Bone Metastasis Phenotype and Growth Undergo Regulation by Micro-Environment Stimuli: Efficacy of Early Therapy with HGF or TGFβ1-Type I Receptor Blockade


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
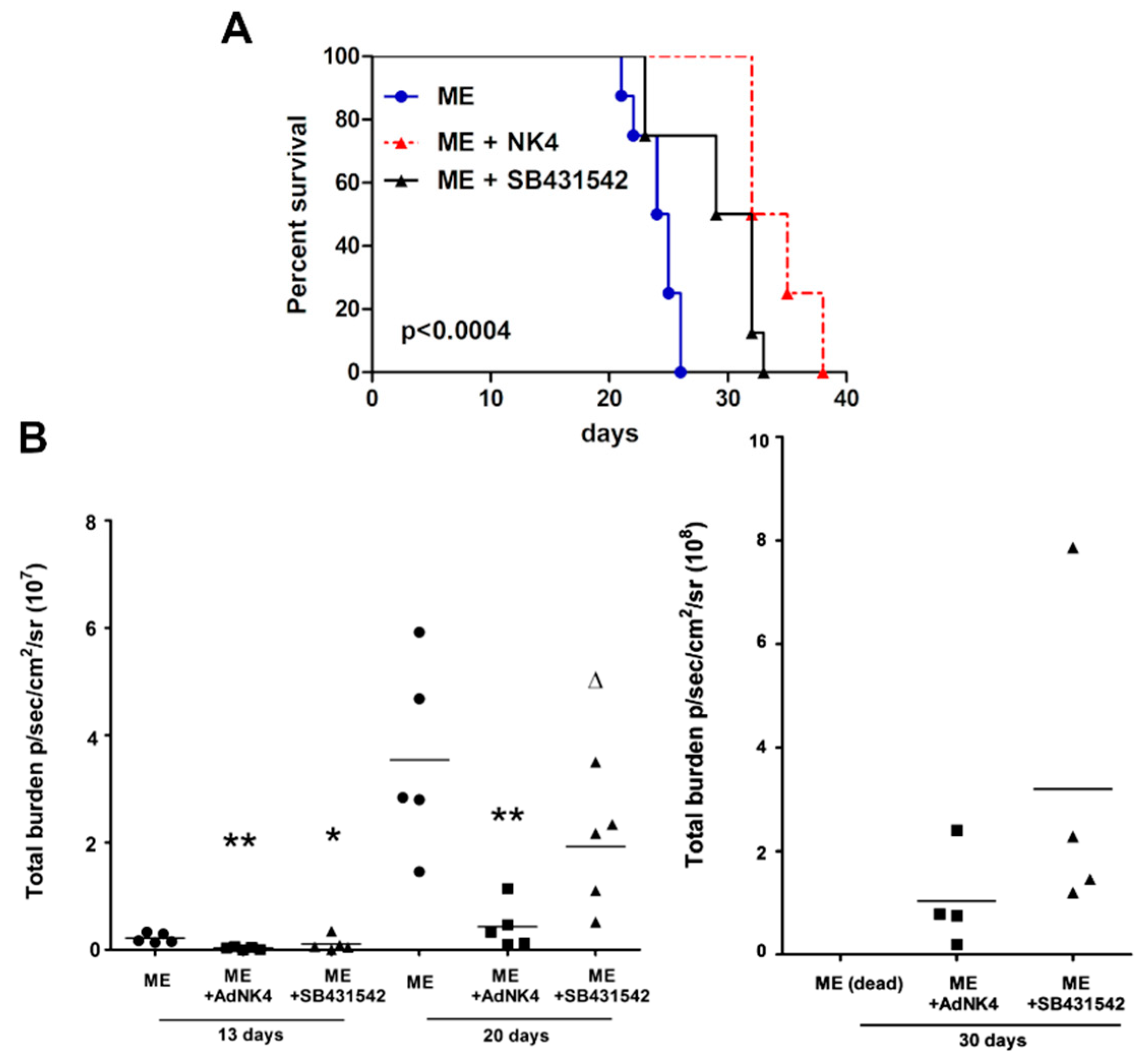
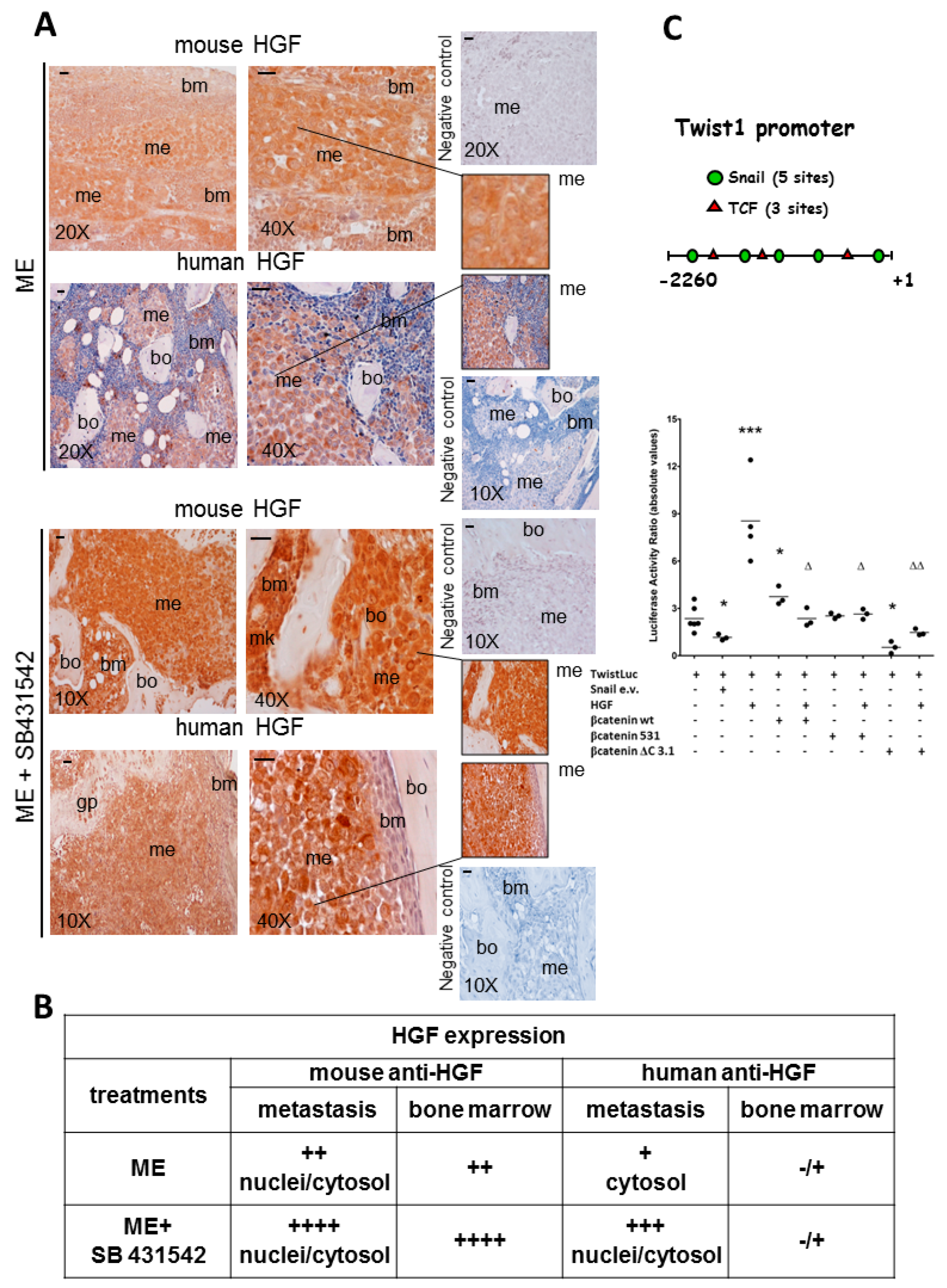
2.1. The Blockade of Hepatocyte Growth Factor (HGF) or Transforming Growth Factor β1 (TGFβ1) Signaling Pathway Impaired Bone Metastasis Outgrowth and Prolonged Mice Survival
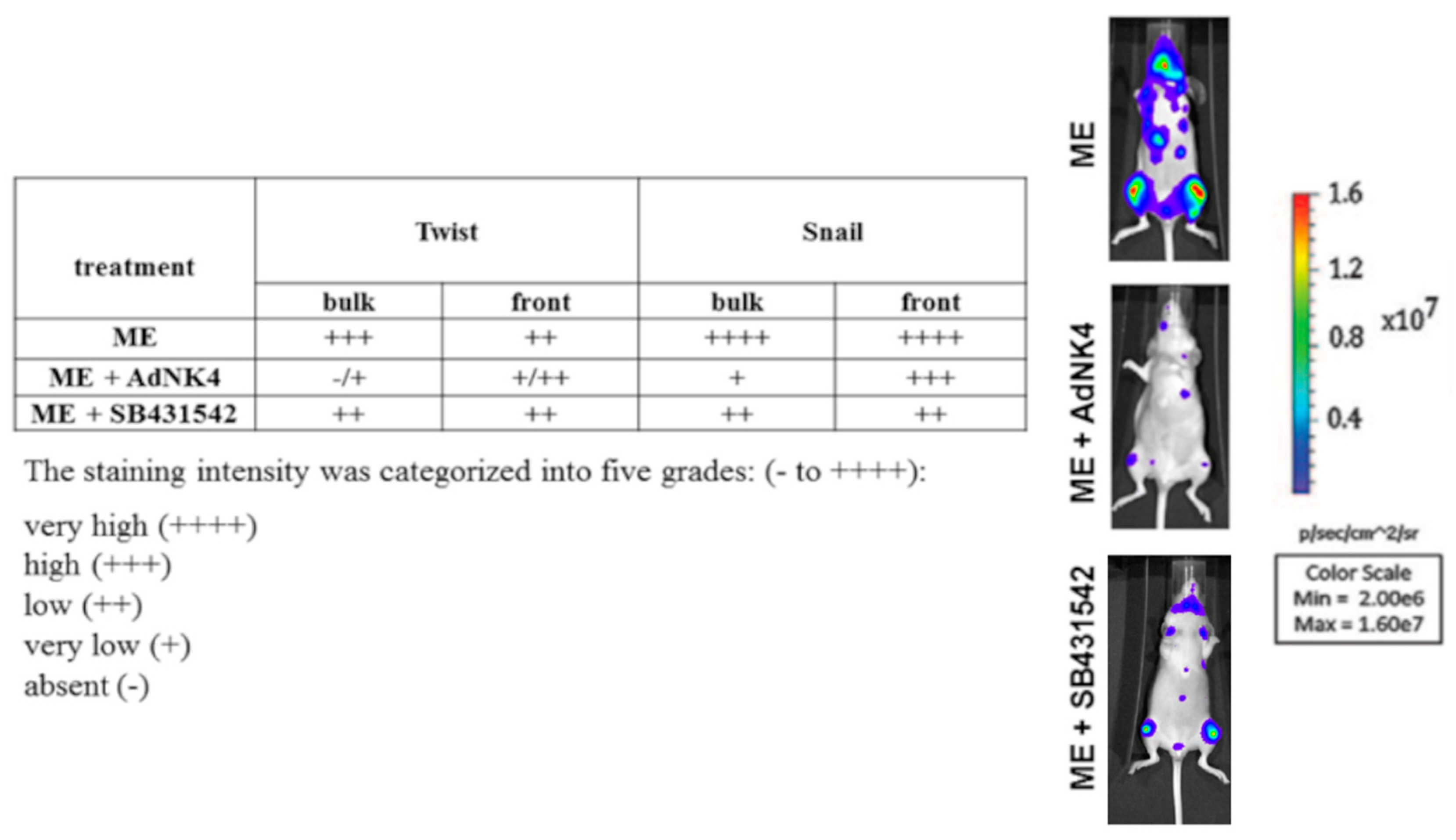
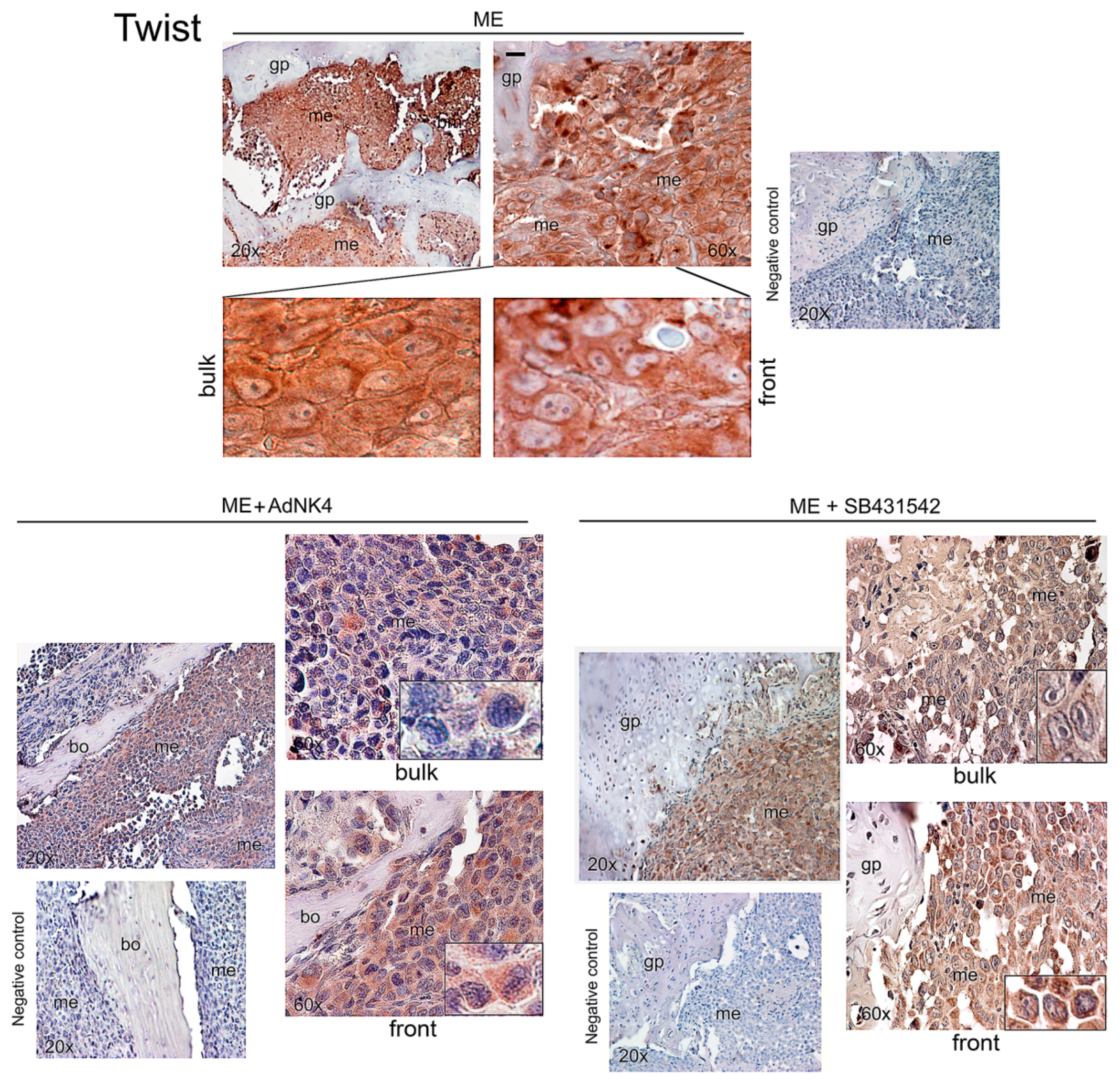
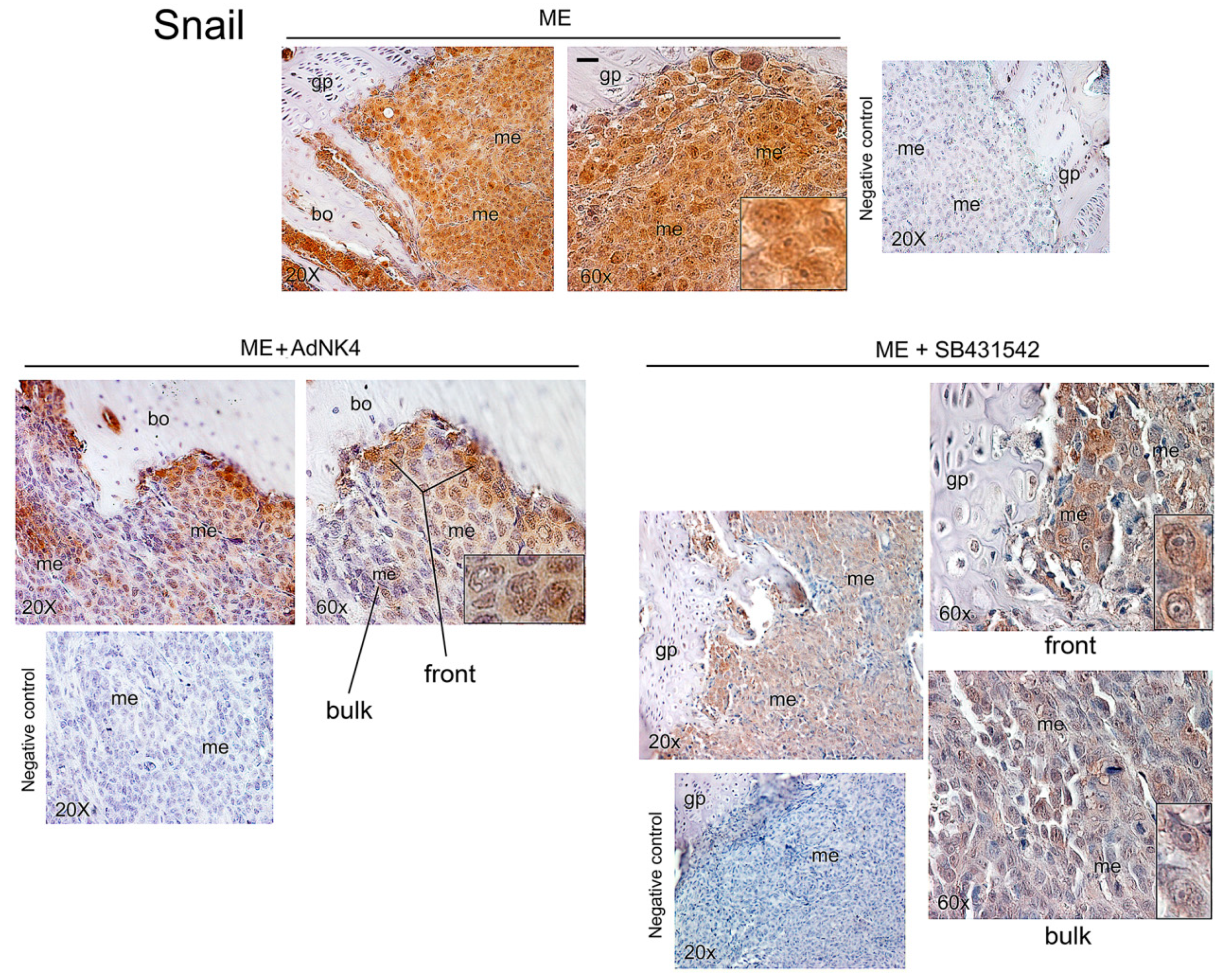
2.2. Effects of the Blockade of HGF or TGFβ1 Signaling Pathway on Twist, Snail and HGF Expression in Xenograft Mice, and Regulation of Twist Transactivating Activity by Snail and HGF
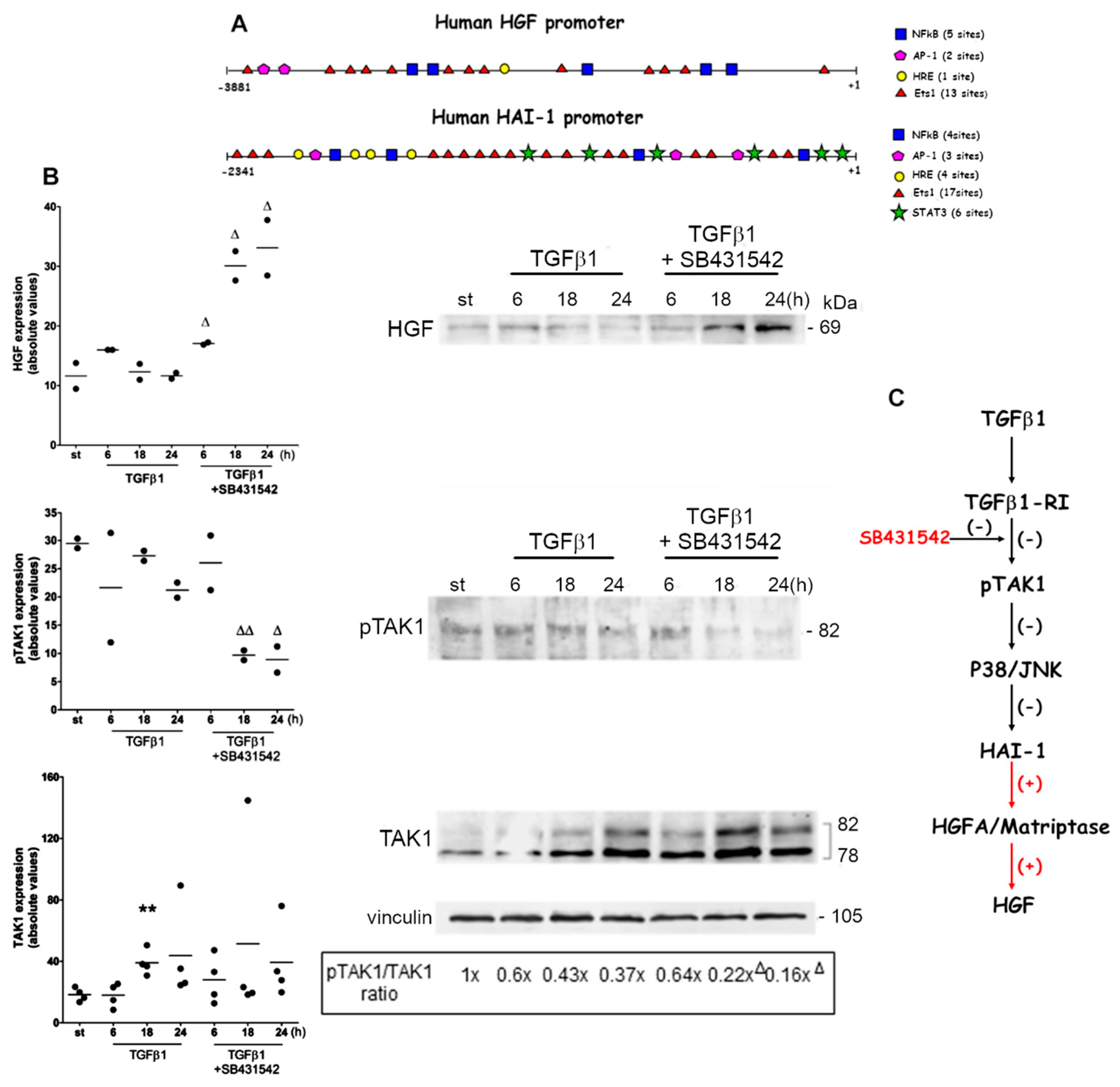
2.3. Auxiliary Pathway Responsible for HGF Accumulation under TGFβ1-RI Blockade
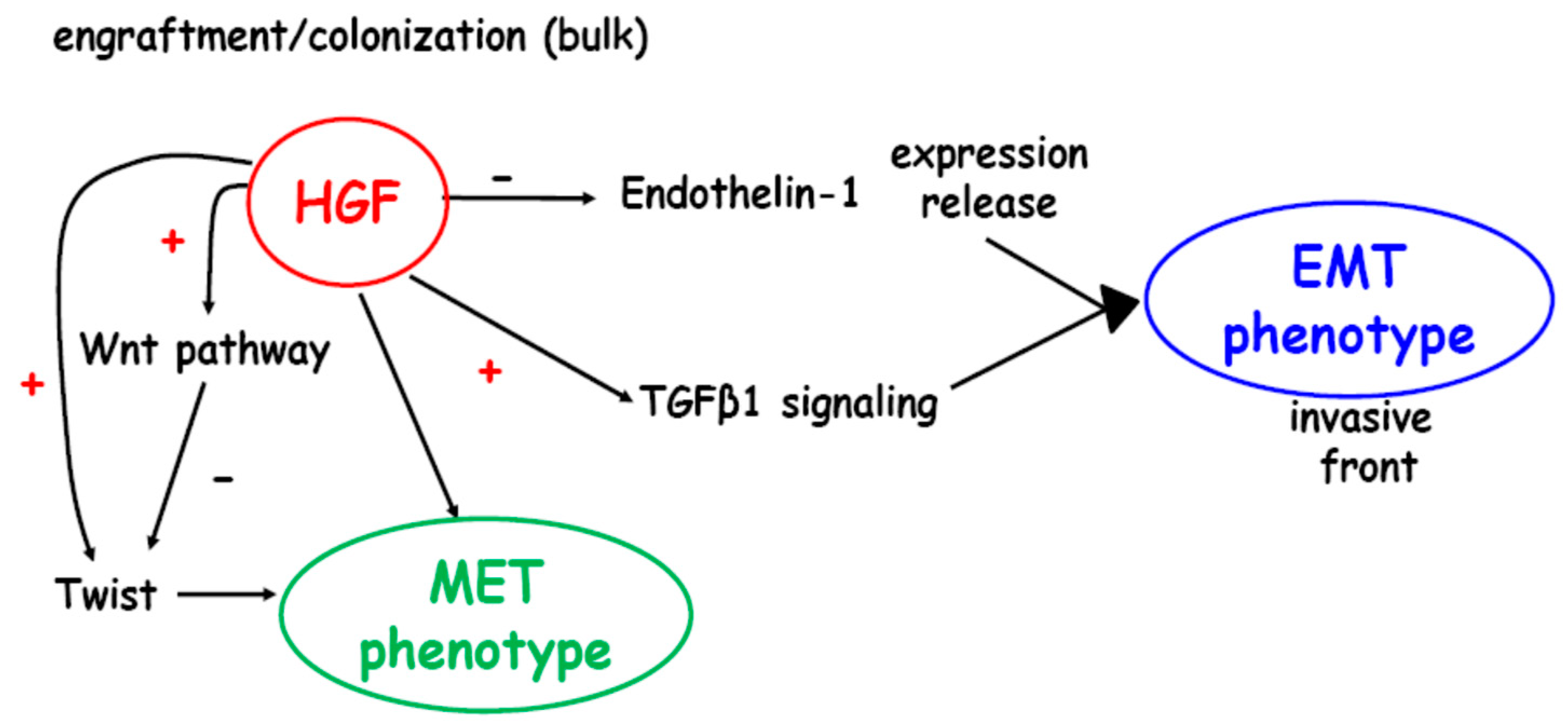
3. Discussion
4. Materials and Methods
4.1. Reagents and Plasmids
4.2. Xenograft Model Preparation
4.3. Immunohistochemistry Assay
4.4. Cell Cultures and Treatments
4.5. Western Blot Analysis
4.6. Quantitative Reverse-Transcription Polymerase Chain Reaction (RT-PCR) Detection of mRNAs
4.7. Transient Transfection and Luciferase Reporter Assay
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| EMT | Epithelial-mesenchymal transition |
| MET | Mesenchymal-epithelial transition |
| TGFβ1 | transforming growth factor β1 |
| HGF | hepatocyte growth factor |
| VEGF | vascular endothelial growth factor |
| TGFβ1-RI | TGFβ1 type I receptor |
| TAK1 | TGFβ-activated kinase1 |
| JNK | c-Jun N-terminal kinase |
| HAI-1 | HGF activator inhibitor-1 |
| HAI-2 | HGF activator inhibitor-2 |
References
- Croucher, P.I.; McDonald, M.M.; Martin, T.J. Bone metastasis: The importance of the neighbourhood. Nat. Rev. Cancer 2016, 16, 373–386. [Google Scholar] [CrossRef] [PubMed]
