Crossroads between Different Types of HDAC Inhibitor-Mediated Cancer Cell Death



Abstract
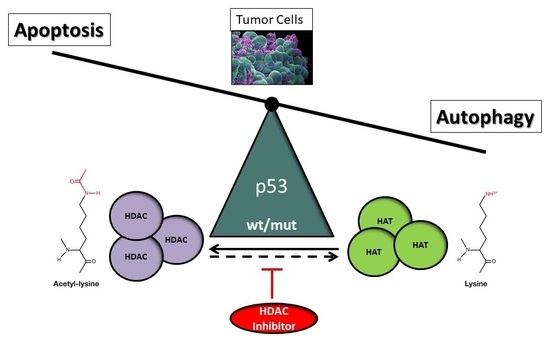
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction: Cell Death and Cancer
2. Post-Translational Acetylation Defects in Cancer
3. HATs and HDACs, the Effectors of Post-Translational Acetylation and Deacetylation
4. Inhibition of Post-Translational Deacetylation in Cancer Cells
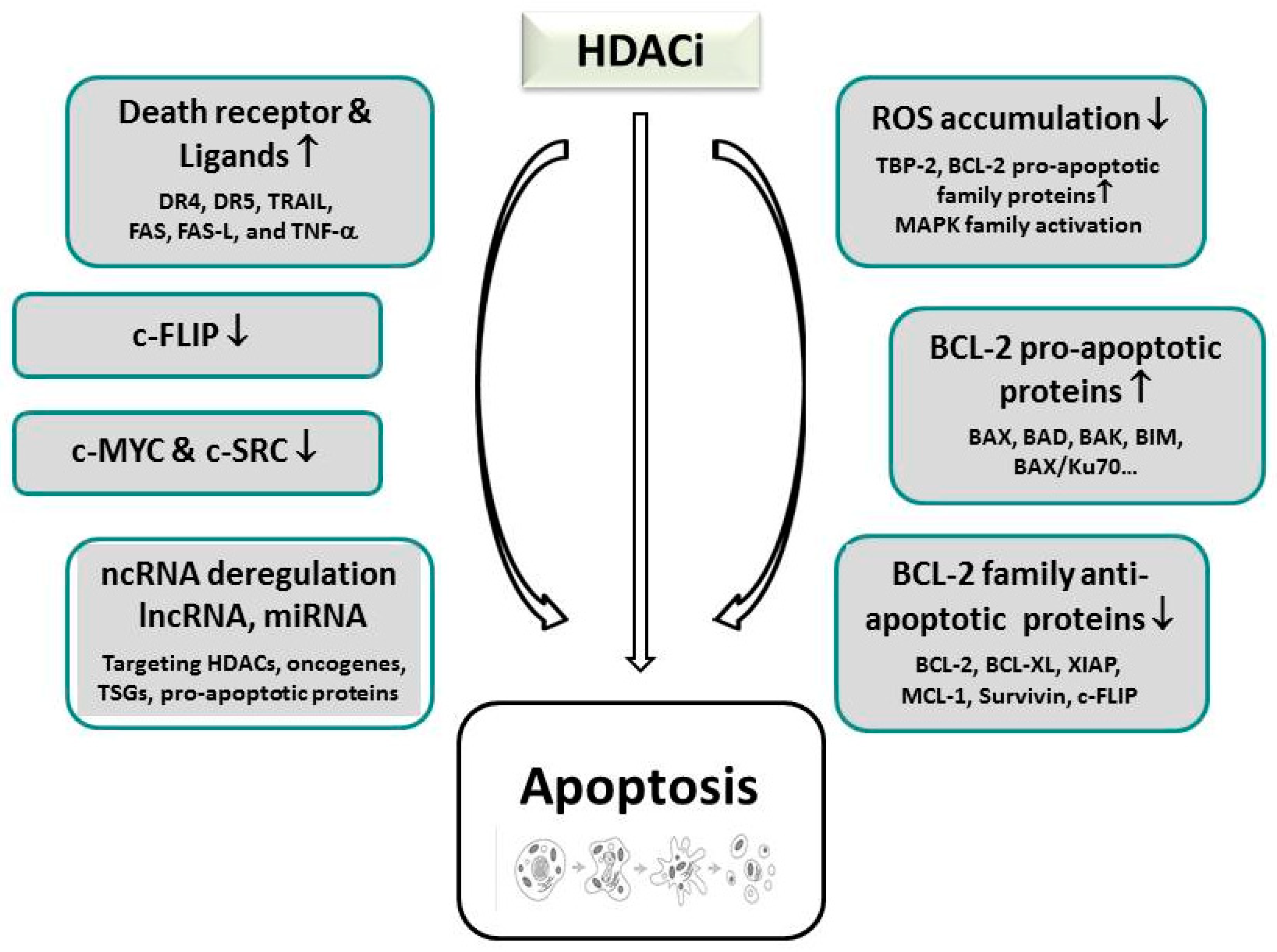
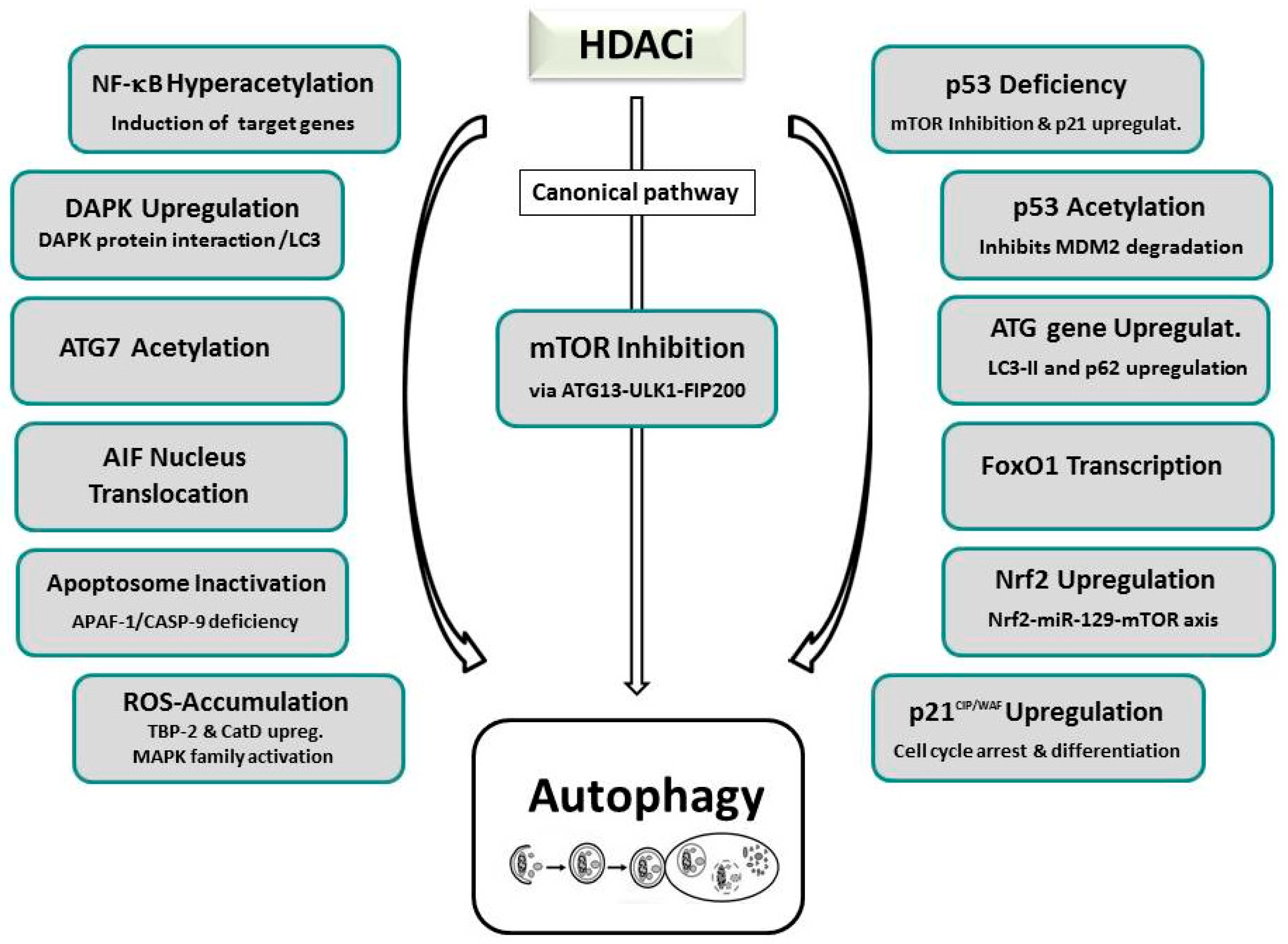
5. Effector Mechanisms of HDACi-Induced Cell Death
5.1. HDAC Inhibitor-Induced Apoptosis
5.2. HDAC Inhibitor-Induced Autophagy
6. The Role of p53 in HDACi-Mediated Cancer Cell Death
6.1. Post-Translational Regulation of Wild-Type and Mutant p53 by Acetylation
6.2. HDACi-Mediated Apoptosis and Autophagy by p53
6.3. Balanced Control of HDACi-Mediated Apoptosis and Autophagy by p53
7. Perspectives and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Lockshin, R.; Williams, C. Programmed cell death—II. Endocrine potentiation of the breakdown of the intersegmental muscles of silkmoths. J. Insect Physiol. 1964, 10, 643–649. [Google Scholar] [CrossRef]
- Lockshin, R.A.; Zakeri, Z. Apoptosis, autophagy, and more. Int. J. Biochem. Cell Biol. 2004, 36, 2405–2419. [Google Scholar] [CrossRef]
- Tan, M.; Ooi, J.; Ismail, N.; Moad, A.; Muhammad, T. Programmed cell death pathways and current antitumor targets. Pharmacol. Res. 2009, 26, 1547–1560. [Google Scholar] [CrossRef] [PubMed]
- Vanden Berghe, T.; Linkermann, A.; Jouan-Lanhouet, S.; Walczak, H.; Vandenabeele, P. Regulated necrosis: The expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol. 2014, 15, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Schweichel, J.-U.; Merker, H.-J. The morphology of various types of cell death in prenatal tissues. Teratology 1973, 7, 253–256. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Salehi, B.; Sharifi-Rad, M.; Pezzani, R.; Kobarfard, F.; Sharifi-Rad, J.; Nigam, M. Programmed Cell Death from a Cancer Perspective: An Overview. Mol. Diagn. Ther. 2018, 22, 281–295. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Sarkar, S.; Horn, G.; Moulton, K.; Oza, A.; Byler, S.; Kokolus, S.; Longacre, M. Cancer development, progression, and therapy: An epigenetic overview. Int. J. Mol. Sci. 2013, 14, 21087–21113. [Google Scholar] [CrossRef]
- Garraway, L.A.; Lander, E.S. Lessons from the Cancer Genome. Cell 2013, 153, 17–37. [Google Scholar] [CrossRef] [Green Version]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.J.; Kinzler, K.W. Cancer Genome Landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, A.G.; Piluso, L.G.; Cai, X.; Gadd, B.J.; Ladurner, A.G.; Liu, X. An Acetylation Switch in p53 Mediates Holo-TFIID Recruitment. Mol. Cell 2007, 28, 408–421. [Google Scholar] [CrossRef]
- Fröhlich, L.F.; Mrakovcic, M.; Smole, C.; Zatloukal, K. Molecular mechanism leading to SAHA-induced autophagy in tumor cells: Evidence for a p53-dependent pathway. Cancer Cell Int. 2016, 16, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Mrakovcic, M.; Kleinheinz, J.; Fröhlich, L.F. Histone deacetylase inhibitor-induced autophagy in tumor cells: Implications for p53. Int. J. Mol. Sci. 2017, 18, 1883. [Google Scholar] [CrossRef] [PubMed]
- Mrakovcic, M.; Bohner, L.; Hanisch, M.; Fröhlich, L.F. Epigenetic Targeting of Autophagy via HDAC Inhibition in Tumor Cells: Role of p53. Int. J. Mol. Sci. 2018, 19, 3952. [Google Scholar] [CrossRef] [PubMed]
- Kornberg, R.D. Chromatin structure: A repeating unit of histones and DNA. Science 1974, 184, 868–871. [Google Scholar] [CrossRef] [PubMed]
- Grunstein, M. Histone acetylation in chromatin structure and transcription. Nature 1997, 389, 349–352. [Google Scholar] [CrossRef] [PubMed]
- Spange, S.; Wagner, T.; Heinzel, T.; Krämer, O.H. Acetylation of non-histone proteins modulates cellular signalling at multiple levels. Int. J. Biochem. Cell Biol. 2009, 41, 185–198. [Google Scholar] [CrossRef]
- Lee, D.Y.; Hayes, J.J.; Pruss, D.D.; Wolffe, A.P. A positive role for histone acetylation in transcription factor access to nucleosomal DNA. Cell 1993, 72, 73–84. [Google Scholar] [CrossRef]
- Roth, S.Y.; Allis, C.D. Histone acetylation and chromatin assembly: A single escort, multiple dances? Cell 1996, 87, 5–8. [Google Scholar] [CrossRef]
- Kazanets, A.; Shorstova, T.; Hilm, I.K.; Marques, M.; Witcher, M. Epigenetic silencing of tumor suppressor genes: Paradigms, puzzles, and potential. Biochim. Biophys. Acta Rev. Cancer. 2016, 1865, 275–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, W.; Roeder, R.G. Activation of p53 Sequence-Specific DNA Binding by Acetylation of the p53 C-Terminal Domain. Cell 1997, 90, 595–606. [Google Scholar] [CrossRef] [Green Version]
- Glozak, M.A.; Seto, E. Histone deacetylases and cancer. Oncogene 2007, 26, 5420–5432. [Google Scholar] [CrossRef] [Green Version]
- Ropero, S.; Esteller, M. The role of histone deacetylases (HDACs) in human cancer. Mol. Oncol. 2007, 1, 19–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Seto, E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb. Perspect. Med. 2016, 6, a026831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chrun, E.; Modolo, F.; Ivan, F.D. Histone modifications: A review about the presence of this epigenetic phenomenon in carcinogenesis. Pathol. Pract. 2017, 213, 1329–1339. [Google Scholar] [CrossRef] [PubMed]
- Barneda-Zahonero, B.; Parra, M. Histone deacetylases and cancer. Mol. Oncol. 2012, 6, 579–589. [Google Scholar] [CrossRef] [Green Version]
- Fraga, M.; Ballestar, E.; Villar-Garea, A.; Boix-Chornet, M.; Espada, J.; Schotta, G.; Bonaldi, T.; Haydon, C.; Ropero, S.; Petrie, K.; et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat. Genet. 2005, 37, 391–400. [Google Scholar] [CrossRef]
- Toh, Y.; Ohga, T.; Endo, K.; Adachi, E.; Kusumoto, H.; Haraguchi, M.; Okamura, T.; Nicolson, G.L. Expression of the metastasis-associated MTA1 protein and its relationship to deacetylation of the histone H4 in esophageal squamous cell carcinomas. Int. J. Cancer 2004, 110, 362–367. [Google Scholar] [CrossRef] [Green Version]
- Seligson, D.; Horvath, S.; Shi, T.; Yu, H.; Tze, S.; Grunstein, M.; Kurdistani, S. Global histone modification patterns predict risk of prostate cancer recurrence. Nature 2005, 435, 1262–1266. [Google Scholar] [CrossRef]
- Seligson, D.; Horvath, S.; McBrian, M.; Mah, V.; Yu, H.; Tze, S.; Wang, Q.; Chia, D.; Goodglick, L.; Kurdistani, S. Global levels of histone modifications predict prognosis in different cancers. Am. J. Pathol. 2009, 174, 1619–1628. [Google Scholar] [CrossRef] [PubMed]
- Gayther, S.; Batley, S.; Linger, L.; Bannister, A.; Thorpe, K.; Chin, S.; Daigo, Y.; Russell, P.; Wilson, A.; Sowter, H.; et al. Mutations truncating the EP300 acetylase in human cancers. Nat. Genet. 2000, 24, 300–303. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Gural, A.; Sun, X.; Zhao, X.; Perna, F.; Huang, G.; Hatlen, M.; Vu, L.; Liu, F.; Xu, H.; et al. The leukemogenicity of AML1-ETO is dependent on site-specific lysine acetylation. Science 2011, 333, 765–769. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Wu, L.; Liang, T.; Liu, Z.; Li, J.; Li, D.; Xie, H.; Yin, S.; Yu, J.; Lin, Q.; et al. Overexpression of myocyte enhancer factor 2 and histone hyperacetylation in hepatocellular carcinoma. J. Cancer Res. Clin. Oncol. 2008, 134, 83–91. [Google Scholar] [CrossRef]
- Zhong, J.; Ding, L.; Bohrer, L.; Pan, Y.; Liu, P.; Zhang, J.; Sebo, T.; Karnes, R.; Tindall, D.; van Deursen, J.; et al. p300 acetyltransferase regulates androgen receptor degradation and PTEN-deficient prostate tumorigenesis. Cancer Res. 2014, 74, 1870–1880. [Google Scholar] [CrossRef] [PubMed]
- Debes, J.; Sebo, T.; Lohse, C.; Murphy, L.; Haugen, D.; Tindall, D. p300 in prostate cancer proliferation and progression. Cancer Res. 2003, 63, 7638–7640. [Google Scholar]
- Iyer, N.G.; Ozdag, H.; Caldas, C. p300/CBP and cancer. Oncogene 2004, 23, 4225–4231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishihama, K.; Yamakawa, M.; Semba, S.; Takeda, H.; Kawata, S.; Kimura, S.; Kimura, W. Expression of HDAC1 and CBP/p300 in human colorectal carcinomas. J. Clin. Pathol. 2007, 60, 1205–1210. [Google Scholar] [CrossRef]
- Patel, H.; Du, Y.; Ard, P.; Phillips, C.; Carella, B.; Chen, C.; Rakowski, C.; Chatterjee, C.; Liebermann, P.; Lane, W.; et al. The c-MYC oncoprotein is a substrate of the acetyltransferases hGCN5/PCAF and TIP60. Mol. Cell. Biol. 2004, 24, 10826–10834. [Google Scholar] [CrossRef] [PubMed]
- Grossmann, S. p300/CBP/p53 interaction and regulation of the p53 response. Eur. J. Biochem. 2001, 268, 2773–2778. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Scolnick, D.; Trievel, R.; Zhang, H.; Marmorstein, R.; Halazonetis, T. p53 sites acetylated in vitro by PCAF and p300 are acetylated in vivo in response to DNA damage. Mol. Cell. Biol. 1999, 19, 1202–1209. [Google Scholar] [CrossRef] [PubMed]
- Tessarz, P.; Kouzarides, T. Histone core modifications regulating nucleosome structure and dynamics. Nat. Rev. Mol. Cell Biol. 2014, 15, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Shilatifard, A. Chromatin modifications by methylation and ubiquitination: Implications in the regulation of gene expression. Annu. Rev. Biochem. 2006, 75, 243–269. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Lane, D.P. p53 in health and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Sarma, K.; Reinberg, D. Histone variants meet their match. Nat. Rev. Mol. Cell Biol. 2005, 6, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Strahl, B.; Allis, C. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef]
- Huang, H.; Sabari, B.R.; Garcia, B.A.; Allis, C.D.; Zhao, Y. SnapShot: Histone modifications. Cell 2014, 159, 458. [Google Scholar] [CrossRef] [PubMed]
- Roth, S.Y.; Denu, J.M.; Allis, C.D. Histone acetyltransferases. Annu. Rev. Biochem. 2001, 70, 81–120. [Google Scholar] [CrossRef] [PubMed]
- de Ruijter, A.J.; van Gennip, A.H.; Caron, H.N.; Kemp, S.; van Kuilenburg, A.B. Histone deacetylases (HDACs): Characterization of the classical HDAC family. Biochem. J. 2003, 370, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Morales, V.; Richard-Foy, H. Role of histone N-terminal tails and their acetylation in nucleosome dynamics. Mol. Cell. Biol. 2000, 20, 7230–7237. [Google Scholar] [CrossRef] [PubMed]
- Gregoretti, I.V.; Lee, Y.-M.; Goodson, H.V. Molecular evolution of the histone deacetylase family: Functional implications of phylogenetic analysis. J. Mol. Biol. 2004, 338, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Glozak, M.A.; Sengupta, N.; Zhang, X.; Seto, E. Acetylation and deacetylation of non-histone proteins. Gene 2005, 363, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Buchwald, M.; Krämer, O.H.; Heinzel, T. HDACi - Targets beyond chromatin. Cancer Lett. 2009, 280, 160–167. [Google Scholar] [CrossRef]
- Haberland, M.; Montgomery, R.L.; Olson, E.N. The many roles of histone deacetylases in development and physiology: Implications for disease and therapy. Nat. Rev. Genet. 2011, 10, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Workman, J. Histone acetyltransferase complexes: One size doesn’t fit all. Nat. Rev. Mol. Cell Biol. 2007, 8, 284–295. [Google Scholar] [CrossRef] [PubMed]
- Marmorstein, R.; Roth, S.Y. Histone acetyltransferases: Function, structure, and catalysis. Curr. Opin. Genet. Dev. 2001, 11, 155–161. [Google Scholar] [CrossRef]
- Wapenaar, H.; Dekker, F. Histone acetyltransferases: Challenges in targeting bi-substrate enzymes. Clin. Epigenetics 2016, 8, 59. [Google Scholar] [CrossRef]
- Santer, F.; Höschele, P.; Oh, S.; Erb, H.; Bouchal, J.; Cavaretta, I.; Parson, W.; Meyers, D.; Cole, P.; Culig, Z. Inhibition of the acetyltransferases p300 and CBP reveals a targetable function for p300 in the survival and invasion pathways of prostate cancer cell lines. Mol. Cancer Ther. 2011, 10, 1644–1655. [Google Scholar] [CrossRef]
- Weidle, U.H.; Grossmann, A. Inhibition of histone deacetylases: A new strategy to target epigenetic modifications for anticancer treatment. Anticancer Res. 2000, 20, 1471–1485. [Google Scholar]
- Bolden, J.; Peart, M.; Johnstone, R. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 2006, 5, 769–784. [Google Scholar] [CrossRef]
- Lee, J.-H.; Choy, M.L.; Marks, P.A. Mechanism of resistance to histone deacetylase inhibitors. Adv. Cancer Res. 2012, 116, 39–86. [Google Scholar] [CrossRef] [PubMed]
- Dokmanovic, M.; Clarke, C.; Marks, P.A. Histone Deacetylase Inhibitors: Overview and Perspectives. Mol. Cancer Res. 2007, 5, 981–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balasubramanian, S.; Verner, E.; Buggy, J.J. Isoform-specific histone deacetylase inhibitors: The next step? Cancer Lett. 2009, 280, 211–221. [Google Scholar] [CrossRef]
- Haggarty, S.J.; Koeller, K.M.; Wong, J.C.; Grozinger, C.M.; Schreiber, S.L. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc. Natl. Acad. Sci. USA 2003, 100, 4389–4394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richon, V.; Emiliani, S.; Verdin, E.; Webb, Y.; Breslow, R.; Rifkind, R.; Marks, P. A class of hybrid polar inducers of transformed cell differentiation inhibits histone deacetylases. Proc. Natl. Acad. Sci. USA 1998, 95, 3003–3007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coffey, D.C.; Kutko, M.C.; Glick, R.D.; Butler, L.M.; Heller, G.; Rifkind, R.A.; Marks, P.A.; Richon, V.M.; La Quaglia, M.P. The Histone Deacetylase Inhibitor, CBHA, Inhibits Growth of Human Neuroblastoma Xenografts in Vivo, Alone and Synergistically with All-Trans Retinoic Acid. Cancer Res. 2001, 61, 3591–3594. [Google Scholar]
- Plumb, J.A.; Finn, P.W.; Williams, R.J.; Bandara, M.J.; Romero, M.R.; Watkins, C.J.; La Thangue, N.B.; Brown, R. Pharmacodynamic Response and Inhibition of Growth of Human Tumor Xenografts by the Novel Histone Deacetylase Inhibitor PXD101. Mol. Cancer Ther. 2003, 2, 721–728. [Google Scholar] [PubMed]
- Furumai, R.; Matsuyama, A.; Kobashi, N.; Lee, K.; Nishiyama, M.; Nakajima, H.; Tanaka, A.; Komatsu, Y.; Nishino, N.; Yoshida, M.; et al. FK228 (Depsipeptide) as a Natural Prodrug That Inhibits Class I Histone Deacetylases. Cancer Res. 2002, 62, 4916–4921. [Google Scholar] [PubMed]
- Liu, T.; Kapustin, G.; Etzkorn, F.A. Design and Synthesis of a Potent Histone Deacetylase Inhibitor. J. Med. Chem. 2007, 50, 2003–2006. [Google Scholar] [CrossRef]
- Jose, B.; Oniki, Y.; Kato, T.; Nishino, N.; Sumida, Y. Novel histone deacetylase inhibitors: Cyclic tetrapeptide with trifluoromethyl and pentafluoroethyl ketones. Bioorg. Med. Chem. Lett. 2004, 14, 5343–5346. [Google Scholar] [CrossRef]
- Maggio, S.C.; Rosato, R.R.; Kramer, L.B.; Dai, Y.; Rahmani, M.; Paik, D.S.; Czarnik, A.C.; Payne, S.G.; Spiegel, S.; Grant, S. The Histone Deacetylase Inhibitor MS-275 Interacts Synergistically with Fludarabine to Induce Apoptosis in Human Leukemia Cells. Cancer Res. 2004, 64, 2590–2600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blum, K.A.; Advani, A.; Fernandez, L.; Van Der Jagt, R.; Kambhampati, S.; Kassis, J.; Davis, M.; Bonfils, C.; Dubay, M.; Dumouchel, J.; et al. Phase II study of the histone deacetylase inhibitor MGCD0103 in patients with previously treated chronic lymphocytic leukaemia. Br. J. Haematol. 2010, 147, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Boumber, Y.; Younes, A.; Garcia-Manero, G. Mocetinostat (MGCD0103): A review of an isotype-specific histone deacetylase inhibitor. Expert. Opin. Investig. Drugs 2011, 20, 823–829. [Google Scholar] [CrossRef] [PubMed]
- Minucci, S.; Zhu, P.; Kra, O.H.; Schimpf, A.; Giavara, S.; Sleeman, J.P.; Coco, F.L.; Nervi, C.; Pelicci, P.G.; Heinzel, T. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 2001, 20, 6969–6978. [Google Scholar] [Green Version]
- Rasheed, W.K.; Johnstone, R.W.; Prince, H.M. Histone deacetylase inhibitors in cancer therapy. Expert. Opin. Investig. Drugs 2017, 16, 659–678. [Google Scholar] [CrossRef] [PubMed]
- Krämer, O.H.; Göttlicher, M.; Heinzel, T. Histone deacetylase as a therapeutic target. Trends Endocrinol. Metab. 2001, 12, 294–300. [Google Scholar] [CrossRef]
- Marks, P.A.; Richon, V.M.; Rifkind, R.A. Histone deacetylase inhibitors: Inducers of differentiation or apoptosis of transformed cells. J. Natl. Cancer Inst. 2000, 92, 1210–1216. [Google Scholar] [CrossRef]
- Kim, Y.B.; Ki, S.W.; Yoshida, M.; Horinouchi, S. Mechanism of cell cycle arrest caused by histone deacetylase inhibitors in human carcinoma cells. J. Antibiot. (Tokyo) 2000, 53, 1191–1200. [Google Scholar] [CrossRef]
- Bali, P.; Pranpat, M.; Bradner, J.; Balasis, M.; Fiskus, W.; Guo, F.; Rocha, K.; Kumaraswamy, S.; Boyapalle, S.; Atadja, P.; et al. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: A novel basis for antileukemia activity of histone deacetylase inhibitors. J. Biol. Chem. 2005, 280, 26729–26734. [Google Scholar] [CrossRef]
- Newbold, A.; Falkenberg, K.J.; Prince, H.M.; Johnstone, R.W. How do tumor cells respond to HDAC inhibition? FEBS J. 2016, 283, 4032–4046. [Google Scholar] [CrossRef] [PubMed]
- Gui, C.Y.; Ngo, L.; Xu, W.S.; Richon, V.M.; Marks, P.A. Histone deacetylase (HDAC) inhibitor activation of p21WAF1 involves changes in promoter associated proteins, including HDAC1. Proc. Natl. Acad. Sci. USA 2004, 101, 1241–1246. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.M.; Hackanson, B.; Lübbert, M.; Jung, M. Histone deacetylase (HDAC) inhibitors in recent clinical trials for cancer therapy. Clin. Epigenetics 2010, 1, 117–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, J.; Cang, S.; Ma, Y.; Petrillo, R.L.; Liu, D. Novel histone deacetylase inhibitors in clinical trials as anti-cancer agents. J. Hematol. Oncol. 2010, 3, 1–13. [Google Scholar] [CrossRef] [PubMed]
- O´Connor, O.A.; Heaney, M.L.; Schwartz, L.; Richardson, S.; Willim, R.; MacGregor-Cortelli, B.; Curly, T.; Moskowitz, C.; Portlock, C.; Horwitz, S.; et al. Clinical Experience with Intravenous and Oral Formulations of the Novel Histone Deacetylase Inhibitor Suberoylanilide Hydroxamic Acid in Patients with Advanced Hematologic Malignancies. J. Clin. Oncol. 2017, 24, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Duvic, M.; Vu, J. Update on the treatment of cutaneous T-cell lymphoma (CTCL): Focus on vorinostat. Biologics 2007, 1, 377. [Google Scholar] [PubMed]
- Duvic, M.; Talpur, R.; Ni, X.; Zhang, C.; Hazarika, P.; Kelly, C.; Chiao, J.H.; Reilly, J.F.; Ricker, J.L.; Richon, V.M.; et al. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood 2007, 109, 31–39. [Google Scholar] [CrossRef] [Green Version]
- Bertino, E.M.; Otterson, G.A. Romidepsin: A novel histone deacetylase inhibitor for cancer. Expert. Opin. Investig. Drugs 2011, 20, 1151–1158. [Google Scholar] [CrossRef]
- Rashidi, A.; Cashen, A.F. Belinostat for the treatment of relapsed or refractory peripheral T-cell lymphoma. Futur. Oncol. 2015, 11, 1659–1664. [Google Scholar] [CrossRef] [PubMed]
- Greig, S.L. Panobinostat: A review in relapsed or refractory multiple myeloma. Target. Oncol. 2016, 11, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Slingerland, M.; Guchelaar, H.J.; Gelderblom, H. Histone deacetylase inhibitors: An overview of the clinical studies in solid tumors. Anticancer. Drugs 2014, 25, 140–149. [Google Scholar] [CrossRef]