- Maroni, P.; Bendinelli, P.; Morelli, D.; Drago, L.; Luzzati, A.; Perrucchini, G.; Bonini, C.; Matteucci, E.; Desiderio, M.A. High SPARC expression starting from dysplasia, associated with breast carcinoma, is predictive for bone metastasis without enhancement of plasma levels. Int. J. Mol. Sci. 2015, 16, 28108–28122. [Google Scholar] [CrossRef] [PubMed]
- Emon, B.; Bauer, J.; Jain, Y.; Jung, B.; Saif, T. Biophysics of tumor microenvironment and cancer metastasis. A miniReview. Computational Structural Biotech. J. 2018, 16, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Rossnagl, S.; Ghura, H.; Groth, C.; Altrock, E.; Jakob, F.; Schott, S.; Wimberger, P.; Link, T.; Kuhlmann, J.D.; Stenzl, A.; et al. A subpopulation of stromal cells controls cancer cell homing to the bone marrow. Cancer Res. 2018, 78, 129–142. [Google Scholar] [CrossRef]
- Bendinelli, P.; Maroni, P.; Matteucci, E.; Desiderio, M.A. HGF and TGFβ1 differently influenced Wwox regulatory function on Twist program for mesenchymal-epithelial transition in bone metastatic versus parental breast carcinoma cells. Mol. Cancer 2015, 14, 112. [Google Scholar] [CrossRef]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nature Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Jiang, X.; Hu, S.; Liu, Q.; Qian, C.; Liu, Z.; Luo, D. Exosomal microRNA remodels the tumor microenvironment. Peer J. 2017, 5, e4196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bendinelli, P.; Maroni, P.; Matteucci, E.; Desiderio, M.A. Epigenetic regulation of HGF/Met receptor axis is critical for the outgrowth of bone metastasis from breast carcinoma. Cell Death Dis. 2017, 8, e2578. [Google Scholar] [CrossRef]
- Hiraga, T. Hypoxic microenvironment and metastatic bone disease. Int. J. Mol. Sci. 2018, 19, 3523. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Huang, Z.; Cui, D.; Ouyang, G. The multiaspect functions of periostin in tumor progression. Adv. Exp. Med. Biol. 2019, 1132, 125–136. [Google Scholar]