- Rasheed, W.; Bishton, M.; Johnstone, R.W.; Prince, H.M. Histone deacetylase inhibitors in lymphoma and solid malignancies. Expert Rev. Anticancer. Ther. 2008, 8, 413–432. [Google Scholar] [CrossRef] [PubMed]
- Modesitt, S.; Sill, M.; Hoffman, J.; Bender, D.; Gynecologic Oncology Group. A phase II study of vorinostat in the treatment of persistent or recurrent epithelial ovarian or primary peritoneal carcinoma: A Gynecologic Oncology Group study. Gynecol. Oncol. 2008, 109, 182–186. [Google Scholar] [CrossRef]
- Molife, L.R.; Attard, G.; Fong, P.C.; Karavasilis, V.; Reid, A.H.M.; Patterson, S.; Riggs, C.E.; Higano, C.; Stadler, W.M.; McCulloch, W.; et al. Phase II, two-stage, single-arm trial of the histone deacetylase inhibitor (HDACi) romidepsin in metastatic castration-resistant prostate cancer (CRPC). Ann. Oncol. 2010, 21, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Stadler, W.M.; Margolin, K.; Ferber, S.; McCulloch, W.; Thompson, J.A. A phase II study of depsipeptide in refractory metastatic renal cell cancer. Clin. Genitourin. Cancer. 2006, 5, 57–60. [Google Scholar] [CrossRef]
- Haigentz, M.; Kim, M.; Sarta, C.; Lin, J.; Keresztes, R.S.; Culliney, B.; Gaba, A.G.; Smith, R.V.; Shapiro, G.I.; Chirieac, L.R.; et al. Phase II trial of the histone deacetylase inhibitor romidepsin in patients with recurrent/metastatic head and neck cancer. Oral Oncol. 2012, 48, 1281–1288. [Google Scholar] [CrossRef] [Green Version]
- Luu, T.H.; Morgan, R.J.; Leong, L.; Lim, D.; McNamara, M.; Portnow, J.; Frankel, P.; Smith, D.D.; Doroshow, J.H.; Wong, C.; et al. A phase II trial of vorinostat (suberoylanilide hydroxamic acid) in metastatic breast cancer: A California Cancer Consortium study. Clin. Cancer Res. 2008, 14, 7138–7142. [Google Scholar] [CrossRef] [PubMed]
- Gryder, B.E.; Sodji, Q.H.; Oyelere, A.K. Targeted cancer therapy: Giving histone deacetylase inhibitors all they need to succeed. Futur. Med. Chem. 2012, 4, 505–524. [Google Scholar] [CrossRef] [PubMed]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.; Chen, Z.; Shin, D. Nanoparticle therapeutics: An emerging treatment modality for cancer. Nat. Rev. Drug. Discov. 2008, 7, 771–782. [Google Scholar] [CrossRef] [PubMed]
- Mrakovcic, M.; Absenger, M.; Riedl, R.; Smole, C.; Roblegg, E.; Fröhlich, L.F.; Fröhlich, E. Assessment of long-term effects of nanoparticles in a microcarrier cell culture system. PLoS ONE 2013, 8, e56791. [Google Scholar] [CrossRef]
- Khan, N.; Jeffers, M.; Kumar, S.; Hackett, C.; Boldog, F.; Khramtsov, N.; Qian, X.; Mills, E.; Berghs, S.C.; Carey, N.; et al. Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors. Biochem. J. 2008, 409, 581–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, W.S.; Parmigiani, R.B.; Marks, P.A. Histone deacetylase inhibitors: Molecular mechanisms of action. Oncogene 2007, 26, 5541–5552. [Google Scholar] [CrossRef] [PubMed]
- Marks, P.A. The mechanism of the anti-tumor activity of the histone deacetylase inhibitor, suberoylanilide hydroxamic acid (SAHA). Cell Cycle 2004, 3, 534–535. [Google Scholar] [CrossRef] [PubMed]
- Bose, P.; Dai, Y.; Grant, S. Histone deacetylase inhibitor (HDACI) mechanisms of action: Emerging insights. Pharmacol. Ther. 2014, 143, 323–336. [Google Scholar] [CrossRef] [Green Version]
- Richon, V.M.; Sandhoff, T.W.; Rifkind, R.A.; Marks, P.A. Histone deacetylase inhibitor selectively induces p21 WAF1 expression and gene-associated histone acetylation. Proc. Natl. Acad. Sci. USA 2000, 97, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Sandor, V.; Senderowicz, A.; Mertins, S.; Sackett, D.; Sausville, E.; Blagoskonny, M.V.; Bates, S.E. P21-dependent G1 arrest with downregulation of cyclin D1 and upregulation of cyclin E by the histone deacetylase inhibitor FR901228. Br. J. Cancer 2000, 83, 817–825. [Google Scholar] [CrossRef] [PubMed]
- Butler, L.M.; Zhou, X.; Xu, W.-S.; Scher, H.I.; Rifkind, R.A.; Marks, P.A.; Richon, V.M. The histone deacetylase inhibitor SAHA arrests cancer cell growth, up-regulates thioredoxin-binding protein-2, and down-regulates thioredoxin. Proc. Natl. Acad. Sci. USA 2002, 99, 11700–11705. [Google Scholar] [CrossRef] [Green Version]
- Seo, S.-K.; Jin, H.-O.; Woo, S.-H.; Kim, Y.-S.; An, S.; Lee, J.-H.; Hong, S.-I.; Lee, K.-H.; Choe, T.-B.; Park, I.-C. Histone Deacetylase Inhibitors Sensitize Human Non-small Cell Lung Cancer Cells to Ionizing Radiation Through Acetyl p53-Mediated c-myc Down-Regulation. J. Thorac. Oncol. 2011, 6, 1313–1319. [Google Scholar] [CrossRef] [Green Version]
- Takai, N.; Desmond, J.; Kumagai, T.; Gui, D.; Said, J.; Whittaker, S.; Miyakawa, I.; Koeffler, H. Histone deacetylase inhibitors have a profound antigrowth activity in endometrial cancer cells. Clin. Cancer Res. 2004, 10, 1141–1149. [Google Scholar] [CrossRef]
- Souleimani, A.; Asselin, C. Regulation of c-myc expression by sodium butyrate in the colon carcinoma cell line Caco-2. FEBS Lett. 1993, 326, 45–50. [Google Scholar] [CrossRef] [Green Version]
- Hirsch, C.L.; Smith-Windsor, E.L.; Bonham, K. Src family kinase members have a common response to histone deacetylase inhibitors in human colon cancer cells. Int. J. Cancer 2006, 118, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Vrana, J.; Decker, R.; Johnson, C.; Wang, Z.; Jarvis, W.; Richon, V.; Ehinger, M.; Fisher, P.; Grant, S. Induction of apoptosis in U937 human leukemia cells by suberoylanilide hydroxamic acid (SAHA) proceeds through pathways that are regulated by Bcl-2/Bcl-XL, c-Jun, and p21CIP1, but independent of p53. Oncogene 1999, 18, 7016–7025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nawrocki, S.T.; Carew, J.S.; Douglas, L.; Cleveland, J.L.; Humphreys, R.; Houghton, J.A. Histone Deacetylase Inhibitors Enhance Lexatumumab-Induced Apoptosis via a p21 Cip1-Dependent Decrease in Survivin Levels. Cancer Res. 2007, 67, 6987–6995. [Google Scholar] [CrossRef] [PubMed]
- Rahmani, M.; Yu, C.; Reese, E.; Ahmed, W.; Hirsch, K.; Dent, P.; Grant, S. Inhibition of PI-3 kinase sensitizes human leukemic cells to histone deacetylase inhibitor-mediated apoptosis through p44/42 MAP kinase inactivation and abrogation of p21 CIP1/WAF1 induction rather than AKT inhibition. Oncogene 2003, 22, 6231–6242. [Google Scholar] [CrossRef]
- Burgess, A.J.; Pavey, S.; Warrener, R.; Hunter, L.K.; Piva, T.J.; Musgrove, E.A.; Saunders, N.; Parsons, P.G.; Gabrielli, B.G. Up-Regulation of p21 WAF1/CIP1 by Histone Deacetylase Inhibitors Reduces Their Cytotoxicity. Mol. Pharmacol. 2001, 60, 828–837. [Google Scholar]
- Steinmann, R.; Hoffmann, B.; Iro, A.; Guillouf, C.; Liebermann, D.A.; El-Houseini, M.E. Induction of p21 (WAF-1/CIP1) during differentiation. Oncogene 1994, 9, 3389–3396. [Google Scholar]
- Harper, J.W.; Adami, G.R.; Wei, N.; Keyomarsi, K.; Elledge, S.J. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell 1993, 75, 805–816. [Google Scholar] [CrossRef]
- Qiu, L.; Burgess, A.; Fairlie, D.; Leonard, H.; Parsons, P.; Gabrielli, B. Histone deacetylase inhibitors trigger a G2 checkpoint in normal cells that is defective in tumor cells. Mol. Biol. Cell 2000, 11, 2069–2083. [Google Scholar] [CrossRef] [PubMed]
- Ruefli, A.A.; Ausserlechner, M.J.; Bernhard, D.; Sutton, V.R.; Tainton, K.M.; Kofler, R.; Smyth, M.J.; Johnstone, R.W. The histone deacetylase inhibitor and chemotherapeutic agent suberoylanilide hydroxamic acid (SAHA) induces a cell-death pathway characterized by cleavage of Bid and production of reactive oxygen species. Proc. Natl. Acad. Sci. USA 2001, 98, 10833–10838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Choy, M.L.; Ngo, L.; Foster, S.S.; Marks, P.A. Histone deacetylase inhibitor induces DNA damage, which normal but not transformed cells can repair. Proc. Natl. Acad. Sci. USA 2010, 107, 14639–14644. [Google Scholar] [CrossRef] [Green Version]
- Warrener, R.; Beamish, H.; Burgess, A.; Waterhouse, N.J.; Giles, N.; Fairlie, D.; Gabrielli, B. Tumor cell-selective cytotoxicity by targeting cell cycle checkpoints. FASEB J. 2003, 17, 1550–1552. [Google Scholar] [CrossRef]
- Conrad, E.; Polonio-Vallon, T.; Meister, M.; Matt, S.; Bitomsky, N.; Herbel, C.; Liebl, M.; Greiner, V.; Kriznik, B.; Schumacher, S.; et al. HIPK2 restricts SIRT1 activity upon severe DNA damage by a phosphorylation-controlled mechanism. Cell Death Differ. 2016, 23, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Ungerstedt, J.S.; Sowa, Y.; Xu, W.; Shao, Y.; Dokmanovic, M.; Perez, G.; Ngo, L.; Holmgren, A.; Jiang, X.; Marks, P.A. Role of thioredoxin in the response of normal and transformed cells to histone deacetylase inhibitors. Proc. Natl. Acad. Sci. USA 2005, 102, 673–678. [Google Scholar] [CrossRef] [Green Version]
- Namdar, M.; Perez, G.; Ngo, L.; Marks, P.A. Selective inhibition of histone deacetylase 6 (HDAC6) induces DNA damage and sensitizes transformed cells to anticancer agents. Proc. Natl. Acad. Sci. USA 2010, 107, 20003–20008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Zhu, W.-G. Targeting Histone Deacetylases for Cancer Therapy: From Molecular Mechanisms to Clinical Implications. Int. J. Biol. Sci. 2014, 10, 757–770. [Google Scholar] [CrossRef] [Green Version]
- Miller, K.M.; Tjeertes, J.V.; Coates, J.; Legube, G.; Polo, S.E.; Britton, S.; Jackson, S.P. Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining. Nat. Struct. Mol. Biol. 2010, 17, 1144–1151. [Google Scholar] [CrossRef] [PubMed]
- Bhaskara, S.; Knutson, S.K.; Jiang, G.; Chandrasekharan, M.B.; Wilson, A.J.; Zheng, S.; Yenamandra, A.; Locke, K.; Yuan, J.L.; Bonine-Summers, A.R.; et al. Hdac3 is essential for the maintenance of chromatin structure and genome stability. Cancer Cell 2010, 18, 436–447. [Google Scholar] [CrossRef] [Green Version]
- Thurn, K.T.; Thomas, S.; Raha, P.; Qureshi, I.; Munster, P.N. Histone deacetylase regulation of ATM-mediated DNA damage signaling. Mol. Cancer Ther. 2013, 12, 2078–2087. [Google Scholar] [CrossRef] [PubMed]
- Kao, G.D.; McKenna, W.G.; Guenther, M.G.; Muschel, R.J.; Lazar, M.A.; Yen, T.J. Histone deacetylase 4 interacts with 53BP1 to mediate the DNA damage response. J. Cell Biol. 2003, 160, 1017–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotian, S.; Liyanarachchi, S.; Zelent, A.; Parvin, J.D. Histone deacetylase 9 and 10 are required for homologous recombination. J. Biol. Chem. 2011, 286, 7722–7726. [Google Scholar] [CrossRef] [PubMed]
- Gorospe, M.; de Cabo, R. AsSIRTing the DNA damage response. Trends Cell Biol. 2008, 18, 77–83. [Google Scholar] [CrossRef]
- Kaidi, A.; Weinert, B.T.; Choudhary, C.; Jackson, S.P. Human SIRT6 promotes DNA end resection through CtlP deacetylation. Science 2010, 329, 1348–1353. [Google Scholar] [CrossRef] [PubMed]
- Motoslavsky, R.; Chua, K.F.; Lombard, D.B.; Pang, W.W.; Fischer, M.R.; Gellon, L.; Liu, P.; Motoslavsky, G.; Franco, S.; Murphy, M.M.; et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell 2006, 124, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Zhao, W.; Chen, Y.; Zhao, Y.; Gu, W. Acetylation Is Indispensable for p53 Activation. Cell 2008, 133, 612–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosato, R.R.; Almenara, J.A.; Grant, S. The Histone Deacetylase Inhibitor MS-275 Promotes Differentiation or Apoptosis in Human Leukemia Cells through a Process Regulated by Generation of Reactive Oxygen Species and Induction of p21 CIP1/WAF1. Cancer Res. 2003, 63, 3637–3645. [Google Scholar] [PubMed]
- Lin, K.; Wang, Y.; Chen, C.; Ho, C.; Su, W.; Jou, Y. HDAC inhibitors augmented cell migration and metastasis through induction of PKCs leading to identification of low toxicity modalities for combination cancer therapy. Clin. Cancer Res. 2012, 18, 4691–4701. [Google Scholar] [CrossRef] [PubMed]
- Pulukuri, S.; Gorantla, B.; Rao, J. Inhibition of histone deacetylase activity promotes invasion of human cancer cells through activation of urokinase plasminogen activator. J. Biol Chem. 2007, 282, 35594–35603. [Google Scholar] [CrossRef] [PubMed]
- Schoepflin, Z.R.; Shapiro, I.M.; Risbud, M.V. Class I and IIa HDACs mediate HIF-1a stability through PHD2-dependent mechanism, while HDAC6, a classIIb member, promotes HIF1a transcriptional activity in nucleus pulposus cells of the intervertebral disc. J. Bone Miner. Res. 2016, 31, 1287–1299. [Google Scholar] [CrossRef]
- Deroanne, C.F.; Bonjean, K.; Servotte, S.; Devy, L.; Colige, A.; Clausse, N.; Blacher, S.; Verdin, E.; Foidart, J.M.; Nusgens, B.V.; et al. Histone deacetylases inhibitors as anti-angiogenic agents altering vascular endothelial growth factor signaling. Oncogene 2002, 21, 427–436. [Google Scholar] [CrossRef] [Green Version]
- Jeong, J.W.; Bae, M.K.; Ahn, M.Y.; Kim, S.H.; Sohn, T.K.; Bae, M.H.; Yoo, M.A.; Song, E.J.; Lee, K.J.; Kim, K.W. Regulation and destabilization of HIF-1alpha by ARD1-mediated acetylation. Cell 2002, 111, 709–720. [Google Scholar] [CrossRef]
- Fournel, M.; Bonfils, C.; Hou, Y.; Yan, P.; Trachy-Bourget, M.C.; Kalita, A.; Liu, J.; Lu, A.H.; Zhou, N.Z.; Robert, M.F.; et al. MGCD0103, a novel isotype-selective histone deacetylase inhibitor, has broad spectrum antitumor activity in vitro and in vivo. Mol. Cancer Ther. 2008, 7, 759–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vigushin, D.M.; Ali, S.; Pace, P.E.; Mirsaidi, N.; Ito, K.; Adcock, I.; Coombes, R.C. Trichostatin A is a histone deacetylase inhibitor with potent antitumor activity against breast cancer in vivo. Clin. Cancer Res. 2001, 7, 971–976. [Google Scholar] [PubMed]
- Sato, N.; Ohta, T.; Kitagawa, H.; Kayahara, M.; Ninomiya, I.; Fushida, S.; Fujimura, T.; Nishimura, G.; Shimizu, K.; Miwa, K. FR901228, a novel histone deacetylase inhibitor, induces cell cycle arrest and subsequent apoptosis in refractory human pancreatic cancer cells. Int. J. Oncol. 2004, 24, 679–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, H.J.; Owa, T.; Hassig, C.A.; Shimada, J.; Schreiber, S.L. Depudecin induces morphological reversion of transformed fibroblasts via the inhibition of histone deacetylase. Proc. Natl. Acad. Sci. USA 1998, 95, 3356–3361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosato, R.; Almenara, J.; Dai, Y.; Grant, S. Simultaneous activation of the intrinsic and extrinsic pathways by histone deacetylase (HDAC) inhibitors and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) synergistically induces mitochondrial damage and apoptosis in human leukemia cells. Mol. Cancer Ther. 2003, 2, 1273–1284. [Google Scholar] [PubMed]
- Emanuele, S.; Lauricella, M.; Tesoriere, G. Histone deacetylase inhibitors: Apoptotic effects and clinical implications. Int. J. Oncol. 2008, 33, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Frew, A.J.; Johnstone, R.W.; Bolden, J.E. Enhancing the apoptotic and therapeutic effects of HDAC inhibitors. Cancer Lett. 2009, 280, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhong, Q. Histone deacetylase inhibitors and cell death. Cell. Mol. Life Sci. 2014, 71, 3885–3901. [Google Scholar] [CrossRef] [Green Version]
- Minucci, S.; Pelicci, P.G. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat. Rev. Cancer 2006, 6, 38–51. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Adachi, M.; Kawamura, R.; Imai, K. Bmf is a possible mediator in histone deacetylase inhibitors FK228 and CBHA-induced apoptosis. Cell Death Differ. 2006, 13, 129–140. [Google Scholar] [CrossRef]
- Zhao, Y.; Tan, J.; Zhuang, L.; Jiang, X.; Liu, E.T.; Yu, Q. Inhibitors of histone deacetylases target the Rb-E2F1 pathway for apoptosis induction through activation of proapoptotic protein Bim. Proc. Natl. Acad. Sci. USA 2005, 102, 16090–16095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, X.; Wang, X. Cytochrome C-mediated Apoptosis. Annu. Rev. Biochem. 2004, 73, 87–106. [Google Scholar] [CrossRef]
- Elkholi, R.; Floros, K.; Chipuk, J. The Role of BH3-Only Proteins in Tumor Cell Development, Signaling, and Treatment. Genes Cancer. Genes Cancer 2011, 2, 523–537. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Bae, S.C. Histone deacetylase inhibitors: Molecular mechanisms of action and clinical trials as anti-cancer drugs. Am. J. Transl. Res. 2011, 3, 166–179. [Google Scholar] [PubMed]
- Robert, C.; Rassool, F. V Chapter Three—HDAC Inhibitors: Roles of DNA Damage and Repair. Adv. Cancer Res. 2012, 116, 87–129. [Google Scholar]
- Sade, H.; Sarin, A. Reactive oxygen species regulate quiescent T-cell apoptosis via the BH3-only proapoptotic protein BIM. Cell Death Differ. 2004, 11, 416–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosato, R.R.; Maggio, S.C.; Almenara, J.A.; Payne, S.G.; Atajda, P.; Spiegel, S.; Dent, P.; Grant, S. The histone deacetylase inhibitor LAQ824 induces human leukemia cell death through a process involving XIAP down-regulation, oxidative injury, and the acid shingomyelinase-dependent generation of ceramide. Mol. Pharmacol. 2006, 69, 216–225. [Google Scholar] [CrossRef]
- Fandy, T.E.; Srivastava, R.K. Trichostatin A sensitizes TRAIL-resistant myeloma cells by downregulation of the antiapoptotic Bcl-2 proteins. Cancer Chemother. Pharmacol. 2006, 58, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, C.; Opipari, A.W.; Bian, X.; Castle, V.P.; Kwok, R.P. Ku70 acetylation mediates neuroblastoma cell death induced by histone deacetylase inhibitors. Proc. Natl. Acad. Sci. USA 2005, 102, 4842–4847. [Google Scholar] [CrossRef] [Green Version]
- Fandy, T.E.; Shankar, S.; Ross, D.D.; Sausville, E.; Srivastava, R.K. Interactive Effects of HDAC Inhibitors and TRAIL on Apoptosis Are Associated with Changes in Mitochondrial Functions and Expressions of Cell Cycle Regulatory Genes in Multiple Myeloma. Neoplasia 2005, 7, 646–657. [Google Scholar] [CrossRef] [Green Version]
- Sonnemann, J.; Dreyer, L.; Hartwig, M.; Palani, C.D.; Hong, L.T.T.; Klier, U.; Bröker, B.; Völker, U.; Beck, J.F. Histone deacetylase inhibitors induce cell death and enhance the apoptosis-inducing activity of TRAIL in Ewing’s sarcoma cells. J. Cancer Res. Clin. Oncol. 2007, 133, 847–858. [Google Scholar] [CrossRef]
- Carlisi, D.; Lauricella, M.; D’Anneo, A.; Emanuele, S.; Angileri, L.; Di Fazio, P.; Santulli, A.; Vento, R.; Tesoriere, G. The histone deacetylase inhibitor suberoylanilide hydroxamic acid sensitises human hepatocellular carcinoma cells to TRAIL-induced apoptosis by TRAIL-DISC activation. Eur. J. Cancer 2009, 45, 2425–2438. [Google Scholar] [CrossRef] [PubMed]
- Nakata, S.; Yoshida, T.; Horinaka, M.; Shiraishi, T.; Wakada, M.; Sakai, T. Histone deacetylase inhibitors upregulate death receptor 5/TRAIL-R2 and sensitize apoptosis induced by TRAIL/APO2-L in human malignant tumor cells. Oncogene 2004, 23, 6261–6271. [Google Scholar] [CrossRef] [Green Version]
- Aquilera, D.G.; Das, C.M.; Sinnappah-Kang, N.D.; Joyce, C.; Taylor, P.H.; Wen, S.; Hasselblatt, M.; Paulus, W.; Fuller, G.; Wolff, J.E.; et al. Reactivation of death receptor 4 (DR4) expression sensitizes medulloblastoma cell lines to TRAIL. J. Neurooncol. 2009, 93, 303–318. [Google Scholar] [CrossRef] [Green Version]
- Fulda, S.; Küfer, M.U.; Meyer, E.; van Valen, F.; Dockhorn-Dworniczak, B.; Debatin, K.M. Sensitization for death receptor- or drug-induced apoptosis by re-expression of caspase-8 through demethylation or gene transfer. Oncogene 2001, 20, 5865–5877. [Google Scholar] [CrossRef]
- Fulda, S. Modulation of TRAIL-induced apoptosis by HDAC inhibitors. Curr. Cancer Drug Targets 2008, 8, 132–140. [Google Scholar] [CrossRef]