- Pang, X.; Gong, K.; Zhang, X.; Wu, S.; Cui, Y.; Qian, B.-Z. Osteopontin as a multifaceted driver of bone metastasis and drug resistance. Pharmacol. Res. 2019, 144, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Bendinelli, P.; Maroni, P.; Matteucci, E.; Desiderio, M.A. Cell and signal components of the microenvironment of bone metastasis are affected by hypoxia. Int. J. Mol. Sci. 2016, 17, 706. [Google Scholar] [CrossRef] [PubMed]
- Tam, W.L.; Weinberg, R.A. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat. Med. 2013, 19, 1438–1449. [Google Scholar] [CrossRef] [Green Version]
- Jung, H.Y.; Fattet, L.; Yang, J. Molecular pathways: Linking tumor microenvironment to epithelial-mesenchymal transition in metastasis. Clin. Cancer Res. 2015, 21, 962–968. [Google Scholar] [CrossRef]
- Matteucci, E.; Maroni, P.; Nicassio, F.; Ghini, F.; Bendinelli, P.; Desiderio, M.A. Microenvironment stimuli HGF and hypoxia differently affected miR-125b and Ets-1 function with opposite effects on the invasiveness of bone metastatic cells: A comparison with breast carcinoma cells. Int. J. Mol. Sci. 2018, 19, 258. [Google Scholar] [CrossRef] [PubMed]
- Esposito, M.; Guise, T.; Kang, Y. The biology of bone metastasis. Cold Spring Harb. Perspect. Med. 2018, 8, 1–23. [Google Scholar] [CrossRef]
- Maroni, P.; Puglisi, R.; Mattia, G.; Carè, A.; Matteucci, E.; Bendinelli, P.; Desiderio, M.A. In bone metastasis miR-34a-5p absence inversely correlates with Met expression, while Met oncogene is unaffected by miR-34a-5p in non-metastatic and metastatic breast carcinomas. Carcinogenesis 2017, 38, 492–503. [Google Scholar] [CrossRef]
- Halder, S.K.; Beauchamp, R.D.; Datta, P.K. A specific inhibitor of TGF-beta receptor kinase, SB-431542, as a potent antitumor agent for human cancers. Neoplasia 2005, 7, 509–521. [Google Scholar] [CrossRef]