- Gillenwater, A.M.; Zhong, M.; Lotan, R. Histone deacetylase inhibitor suberoylanilide hydroxamic acid induces apoptosis through both mitochondrial and Fas (Cd95) signaling in head and neck squamous carcinoma cells. Mol. Cancer Ther. 2007, 6, 2967–2975. [Google Scholar] [CrossRef] [Green Version]
- Shankar, S.; Singh, T.; Fandy, T.; Luetrakul, T.; Ross, D.; Srivastava, R. Interactive effects of histone deacetylase inhibitors and TRAIL on apoptosis in human leukemia cells: Involvement of both death receptor and mitochondrial pathways. Int. J. Mol. Med. 2005, 16, 1125–1138. [Google Scholar] [CrossRef]
- Ashkenazi, A.; Dixit, V.M. Death Receptors: Signaling and Modulation. Science 1998, 281, 1305–1309. [Google Scholar] [CrossRef] [PubMed]
- LeBlanc, H.; Ashkenzi, A. Apo2L/TRAIL and its death and decoy receptors. Cell Death Differ. 2003, 10, 66–75. [Google Scholar] [CrossRef] [Green Version]
- Natoni, F.; Diolordi, L.; Santoni, C.; Gilardini Montani, M.S. Sodium butyrate sensitises human pancreatic cancer cells to both the intrinsic and extrinsic apoptotic pathways. Biochim. Biophys. Acta 2005, 1745, 318–329. [Google Scholar] [CrossRef] [PubMed]
- Moutinho, C.; Esteller, M. MicroRNAs and Epigenetics. Adv. Cancer Res. 2017, 135, 189–220. [Google Scholar] [CrossRef] [PubMed]
- Noonan, E.J.; Place, R.F.; Pookot, D.; Basak, S.; Whitson, J.M.; Hirata, H.; Giardina, C.; Dahiya, R. miR-449a targets HDAC-1 and induces growth arrest in prostate cancer. Oncogene 2009, 28, 1714–1724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brest, P.; Lassalle, S.; Hofmann, V.; Bordone, O.; Gavric Tanga, V.; Bonnetaud, C.; Moreilhon, C.; Rios, G.; Santini, J.; Barbry, P.; et al. miR-129-5p is required for histone deacetylase inhibitor-induced cell death in thyroid cancer cells. Endocr. Relat. Cancer 2011, 18, 711–719. [Google Scholar] [CrossRef]
- Adams, C.M.; Hiebert, S.W.; Eischen, C.M. Myc induces miRNA-mediated apoptosis in response to HDAC inhibition in hematologic malignancies. Cancer Res. 2016, 76, 736–748. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.H.; Dimri, M.; Dimri, G.P. MicroRNA-31 is a trascriptional target of histone deacetylase inhibitors and a regulator of cellular senescence. J. Biol. Chem. 2015, 290, 10555–10567. [Google Scholar] [CrossRef]
- Nalls, D.; Tang, S.-N.; Rodova, M.; Srivastava, R.K.; Shankar, S. Targeting epigenetic regulation of miR-34a for treatment of pancreatic cancer by inhibition of pancreatic cancer stem cells. PLoS ONE 2011, 6, e24099. [Google Scholar] [CrossRef]
- Kim, J.; Noh, J.; Eun, J.; Jung, K.; Bae, H.; Shen, Q.; Kim, M.; Chan, Y.; Kim, S.; Park, W.; et al. Targeted inactivation of HDAC2 restores p16INK4a activity and exerts antitumor effects on human gastric cancer. Cancer Res. 2013, 11, 62–73. [Google Scholar] [CrossRef]
- Kang, Y.; Nian, H.; Rajendran, P.; Kim, E.; Dashwood, W.; Pinto, J.; Boardman, L.; Thibodeau, S.; Limburg, P.; Löhr, C.; et al. HDAC8 and STAT3 repress BMF gene activity in colon cancer cells. Cell Death Dis. 2014, 5, e1476. [Google Scholar] [CrossRef]
- Feng, L.; Pan, M.; Sun, J.; Lu, H.; Shen, Q.; Zhang, S.; Jiang, T.; Liu, L.; Jin, W.; Chen, Y.; et al. Histone deacetylase 3 inhibits expression of PUMA in gastric cancer cells. J. Mol. Med. 2013, 91, 49–58. [Google Scholar] [CrossRef]
- Mrakovcic, M.; Fröhlich, L.F. P53-mediated molecular control of autophagy in tumor cells. Biomolecules 2018, 8, 14. [Google Scholar] [CrossRef] [PubMed]
- Li, W.W.; Li, J.; Bao, J. Microautophagy: Lesser-known self-eating. Cell. Mol. Life Sci. 2012, 69, 1125–1136. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. Chaperone-mediated autophagy: A unique way to enter the lysosome world. Trends Cell Biol. 2012, 22, 305–309. [Google Scholar] [CrossRef]
- Youle, R.; Narendra, D. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Noda, T.; Yoshimori, T.; Tanaka, Y.; Ishii, T.; George, M.D.; Klionsky, D.J.; Ohsumi, M.; Ohsumi, Y. A protein conjugation system essential for autophagy. Nature 1998, 395, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Gao, Z.; Marks, P.A.; Jiang, X. Apoptotic and autophagic cell death induced by histone deacetylase inhibitors. Proc. Natl. Acad. Sci. USA 2004, 101, 18030–18035. [Google Scholar] [CrossRef] [Green Version]
- Shintani, T.; Klionsky, D.J. Autophagy in health and disease: A double-edged sword. Science 2004, 306, 990–995. [Google Scholar] [CrossRef]
- Yang, Z.J.; Chee, C.E.; Hunag, S.; Sinicrope, F.A. The role of autophagy in cancer: Therapeutic implications. Mol. Cancer Ther. 2011, 10, 1533–1541. [Google Scholar] [CrossRef]
- Choi, A.M.; Ryter, S.W.; Levine, B. Autophagy in human health and disease. N. Engl. J. Med. 2013, 368, 651–662. [Google Scholar] [CrossRef]
- Rosenfeldt, M.T.; Ryan, K.M. The role of autophagy in tumour development and cancer therapy. Expert Rev. Mol. Med. 2009, 11, e36. [Google Scholar] [CrossRef]
- Matthew, R. The role of autophagy in cancer. Nat. Rev. Cancer 2007, 12, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Baehrecke, E.H. Autophagy, cell death, and cancer. Mol. Cell. Oncol. 2015, 2, e985913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, L.; Wan, F.; Dutta, S.; Welsh, S.; Liu, Z.; Freundt, E.; Baehrecke, E.L.M. Autophagic programmed cell death by selective catalase degradation. Proc. Natl. Acad. Sci. USA 2006, 103, 4952–4957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poillet-Perez, L.; Despouy, G.; Delage-Mourroux, R.; Boyer-Guittaut, M. Interplay between ROS and autophagy in cancer cells, from tumor initiation to cancer therapy. Redox. Biol. 2015, 4, 184–192. [Google Scholar] [CrossRef]
- Yue, Z.; Jin, S.; Yang, C.; Levine, A.; Heintz, N. Beclin-1, an autophagy gene essential for early embryonic development, is a haploinsuffient tumor suppressor. Proc. Natl. Acad. Sci. USA 2003, 100, 15077–15082. [Google Scholar] [CrossRef]
- Mowers, E.E.; Sharifi, M.N.; Macleod, K.F. Autophagy in cancer metastasis. Oncogene 2017, 36, 1619–1630. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Klionsky, D.J. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [CrossRef] [PubMed]
- Lopez, G.; Torres, K.; Lev, D. Autophagy blockade enhances HDAC inhibitors’ pro-apoptotic effects: Potential implications for the treatment of a therapeutic-resistant malignancy. Autophagy 2011, 7, 40–41. [Google Scholar] [CrossRef]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Levine, B.; Green, D.R.; Kroemer, G. Pharmacological modulation of autophagy: Therapeutic potential and persisting obstacles. Nat. Rev. Drug Discov. 2017, 16, 487–511. [Google Scholar] [CrossRef]
- Yi, C.; Ma, M.; Ran, L.; Zheng, J.; Tong, J.; Zhu, J.; Ma, C.; Sun, Y.; Zhang, S.; Feng, W.; et al. Function and Molecular Mechanism of Acetylation in Autophagy Regulation. Science 2012, 336, 474–477. [Google Scholar] [CrossRef]
- Gammoh, N.; Lam, D.; Puente, C.; Ganley, I.; Marks, P.A.; Jiang, X. Role of autophagy in histone deacetylase inhibitor-induced apoptotic and nonapoptotic cell death. Proc. Natl. Acad. Sci. USA 2012, 109, 6561–6565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Liu, R.; Lei, Y.; Wang, K.; Lau, Q.C.; Xie, N.; Zhou, S.; Nie, C.; Chen, L.; Wei, Y.; et al. Proteomic analysis revealed association of aberrant ROS signaling with suberoylanilide hydroxamic acid-induced autophagy in Jurkat T-leukemia cells. Autophagy 2010, 6, 711–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiao, M.; Cheng, W.; Yang, Y.; Shen, C.; Chiao, M.; Cheng, W.; Yang, Y.; Shen, C.; Ko, J. Suberoylanilide hydroxamic acid (SAHA) causes tumor growth slowdown and triggers autophagy in glioblastoma stem cells. Autophagy 2013, 9, 1509–1526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.-L.; Yang, P.-M.; Shun, C.-T.; Wu, M.-S.; Weng, J.-R.; Chen, C.-C. Autophagy potentiates the anti-cancer effects of the histone deacetylase inhibitors in hepatocellular carcinoma. Autophagy 2010, 6, 1057–1065. [Google Scholar] [CrossRef] [Green Version]
- Hrzenjak, A.; Kremser, M.; Strohmeier, B.; Moinfar, F.; Zatloukal, K.; Denk, H. SAHA induces caspase-independent, autophagic cell death of endometrial stromal sarcoma cells by influencing the mTOR pathway. J. Pathol. 2008, 216, 495–504. [Google Scholar] [CrossRef]
- Cao, Q.; Yu, C.; Xue, R.; Hsueh, W.; Pan, P.; Chen, Z.; Wang, S.; McNutt, M.; Gu, J. Autophagy induced by suberoylanilide hydroxamic acid in Hela S3 cells involves inhibition of protein kinase B and up-regulation of Beclin 1. Int. J. Biochem. Cell Biol. 2008, 40, 272–283. [Google Scholar] [CrossRef] [PubMed]
- El-Khoury, V.; Pierson, S.; Szwarcbart, E.; Brons, N.H.C.; Roland, O.; Cherrier-De Wilde, S.; Plawny, L.; Van Dyck, E.; Berchem, G. Disruption of autophagy by the histone deacetylase inhibitor MGCD0103 and its therapeutic implication in B-cell chronic lymphocytic leukemia. Leukemia 2014, 28, 1636–1646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carew, J.S.; Nawrocki, S.T.; Kahue, C.N.; Zhang, H.; Yang, C.; Chung, L.; Houghton, J.A.; Huang, P.; Giles, F.J.; Cleveland, J.L. Targeting autophagy augments the anticancer activity of the histone deacetylase inhibitor SAHA to overcome Bcr-Abl-mediated drug resistance. Blood 2007, 110, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Park, M.A.; Reinehr, R.; Haussinger, D.; Voelkel-Johnson, C.; Ogretmen, B.; Yacoub, A.; Grant, S.; Dent, B. Sorafenib activates CD95 and promotes autophagy and cell death via Src family kinases in gastrointestinal tumor cells. Mol. Cancer Ther. 2010, 9, 2220–2231. [Google Scholar] [CrossRef] [PubMed]
- Shulak, L.; Beljanski, V.; Chiang, C.; Dutta, S.M.; Van Grevenynghe, J.; Belgnaoui, S.M.; Nguyen, T.L.A.; Di Lenardo, T.; Semmes, O.J.; Lin, R.T.; et al. Histone Deacetylase Inhibitors Potentiate Vesicular Stomatitis Virus Oncolysis in Prostate Cancer Cells by Modulating NF-kappa B-Dependent Autophagy. J. Virol. 2014, 88, 2927–2940. [Google Scholar] [CrossRef]
- Long, J.; Zhao, J.; Yan, Z.; Liu, Z.; Wang, N. Antitumor effects of a novel sulfur-containing hydroxamate histone deacetylase inhibitor H40. Int. J. Cancer 2009, 124, 1235–1244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Giacomo, V.; Di Valerio, V.; Rapino, M.; Bosco, D.; Cacciatore, I.; Ciulla, M.; Marrazzo, A.; Fiorito, J.; Di Stefano, A.; Cataldi, A. MRJF4, a novel histone deacetylase inhibitor, induces p21 mediated autophagy in PC3 prostate cancer cells. Cell Mol. Biol 2015, 61, 17–23. [Google Scholar] [PubMed]
- Watanabe, M.; Adachi, S.; Matsubara, H.; Imai, T.; Yui, Y.; Mizushima, Y. Induction of autophagy in malignant rhabdoid tumor cells by the histone deacetylase inhibitor FK228 through AIF translocation. Int. J. Cancer Res. 2009, 67, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ng, S.; Wang, J.; Zhou, J.; Tan, S.; Yang, N.; Lin, Q.; Xia, D.; Shen, H. Histone deacetylase inhibitors induce autophagy through FOXO1-dependent pathways. Autophagy 2015, 11, 629–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandesiri, M.; Chakilam, S.; Ivanovska, J.; Benderska, N.; Ocker, M.; Di Fazio, P.; Feoktistova, M.; Gali-Muhtasib, H.; Rave-Fränk, M.; Prante, O.; et al. DAPK plays an important role in panobinostat-induced autophagy and commits cells to apoptosis under autophagy deficient conditions. Apoptosis 2012, 17, 1300–1315. [Google Scholar] [CrossRef] [PubMed]
- Ellis, L.; Bots, M.; Lindemann, R.K.; Bolden, J.E.; Newbold, A.; Cluse, L.A.; Scott, C.L.; Strasser, A.; Atadja, P.; Lowe, S.W.; et al. The histone deacetylase inhibitors LAQ824 and LBH589 do not require death receptor signaling or a functional apoptosome to mediate tumor cell death or therapeutic efficacy. Blood 2009, 114, 380–393. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.; Yi, Y.; Xia, G.; Zhao, Y.; Yu, Y.; Li, L.; Hua, C.; He, B.; Yang, B.; Yu, C.; et al. Nrf2-miR-129-3p-mTOR Axis Controls an miRNA Regulatory Network Involved in HDACi-Induced Autophagy. Mol. Ther. 2019, 27, 1039–1050. [Google Scholar] [CrossRef] [PubMed]
- Stankov, M.V.; El Khatib, M.; Kumar Thakur, B.; Heitmann, K.; Panayotova-Dimitrova, D.; Schoening, J.; Bourquin, J.P.; Schweitzer, N.; Leverkus, M.; Welte, K.; et al. Histone deacetylase inhibitors induce apoptosis in myeloid leukemia by suppressing autophagy. Leukemia 2014, 28, 577–588. [Google Scholar] [CrossRef]
- Maccallum, S.F.; Groves, M.J.; James, J.; Murray, K.; Appleyard, V.; Prescott, A.R.; Drbal, A.A.; Nicolaou, A.; Cunningham, J.; Haydock, S.; et al. Dysregulation of autophagy in chronic lymphocytic leukemia with the small-molecule Sirtuin inhibitor Tenovin-6. Sci. Rep. 2013, 3. [Google Scholar] [CrossRef] [Green Version]
- Lee, I.H.; Finkel, T. Regulation of autophagy by the p300 acetyltransferase. J. Biol. Chem. 2009, 284, 6322–6328. [Google Scholar] [CrossRef] [PubMed]
- Sebti, S.; Prébois, C.; Pérez-Gracia, E.; Bauvy, C.; Desmots, F.; Pirot, N.; Gongora, C.; Bach, A.; Hubberstey, A.; Palissot, V.; et al. BAT3 modulates p300-dependent acetylation of p53 and autophagy-related protein 7 (ATG7) during autophagy. Proc. Natl. Acad. Sci. USA 2014, 111, 4115–4120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sebti, S.; Prébois, C.; Pérez-Gracia, E.; Bauvy, C.; Desmots, F.; Pirot, N.; Gongora, C.; Bach, A.; Hubberstey, A.; Palissot, V.; et al. BAG6/BAT3 modulates autophagy by affecting EP300/p300 intracellular localization. Autophagy 2014, 10, 1341–1342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.-Y.; Li, T.Y.; Liu, Q.; Zhang, C.; Li, X.; Chen, Y.; Zhang, S.-M.; Lian, G.; Liu, Q.; Ruan, K.; et al. Protein phosphorylation-acetylation cascade connects growth factor deprivation to autophagy. Autophagy 2012, 9, 1385–1386. [Google Scholar] [CrossRef] [PubMed]
- Juengel, E.; Dauselt, A.; Makarevic, J.; Wiesner, C.; Tsaur, I.; Bartsch, G.; Haferkamp, A.; Blaheta, R. Acetylation of histone H3 prevents resistance development caused by chronic mTOR inhibition in renal carcinoma cells. Cancer Lett. 2012, 324, 83–90. [Google Scholar] [CrossRef]
- Koeneke, E.; Witt, O.; Oehme, I. HDAC Family Members Intertwined in the Regulation of Autophagy: A Druggable Vulnerability in Aggressive Tumor Entities. Cells 2015, 4, 135–168. [Google Scholar] [CrossRef]
- Cao, D.J.; Wang, Z.V.; Battiprolu, P.K.; Jiang, N.; Morales, C.R.; Kong, Y.; Rothermel, B.A.; Gillette, T.G.; Hill, J.A. Histone deacetylase (HDAC) inhibitors attenuate cardiac hypertrophy by suppressing autophagy. Proc. Natl. Acad. Sci. USA 2011, 108, 4123–4128. [Google Scholar] [CrossRef] [Green Version]
- Oh, M.; Choi, I.-K.; Kwon, H.J. Inhibition of histone deacetylase1 induces autophagy. Biochem. Biophys. Res. Commun. 2008, 369, 1179–1183. [Google Scholar] [CrossRef]
- Xie, H.; Noh, J.; Kim, J.; Jung, K.; Eun, J.; Bae, H.; Kim, M.; Chang, Y.; Lee, J.; Park, H.; et al. HDAC1 inactivation induces mitotic defect and caspase-independent autophagic cell death in liver cancer. PLoS ONE 2012, 7, e34265. [Google Scholar] [CrossRef] [PubMed]
- Moresi, V.; Carrer, M.; Grueter, C.; Rifki, O.; Shelton, J.; Richardson, J.; Bassel-Duby, R.; Olson, E. Histone deacetylases 1 and 2 regulate autophagy flux and skeletal muscle homeostasis in mice. Proc. Natl. Acad. Sci. USA 2012, 109, 1649–1654. [Google Scholar] [CrossRef] [Green Version]
- Pandey, U.; Nie, Z.; Batlevi, Y.; McCray, B.; Ritson, G.; Nedelsky, N.; Schwartz, S.; DiProspero, N.; Knight, M.; Schuldiner, O.; et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 2007, 447, 859–863. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Nagano, Y.; Taylor, J.; Lim, K.; Yao, T. Disease causing mutations in parkin impair mitochondrial ubiquitination, aggregation, and HDAC6-dependent mitophagy. J. Cell Biol. 2010, 189, 671–679. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Tan, B.; Chen, W.; Lin, Y.; Huang, J.; Chang, H.; Sun, H.; Hsu, P.; Liou, G.; Shen, J.; et al. Reversible acetylation regulates salt-inducible kinase (SIK2) and its function in autophagy. J. Biol. Chem. 2013, 288, 6227–6237. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, Y.; Kovacs, J.J.; McLaurin, A.; Vance, J.M.; Ito, A.; Yao, T.P. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 2003, 115, 727–738. [Google Scholar] [CrossRef]
- Iwata, A.; Riley, B.E.; Johnston, J.A.; Kopito, R.R. HDAC6 and microtubules are required for autophagic degradation of aggregated Huntingtin. J. Biol. Chem. 2005, 280, 40282–40292. [Google Scholar] [CrossRef]
- Ahn, M.-Y.; Yoon, J.-H. Histone deacetylase 7 silencing induces apoptosis and autophagy in salivary mucoepidermoid carcinoma cells. J. Oral Pathol. Med. 2017, 46, 276–283. [Google Scholar] [CrossRef]
- Oehme, I.; Linke, J.-P.; Bock, B.C.; Milde, T.; Lodrini, M.; Hartenstein, B.; Wiegand, I.; Eckert, C.; Roth, W.; Kool, M.; et al. Histone deacetylase 10 promotes autophagy-mediated cell survival. Proc. Natl. Acad. Sci. USA 2013, 110, E2592–E2601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, I.H.; Cao, L.; Mostoslavsky, R.; Lombard, D.B.; Liu, J.; Bruns, N.E.; Tsokos, M.; Alt, F.W.; Finkel, T. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc. Natl. Acad. Sci. USA 2008, 105, 3374–3379. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Li, X.; He, Z.; Pan, S.; Yang, Y.; Zhang, X.; Chow, K.; Yang, T.; Qiu, J.; Zhou, Q.; et al. Induction of apoptosis and autophagy via sirtuin1- and PI3K/Akt/mTOR-mediated pathways by plumbagin in human prostate cancer cells. Drug Des. Dev. Ther. 2015, 9, 1511–1554. [Google Scholar] [CrossRef]
- Ou, X.; Lee, M.; Huang, X.; Messina-Graham, S.; Broxmeyer, H. SIRT1 positively regulates autophagy and mitochondria function in embryonic stem cells under oxidative stress. Stem Cells 2014, 32, 1183–1194. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Yang, J.; Liao, W.; Liu, X.; Zhang, H.; Wang, S.; Wang, D.; Feng, J.; Yu, L.; Zhu, W.-G. Cytosolic FoxO1 is essential for the induction of autophagy and tumour suppressor activity. Nat. Cell Biol. 2010, 12, 665–675. [Google Scholar] [CrossRef] [PubMed]
- Tseng, A.; Shieh, S.; Wang, D. SIRT3 deacetylates FOXO3 to protect mitochondria against oxidative damage. Free Radic. Biol. Med. 2013, 63, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Webster, B.; Scott, I.; Han, K.; Li, J.; Lu, Z.; Stevens, M.; Malide, D.; Chen, Y.; Samsel, L.; Connelly, P.; et al. Restricted mitochondrial protein acetylation initiates mitochondrial autophagy. J. Cell Sci. 2013, 126, 4843–4849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papa, L.; Germain, D. SirT3 regulates the mitochondrial unfolded protein response. Mol. Cell. Biol. 2014, 34, 699–710. [Google Scholar] [CrossRef] [PubMed]
- Polletta, L.; Vernucci, E.; Carnevale, I.; Arcangeli, T.; Rotili, D.; Palmerio, S.; Steegborn, C.; Nowak, T.; Schutkowski, M.; Pellegrini, L.; et al. SIRT5 regulation of ammonia-induced autophagy and mitophagy. Autophagy 2015, 11, 253–270. [Google Scholar] [CrossRef] [Green Version]
- Takasaka, N.; Araya, J.; Hara, H.; Ito, S.; Kobayashi, K.; Kurita, Y.; Wakui, H.; Tsurushige, C.; Kojima, J.; Numata, T.; et al. Autophagy Induction by SIRT6 through Attenuation of Insulin-like Growth Factor Signaling Is Involved in the Regulation of Human Bronchial Epithelial Cell Senescence. J. Immunol. 2014, 192, 958–968. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.; Yang, X.; Liu, T.; Zhang, T.; Xie, Q.R.; Xia, W. Autophagy induction by SIRT6 is involved in oxidative stress-induced neuronal damage. Protein Cell 2016, 7, 281–290. [Google Scholar] [CrossRef] [Green Version]