- Kataoka, H.; Kawaguchi, M.; Fukushima, T.; Shimomura, T. Hepatocyte growth factor activator inhibitors (HAI-1 and HAI-2): Emerging key players in epithelial integrity and cancer. Pathol. Int. 2018, 68, 145–158. [Google Scholar] [CrossRef]
- Lee, S.Y.; Jeong, E.K.; Ju, M.K.; Jeon, H.M.; Kim, M.Y.; Kim, C.H.; Park, H.G.; Han, S.I.; Kang, H.S. Induction of metastasis, cancer stem cell phenotype, and oncogenic metabolism in cancer cells by ionizing radiation. Mol. Cancer 2017, 16, 10. [Google Scholar] [CrossRef]
- Li, Z.; Dong, M.; Fan, D.; Hou, P.; Li, H.; Liu, L.; Lin, C.; Liu, J.; Su, L.; Wu, L.; et al. LncRNA ANCR down-regulation promotes TGF-β-induced EMT and metastasis in breast cancer. Oncotarget 2017, 8, 67329–67343. [Google Scholar] [CrossRef]
- Previdi, S.; Maroni, P.; Matteucci, E.; Broggini, M.; Bendinelli, P.; Desiderio, M.A. Interaction between human-breast cancer metastasis and bone microenvironment through activated hepatocyte growth factor/Met and beta-catenin/Wnt pathways. Eur. J. Cancer 2010, 46, 1679–1691. [Google Scholar] [CrossRef]
- Bendinelli, P.; Maroni, P.; Matteucci, E.; Luzzati, A.; Perrucchini, G.; Desiderio, M.A. Microenvironmental stimuli affect Endothelin-1 signaling responsible for invasiveness and osteomimicry of bone metastasis from breast cancer. Biochim. Biophys. Acta 2014, 1843, 815–826. [Google Scholar] [CrossRef] [Green Version]
- Malladi, S.; Macalinao, D.G.; Jin, X.; Zou, Y.; de Stanchina, E.; Massagué, J. Metastatic latency and immune evasion through autocrine inhibition of Wnt. Cell 2016, 165, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Maroni, P.; Bendinelli, P.; Matteucci, E.; Locatelli, A.; Nakamura, T.; Scita, G.; Desiderio, M.A. Osteolytic bone metastasis is hampered by impinging on the interplay among autophagy, anoikis and ossification. Cell Death Dis. 2014, 5, e1005. [Google Scholar] [CrossRef]