- Vousden, K.H.; Lu, X. Live or let die: The cell´s response to p53. Nat. Rev. Cancer 2002, 2, 594–604. [Google Scholar] [CrossRef]
- Brooks, C.L.; Gu, W. p53 Ubiquitination: Mdm2 and Beyond Review. Mol. Cell 2006, 21, 307–315. [Google Scholar] [CrossRef]
- Schuler, M.; Bossy-wetzel, E.; Goldstein, J.C.; Fitzgerald, P.; Green, D.R. p53 Induces Apoptosis by Caspase Activation through Mitochondrial Cytochrome c Release. J. Biol. Chem. 2000, 275, 7337–7342. [Google Scholar] [CrossRef] [Green Version]
- Chipuk, J.E.; Green, D.R. Dissecting p53-dependent apoptosis. Cell Death Differ. 2006, 13, 994–1002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, J.; Li, M.; Tang, Y.; Laszkowska, M.; Roeder, R.G.; Gu, W. Acetylation of p53 augments its site-specific DNA binding both in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2003, 101, 2259–2264. [Google Scholar] [CrossRef] [PubMed]
- Beckerman, R.; Prives, C. Transcriptional regulation by p53. Cold Spring Harb. Perspect. Biol. 2010, 2, a000935. [Google Scholar] [CrossRef] [PubMed]
- Brooks, C.L.; Gu, W. The impact of acetylation and deacetylation on the p53 pathway. Protein Cell 2011, 2, 456–462. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, H.; Dessain, S.; Ng Eaton, E.; Imai, S.; Frye, R.; Pandita, T.; Guarente, L.; Weinberg, R. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 2001, 107, 149–159. [Google Scholar] [CrossRef]
- Sykes, S.M.; Mellert, H.S.; Holbert, M.A.; Li, K.; Lane, W.S.; Mcmahon, S.B. Acetylation of the p53 DNA binding domain regulates apoptosis induction. Mol. Cell 2006, 24, 841–851. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Luo, J.; Zhang, W. Tip60-Dependent Acetylation of p53 Modulates the Decision between Cell-Cycle Arrest and Apoptosis. Mol. Cell 2006, 24, 827–839. [Google Scholar] [CrossRef]
- Luo, J.; Su, F.; Chen, D.; Shiloh, A.; Gu, W. Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature 2000, 408, 377–381. [Google Scholar] [CrossRef]
- Luo, J.; Nikolaev, A.Y.; Imai, S.; Chen, D.; Su, F.; Shiloh, A.; Guarente, L.; Gu, W. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell 2001, 107, 137–148. [Google Scholar] [CrossRef]
- Brandl, A.; Wagner, T.; Uhlig, K.M.; Knauer, S.K.; Stauber, R.H.; Melchior, F.; Schneider, G.; Heinzel, T.; Krämer, O.H. Dynamically regulated sumoylation of HDAC2 controls p53 deacetylation and restricts apoptosis following genotoxic stress. Mol. Cell. Biol. 2012, 4, 284–293. [Google Scholar] [CrossRef] [Green Version]
- Wagner, T.; Brand, P.; Heinzel, T.; Krämer, O.H. Histone deacetylase 2 controls p53 and is a critical factor in tumorigenesis. Biochim. Biophys. Acta 2012, 1846, 524–538. [Google Scholar] [CrossRef] [PubMed]
- Carlisi, D.; Vassallo, B.; Lauricella, M.; Emanuele, S.; D’Anneo, A.; Di Leonardo, E.; Di Fazio, P.; Vento, R.; Tesoriere, G. Histone deacetylase inhibitors induce in human hepatoma HepG2 cells acetylation of p53 and histones in correlation with apoptotic effects. Int. J. Oncol. 2008, 32, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-Y.; Zhou, L.-M. Sirt3 inhibits hepatocellular carcinoma cell growth through reducing Mdm2-mediated p53 degradation. Biochem. Biophys. Res. Commun. 2012, 423, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Barlev, N.A.; Liu, L.; Chehab, N.H.; Mansfield, K.; Harris, K.G.; Halazonetis, T.D.; Berger, S.L. Acetylation of p53 Activates Transcription through Recruitment of Coactivators/Histone Acetyltransferases. Mol. Cell 2001, 8, 1243–1254. [Google Scholar] [CrossRef]
- Wang, X.; Taplick, J.; Geva, N.; Oren, M. Inhibition of p53 degradation by Mdm2 acetylation. FEBS Lett. 2004, 561, 195–201. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Lu, S.; Wu, L.; Chai, G.; Wang, H.; Chen, Y.; Sun, J.; Yu, Y.; Zhou, W.; Zheng, Q.; et al. Acetylation of p53 at Lysine 373/382 by the Histone Deacetylase Inhibitor Depsipeptide Induces Expression of p21 Waf1/Cip1. Mol. Cell. Biol. 2006, 26, 2782–2790. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y. Regulation of p53 responses by post-translational modifications. Cell Death Differ. 2003, 10, 400–403. [Google Scholar] [CrossRef]
- Roy, S.; Packman, K.; Jeffrey, R.; Tenniswood, M. Histone deacetylase inhibitors differentially stabilize acetylated p53 and induce cell cycle arrest or apoptosis in prostate cancer cells. Cell Death Differ. 2005, 12, 482–491. [Google Scholar] [CrossRef] [Green Version]
- Muller, P.A.J.; Vousden, K.H. P53 Mutations in Cancer. Nat. Cell Biol. 2013, 15, 2–8. [Google Scholar] [CrossRef]
- Liu, J.; Ma, Q.; Zhang, M.; Wang, X.; Zhang, D.; Li, W.; Wang, F.; Wu, E. Alterations of TP53 are associated with a poor outcome for patients with hepatocellular carcinoma: Evidence from a systematic review and metaanalysis. Eur. J. Cancer 2012, 48, 2328–2338. [Google Scholar] [CrossRef]
- Zheng, T.; Wang, J.; Zhao, Y.; Zhang, C.; Lin, M.; Wang, X.; Yu, H.; Liu, L.; Feng, Z.; Hu, W. Spliced MDM2 isoforms promote mutant p53 accumulation and gain-of-function in tumorigenesis. Nat. Commun. 2013, 4, 2996. [Google Scholar] [CrossRef] [PubMed]
- Yue, X.; Zhao, Y.; Xu, Y.; Zheng, M. Mutant p53 in Cancer: Accumulation, Gain-of-Function, and Therapy. J. Mol. Biol. 2017, 429, 1595–1606. [Google Scholar] [CrossRef] [PubMed]
- Dittmer, D.; Pati, S.; Zambetti, G.; Chu, S.; Teresky, A.K.; Moore, M.; Finlay, C.; Levine, A.J. Gain of function mutations in p53. Nat. Genet. 1993, 4, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Santoro, R.; Strano, S.; Blandino, G. Transcriptional regulation by mutant p53 and oncogenesis. Subcell. Biochem. 2014, 85, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Willis, A.; Jung, E.J.; Wakefield, T.; Chen, X. Mutant p53 exerts a dominant negative effect by preventing wild-type from binding to the promoter of its target genes. Oncogene 2004, 23, 2330–2338. [Google Scholar] [CrossRef]
- Li, D.; Marchenko, N.D.; Schulz, R. Functional Inactivation of Endogenous MDM2 and CHIP by HSP90 Causes Aberrant Stabilization of Mutant p53 in Human Cancer Cells. Mol. Cancer Res. 2011, 9, 577–588. [Google Scholar] [CrossRef] [PubMed]
- Blagosklonny, M.V.; Trostel, S.; Kayastha, G.; Demidenko, Z.N.; Vassilev, L.T.; Romanova, L.Y.; Bates, S.; Fojo, T. Depletion of Mutant p53 and Cytotoxicity of Histone Deacetylase Inhibitors. Cancer Res. 2005, 65, 7386–7392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, W.; Liu, S.; Xu, E.; Zhang, J.; Zhang, Y.; Chen, X.; Chen, X. Histone deacetylase inhibitors suppress mutant p53 transcription via histone deacetylase 8. Oncogene 2013, 32, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Marchenko, N.; Moll, U. SAHA shows preferential cytotoxicity in mutant p53 cancer cells by destabilizing mutant p53 through inhibition of the HDAC6-Hsp90 chaperone axis. Cell Death Diff. 2011, 18, 1904–1913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garufi, A.; Pucci, D.; D’Orazi, V.; Circone, M.; Bossi, G.; Avantaggiati, M.L.; D’Orazi, G. Degradation of mutant p53H175 protein by Zn(II) through autophagy. Cell Death Dis. 2014, 5, e1271. [Google Scholar] [CrossRef]
- Saveria, M.; Montani, G.; Granato, M.; Santoni, C.; Del Porto, P.; Merendino, N.; Orazi, G.D.; Faggioni, A.; Cirone, M. Histone deacetylase inhibitors VPA and TSA induce apoptosis and autophagy in pancreatic cancer cells. Cell Oncol. 2017, 40, 167–180. [Google Scholar] [CrossRef]
- Ahn, M.-Y.; Ahn, S.-G.; Yoon, J.-H. Apicidin, a histone deaceylase inhibitor, induces both apoptosis and autophagy in human oral squamous carcinoma cells. Oral Oncol. 2011, 47, 1032–1038. [Google Scholar] [CrossRef] [PubMed]
- Kloster, M.; Naderi, E.; Haaland, I.; Gjertsen, B.; Blomhoff, H.; Naderi, S. cAMP signalling inhibits p53 acetylation and apoptosis via HDAC and SIRT deacetylases. Int. J. Oncol. 2013, 42, 1815–1821. [Google Scholar] [CrossRef] [Green Version]
- Sonnemann, J.; Hartwig, M.; Plath, A.; Saravana Kumar, K.; Müller, C.; Beck, J.F. Histone deacetylase inhibitors require caspase activity to induce apoptosis in lung and prostate carcinoma cells. Cancer Lett. 2006, 232, 148–160. [Google Scholar] [CrossRef]
- Sonnemann, J.; Marx, C.; Becker, S.; Wittig, S.; Palani, C.; Kramer, O.; Beck, J. p53–dependent and p53- independent anticancer effects of different histone deacetylase inhibitors. Br. J. Cancer 2014, 110, 656–667. [Google Scholar] [CrossRef] [PubMed]
- Chi, X.Z.; Yang, J.O.; Lee, K.Y.; Ito, K.; Sakakura, C.; Li, Q.L.; Kim, H.R.; Cha, E.J.; Lee, Y.H.; Kaneda, A.; et al. RUNX3 suppresses gastric epithelial cell growth by inducing p21(WAF1/cip1) expression in cooperation with transforming factor (beta)-activated SMAD. Mol. Cell. Biol. 2005, 25, 8097–8107. [Google Scholar] [CrossRef] [PubMed]
- Yano, T.; Ito, K.; Fukamachi, H.; Chi, X.Z.; Wee, H.J.; Inoue, K.; Ida, H.; Bouillet, P.; Strasser, A.; Bae, S.C.; et al. The RUNX3 tumor suppressor upregulates Bim in gastric epithelial cells undergoing transforming growth factor beta induced apoptosis. Mol. Cell. Biol. 2006, 26, 4474–4488. [Google Scholar] [CrossRef]
- Jin, Y.H.; Jeon, E.J.; Li, Q.L.; Lee, Y.H.; Choi, J.K.; Kim, W.J.; Lee, K.Y.; Bae, S.C. Transforming growth factor-beta stimulates p300-dependent RUNX3 acetylation, which inhibits ubiquitination-mediated degradation. J. Biol. Chem. 2004, 279, 29409–29417. [Google Scholar] [CrossRef]
- Robert, T.; Vanoli, F.; Chiolo, I.; Shubassi, G.; Bernstein, K.; Rothstein, R.; Botrugno, O.; Parazzoli, D.; Oldani, A.; Minucci, S.; et al. HDACs link the DNA damage response, processing of double-strand breaks and autophagy. Nature 2011, 471, 74–79. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, S.; Tanaka, K.; Sakimura, R.; Okada, T.; Nakamura, T.; Li, Y.; Takasaki, M.; Nakabeppu, Y.; Iwamoto, Y. Suberoylanilide hydroxamic acid (SAHA) induces apoptosis or autophagy-associated cell death in chondrosarcoma cell lines. Anticancer Res. 2008, 28, 1585–1591. [Google Scholar] [PubMed]
- Fröhlich, L.F.; Mrakovcic, M.; Smole, C.; Lahiri, P.; Zatloukal, K. Epigenetic silencing of apoptosis-inducing gene expression can be efficiently overcome by combined SAHA and TRAIL treatment in uterine sarcoma cells. PLoS ONE 2014, 9, e91558. [Google Scholar] [CrossRef]
- Wang, J.; Kim, T.H.; Ahn, M.Y.; Lee, J.; Jung, J.H.; Choi, W.S.; Lee, B.M.; Yoon, K.S.; Yoon, S.; Kim, H.S. Sirtinol, a class III HDAC inhibitor, induces apoptotic and autophagic cell death in MCF-7 human breast cancer cells. Int. J. Oncol. 2012, 41, 1101–1109. [Google Scholar] [CrossRef]
- Park, E.Y.; Woo, Y.; Kim, S.J.; Kim, D.H.; Lee, E.K.; De, U.; Kim, K.S.; Lee, J.; Jung, J.H.; Ha, K.T.; et al. Anticancer effects of a new SIRT inhibitor, MHY2256, against human breast cancer MCF-7 cells via regulation of MDM2-p53 binding. Int. J. Biol. Sci. 2016, 12, 1555–1567. [Google Scholar] [CrossRef]
- De, U.; Son, J.Y.; Sachan, R.; Park, Y.J.; Kang, D.; Yoon, K.; Lee, B.M.; Kim, I.S.; Moon, H.R.; Kim, H.S. A New Synthetic Histone Deacetylase Inhibitor, MHY2256, Induces Apoptosis and Autophagy Cell Death in Endometrial Cancer Cells via p53 Acetylation. Int. J. Mol. Sci. 2018, 19, 2743. [Google Scholar] [CrossRef] [PubMed]
- Tasdemir, E.M.; Maiuri, C.; Galluzzi, L.; Vitale, I.; Djavaheri-Mergny, M.; D’Amelio, M.; Criollo, A.; Morselli, E.; Zhu, C.; Harper, F.; et al. Regulation of autophagy by cytoplasmic p53. Nat. Cell Biol. 2008, 10, 676–687. [Google Scholar] [CrossRef] [Green Version]
- Morselli, E.; Shen, S.; Ruckenstuhl, C.; Bauer, M.; Mariño, G.; Galluzzi, L.; Criollo, A.; Michaud, M.; Maiuri, M.; Chano, T.; et al. p53 inhibits autophagy by interacting with the human ortholog of yeast Atg17, RB1CC1/FIP200. Cell Cycle 2011, 10, 2763–2769. [Google Scholar] [CrossRef]
- Tripathi, R.; Ash, D.; Shaha, C. Beclin-1–p53 interaction is crucial for cell fate determination in embryonal carcinoma cells. J. Cell Mol. Med. 2014, 18, 2275–2286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaid, S.; Brandts, C.; Serve, H.; Dikic, I. Ubiquitination and selective autophagy. Cell Death Differ. 2013, 20, 21–30. [Google Scholar] [CrossRef]
- Shi, C.; Pan, B.-Q.; Shi, F.; Xie, Z.-H.; Jiang, Y.-Y.; Shang, L.; Zhang, Y.; Xu, X.; Cai, Y.; Hao, J.-J. Sequestosome 1 protects esophageal squamous carcinoma cells from apoptosis via stabilizing SKP2 under serum starvation condition. Oncogene 2018, 37, 3260–3274. [Google Scholar] [CrossRef]
- Seibenhener, M.L.; Geetha, T.; Wooten, M.W. Sequestosome 1/p62 - more than just a scaffold. FEBS Lett. 2007, 581, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Seibenhener, M.L.; Calderilla-Barbosa, L.; Diaz-Meco, M.-T.; Moscat, J.; Jiang, J.; Wooten, M.W.; Wooten, M.C. SQSTM1/p62 interacts with HDAC6 and regulates deacetylase activity. PLoS ONE 2013, 8, e76016. [Google Scholar] [CrossRef]
- Lahiri, P.; Schmidt, V.; Smole, C.; Kufferath, I.; Denk, H.; Strnad, P.; Rölicke, T.; Fröhlich, L.F.; Zatloukal, K. P62/Sequestosome-1 Is indispensable for maturation and stabilization of Mallory-Denk bodies. PLoS ONE 2016, 11. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Ni, H.-M.; Guo, F.; Ding, Y.; Shi, Y.-H.; Lahiri, P.; Fröhlich, L.F.; Rülicke, T.; Smole, C.; Schmidt, V.C.; et al. Sequestosome 1/p62 protein is associated with autophagic removal of excess hepatic endoplasmic reticulum in mice. J. Biol. Chem. 2016, 291. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.; Le Toumelin, G.; Criollo, A.; Rain, J.; Gautier, F.; Juin, P.; Tasdemir, E.; Pierron, G.; Troulinaki, K.; Tavernarakis, N.; et al. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J. 2007, 26, 2527–2539. [Google Scholar] [CrossRef]
- Zalckvar, E.; Berissi, H.; Mizrachy, L.; Idelchuk, Y.; Koren, I.; Eisenstein, M.; Sabanay, H.; Pinkas-Kramarski, R.; Kimchi, A. DAP-kinase mediated phosphorylation on the BH3 domain of beclin 1 promotes dissociation of beclin 1 from Bcl-XL and induction of autophagy. EMBO Rep. 2009, 10, 285–292. [Google Scholar] [CrossRef]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.; Mizushima, N.; Packer, M.; Schneider, M.; Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.Y.; Gang, H.; Aviv, Y.; Dhingra, R.; Margulets, V.; Kirshenbaum, L.A. p53 Mediates Autophagy and Cell Death by a Mechanism Contingent on Bnip3. Hypertension 2013, 62, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Yee, K.S.; Wilkinson, S.; James, J.; Ryan, K.M.; Vousden, K.H. PUMA and Bax-induced Autophagy Contributes to Apoptosis. Cell Death Differ. 2010, 16, 1135–1145. [Google Scholar] [CrossRef] [PubMed]
- Lorrin, S.; Pierron, G.; Ryan, K.; Codogno, P.; Djavaheri-Mergny, M. Evidence for the interplay between JNK and p53-DRAM signaling pathways in the regulation of autophagy. Autophagy 2010, 6, 153. [Google Scholar] [CrossRef]
- Maiuri, M.; Criollo, A.; Kroemer, G. Crosstalk between apoptosis and autophagy within the Beclin 1 interactome. EMBO J. 2010, 29, 515–516. [Google Scholar] [CrossRef] [Green Version]
- Lindqvist, L.; Heinlein, M.; Huang, D.; Vaux, D. Prosurvival Bcl-2 family members affect autophagy only indirectly, by inhibiting Bax and Bak. Proc. Natl. Acad. Sci. USA 2014, 111, 8512–8517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crighton, D.; Wilkinson, S.; Ryan, K. DRAM links autophagy to p53 and programmed cell death. Autophagy 2007, 3, 72–74. [Google Scholar] [CrossRef]
- Crighton, D.; Wilkinson, S.; O’Prey, J.; Syed, N.; Smith, P.; Harrison, P.R.; Gasco, M.; Garrone, O.; Crook, T.; Ryan, K.M. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 2006, 126, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Lorin, S.; Borges, A.; Ribeiro Dos Santos, L.; Souquère, S.; Pierron, G.; Ryan, K.; Codogno, P.; Djavaheri-Mergny, M. c-Jun NH2-terminal kinase activation is essential for DRAM-dependent induction of autophagy and apoptosis in 2-methoxyestradiol-treated Ewing sarcoma cells. Cancer Res. 2009, 69, 6924–6931. [Google Scholar] [CrossRef] [PubMed]
- Yousefi, S.; Perozzo, R.; Schmid, I.; Ziemiecki, A.; Schaffner, T.; Scapozza, L.; Brunner, T.; Simon, H.-U. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat. Cell Biol. 2006, 8, 1124–1132. [Google Scholar] [CrossRef]
- Francisco, R.; Pérez-Perarnau, A.; Cortés, C.; Gil, J.; Tauler, A.; Ambrosio, S. Histone deacetylase inhibition induces apoptosis and autophagy in human neuroblastoma cells. Cancer Lett. 2012, 318, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Ahn, M.Y.; Kim, T.H.; Yoon, S.; Kang, K.W.; Lee, J.; Moon, H.R.; Jung, J.H.; Chung, H.Y.; Kim, H.S. A new synthetic HDAC inhibitor, MHY218, induces apoptosis or autophagy-related cell death in tamoxifen-resistant MCF-7 breast cancer cells. Investig. New Drugs 2012, 30, 1887–1898. [Google Scholar] [CrossRef]
- Martin, A.; Park, M.; Mitchell, C.; Walker, T.; Rahmani, M.; Thorburn, A.; Häussinger, D.; Reinehr, R.; Grant, S.; Dent, P. BCL-2 family inhibitors enhance histone deacetylase inhibitor and sorafenib lethality via autophagy and overcome blockade of the extrinsic pathway to facilitate killing. Mol. Pharmacol. 2009, 76, 327–341. [Google Scholar] [CrossRef]
- Chen, S.; Zhang, Y.; Zhou, L.; Leng, Y.; Lin, H.; Kmieciak, M.; Pei, X.-Y.; Jones, R.; Orlowski, R.Z.; Dai, Y.; et al. A Bim-targeting strategy overcomes adaptive bortezomib resistance in myeloma through a novel link between autophagy and apoptosis. Blood 2014, 124, 2687–2697. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; Goldstein, L.A.; Hou, W.; Gastman, B.R.; Rabinowich, H. Regulation of Mitochondrial Apoptotic Events by p53-mediated Disruption of Complexes between Antiapoptotic Bcl-2 Members and Bim. J. Biol. Chem. 2010, 285, 22473–22483. [Google Scholar] [CrossRef]
- Zhan, Y.; Gong, K.; Chen, C.; Wang, H.; Li, W. P38 MAP kinase functions as a switch in MS-275-induced reactive oxygen species-dependent autophagy and apoptosis in Human colon cancer cells. Free Radic. Biol. Med. 2012, 53, 532–543. [Google Scholar] [CrossRef] [PubMed]
- Torgersen, M.L.; Engedal, N.; Bøe, S.; Hokland, P.; Simonsen, A. Targeting autophagy potentiates the apoptotic effect of histone deacetylase inhibitors in t ( 8; 21 ) AML cells. Blood 2018, 122, 2467–2477. [Google Scholar] [CrossRef] [PubMed]
- Butler, L.M.; Webb, Y.; Agus, D.B.; Higgins, B.; Tolentino, T.R.; Kutko, M.C.; LaQuaglia, M.P.; Drobnjak, M.; Cordon-Cardo, C.; Scher, H.I.; et al. Inhibition of transformed cell growth and induction of cellular differentiation by pyroxamide, an inhibitor of histone deacetylase. Clin. Cancer Res. 2001, 7, 962–970. [Google Scholar] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mrakovcic, M.; Kleinheinz, J.; Fröhlich, L.F. p53 at the Crossroads between Different Types of HDAC Inhibitor-Mediated Cancer Cell Death. Int. J. Mol. Sci. 2019, 20, 2415. https://doi.org/10.3390/ijms20102415
Mrakovcic M, Kleinheinz J, Fröhlich LF. p53 at the Crossroads between Different Types of HDAC Inhibitor-Mediated Cancer Cell Death. International Journal of Molecular Sciences. 2019; 20(10):2415. https://doi.org/10.3390/ijms20102415
Chicago/Turabian StyleMrakovcic, Maria, Johannes Kleinheinz, and Leopold F. Fröhlich. 2019. "p53 at the Crossroads between Different Types of HDAC Inhibitor-Mediated Cancer Cell Death" International Journal of Molecular Sciences 20, no. 10: 2415. https://doi.org/10.3390/ijms20102415
APA StyleMrakovcic, M., Kleinheinz, J., & Fröhlich, L. F. (2019). p53 at the Crossroads between Different Types of HDAC Inhibitor-Mediated Cancer Cell Death. International Journal of Molecular Sciences, 20(10), 2415. https://doi.org/10.3390/ijms20102415