- Maroni, P.; Matteucci, E.; Drago, L.; Banfi, G.; Bendinelli, P.; Desiderio, M.A. Hypoxia induced E-cadherin involving regulators of Hippo pathway due to HIF-1α stabilization/nuclear translocation in bone metastasis from breast carcinoma. Exp. Cell Res. 2015, 330, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Papageorgis, P.; Stylianopoulos, T. Role of TGFβ in regulation of the tumor microenvironment and drug delivery (review). Int. J. Oncol. 2015, 46, 933–943. [Google Scholar] [CrossRef]
- Iyengar, P.V. Regulation of ubiquitin enzymes in the TGF-β pathway. Int. J. Mol. Sci. 2017, 18, 877. [Google Scholar] [CrossRef] [PubMed]
- Gherardi, E.; Birchmeier, W.; Birchmeier, C.; Vande Woude, G. Targeting Met in cancer: Rationale and progress. Nat. Rev. Cancer 2012, 12, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Wang, L.; Li, H.; Li, Y.; Ruan, Y.; Lin, D.; Yang, M.; Jin, X.; Guo, Y.; Zhang, X.; et al. SMAD2 inactivation inhibits CLDN6 methylation to suppress migration and invasion of breast cancer cells. Int. J. Mol. Sci. 2017, 18, 1863. [Google Scholar] [CrossRef] [PubMed]
- Usami, N.; Sekido, Y.; Maeda, O.; Yamamoto, K.; Minna, J.D.; Hasegawa, Y.; Yoshioka, H.; Imaizumi, M.; Ueda, Y.; Takahashi, M.; et al. β-catenin inhibits cell growth of a malignant mesothelioma cell line, NCI-H28, with a 3p21.3 homozygous deletion. Oncogene 2003, 22, 7922–7930. [Google Scholar] [CrossRef]
- Matteucci, E.; Ridolfi, E.; Maroni, P.; Bendinelli, P.; Desiderio, M.A. c-Src/histone deacetylase 3 interaction is crucial for hepatocyte growth factor-dependent decrease of CXCR4 expression in highly invasive breast tumour cells. Mol. Cancer Res. 2007, 5, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Jurjus, A.; Eid, A.; Al Kattar, S.; Zeenny, M.N.; Gerges-Geagea, A.; Haydar, H.; Hilal, A.; Oueidat, D.; Matar, M.; Tawilah, J.; et al. Inflammatory bowel disease, colorectal cancer and type 2 diabetes mellitus: The links. BBA Clin. 2016, 5, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Gkouveris, I.; Nikitakis, N.; Sklavounou, A. p38 Expression and Modulation of STAT3 Signaling in Oral Cancer. Pathol. Oncol. Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-W.; Xu, Z.; Baksh, A.N.; Wang, J.-K.; Chen, C.-Y.; Swanson, R.; Olson, S.T.; Kataoka, H.; Johnson, M.D.; Lin, C.-Y. Antithrombin regulates matriptase activity involved in plasmin generation, syndecan shedding, and HGF activation in ketatinocytes. Plos ONE 2013, 8, 1–11. [Google Scholar] [CrossRef]
- Hao, N.-B.; Tang, B.; Wang, G.-Z.; Xie, R.; Hu, C.-J.; Wang, S.-N.; Wu, Y.-Y.; Liu, E.; Xie, X.; Yang, S.-M. Hepatocyte growth factor (HGF) upregulates heparanase expression via PI3K/Akt/NF-kB signaling pathway for gastric cancer metastasis. Cancer Lett. 2015, 361, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Solís-Calero, C.; Carvalho, H.F. KLK14 interactions with HAI-1 and HAI-2 serine protease inhibitors: A molecular dynamics and relative free-energy calculations study. Cell Biol. Int. 2017, 41, 1246–1264. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.H.; Donaher, J.L.; Murphy, D.A.; Chau, S.; Yang, J. Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell 2012, 22, 725–736. [Google Scholar] [CrossRef]
- Rotoli, D.; Morales, M.; Maeso, M.D.C.; García, M.D.P.; Gutierrez, R.; Valladares, F.; Ávila, J.; Díaz-Flores, L.; Mobasheri, A.; Martín-Vasallo, P. Alterations in IQGAP1 expression and localization in colorectal carcinoma and liver metastases following oxaliplatin-based chemotherapy. Oncol. Lett. 2017, 14, 2621–2628. [Google Scholar] [CrossRef] [Green Version]
- Hong, J.; Zhou, J.; Fu, J.; He, T.; Qin, J.; Wang, L.; Liao, L.; Xu, J. Phosphorylation of serine 68 of Twist1 by MAPKs stabilizes Twist1 protein and promotes breast cancer cell invasiveness. Cancer Res. 2011, 71, 3980–3990. [Google Scholar] [CrossRef]
- Angelini, F.; Ionta, V.; Rossi, F.; Miraldi, F.; Messina, E.; Giacomello, A. Foetal bovine serum-derived exosomes affect yield and phenotype of human cardiac progenitor cell culture. BioImpacts 2016, 6, 15–24. [Google Scholar] [CrossRef]
- Wei, S.C.; Fattet, L.; Tsai, J.H.; Guo, Y.; Pai, V.H.; Majeski, H.E.; Chen, A.C.; Sah, R.L.; Taylor, S.S.; Engler, A.J.; et al. Matrix stiffness drives epithelial-mesenchymal transition and tumour metastasis through a TWIST1-G3BP2 mechanotransduction pathway. Nat. Cell Biol. 2015, 17, 678–688. [Google Scholar] [CrossRef]
- Cheng, J.-C.; Klausen, C.; Leung, P.C.K. Hypoxia-inducible factor 1 alpha mediates epidermal growth factor-induced down-regulation of E-cadherin expression and cell invasion in human ovarian cancer cells. Cancer Lett. 2013, 329, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Gunasinghe, N.P.; Wells, A.; Thompson, E.W.; Hugo, H.J. Mesenchymal-epithelial transition (MET) as a mechanism for metastatic colonization in breast cancer. Cancer Metastasis 2012, 31, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, N.A.; Chytil, A.; Plieth, D.; Gorska, A.E.; Dumont, N.; Shappell, S.; Washington, M.K.; Neilson, E.G.; Moses, H.L. TGF-β signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science 2004, 303, 848–851. [Google Scholar] [CrossRef] [PubMed]
- Owusu, B.Y.; Thomas, S.; Venukadasula, P.; Han, Z.; Janetka, J.W.; Galemmo, R.A., Jr.; Klampfer, L. Targeting the tumor-promoting microenvironment in MET-amplified NSCLC cells with a novel inhibitor of pro-HGF activation. Oncotarget 2017, 8, 63014–63025. [Google Scholar] [CrossRef]
- Franco, H.L.; Casasnovas, J.; Rodríguez-Medina, J.R.; Cadilla, C.L. Redundant or separate entities? -roles of Twist1 and Twist2 as molecular switches during gene transcription. Nucleic Acids Res. 2011, 39, 1177–1186. [Google Scholar] [CrossRef]
- Chiechi, A.; Waning, D.L.; Stayrook, K.R.; Buijs, J.T.; Guise, T.A.; Mohammad, K.S. Role of TGF-b in breast cancer bone metastases. Adv. Biosci. Biotechnol. 2013, 4, 15–30. [Google Scholar] [CrossRef]
- Ikushima, H.; Miyazono, K. TGFbeta signaling: A complex web in cancer progression. Nat. Rev. Cancer 2010, 10, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Siegel, P.M.; Shu, W.; Drobnjak, M.; Kakonen, S.M.; Cordón-Cardo, C.; Guise, T.A.; Massagué, J. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 2003, 3, 537–549. [Google Scholar] [CrossRef] [Green Version]
- Abulaiti, A.; Shintani, Y.; Funaki, S.; Nakagiri, T.; Inoue, M.; Sawabata, N.; Minami, M.; Okumura, M. Interaction between non-small-cell lung cancer cells and fibroblasts via enhancement of TGFb signaling by IL-6. Lung Cancer 2013, 82, 204–213. [Google Scholar] [CrossRef]
- Maroni, P.; Matteucci, E.; Luzzati, A.; Perrucchini, G.; Bendinelli, P.; Desiderio, M.A. Nuclear co-localization and functional interaction of COX-2 and HIF-1α characterize bone metastasis of human breast carcinoma. Breast Cancer Res. Treat. 2011, 129, 433–450. [Google Scholar] [CrossRef]
- Chang, M.-C.; Chang, H.H.; Lin, P.-S.; Huang, Y.-A.; Chan, C.-P.; Tsai, Y.-L.; Lee, S.-Y.; Jeng, P.-Y.; Kuo, H.-Y.; Yeung, S.-Y.; et al. Effects of TGF-β1 on plasminogen activation in human dental pulp cells: Role of ALK5/Smad2, TAK1 and MEK/ERK signaling. J. Tissue Eng. Regen. Med. 2018, 12, 854–863. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bendinelli, P.; Maroni, P.; Dall’Olio, V.; Matteucci, E.; Desiderio, M.A. Bone Metastasis Phenotype and Growth Undergo Regulation by Micro-Environment Stimuli: Efficacy of Early Therapy with HGF or TGFβ1-Type I Receptor Blockade. Int. J. Mol. Sci. 2019, 20, 2520. https://doi.org/10.3390/ijms20102520
Bendinelli P, Maroni P, Dall’Olio V, Matteucci E, Desiderio MA. Bone Metastasis Phenotype and Growth Undergo Regulation by Micro-Environment Stimuli: Efficacy of Early Therapy with HGF or TGFβ1-Type I Receptor Blockade. International Journal of Molecular Sciences. 2019; 20(10):2520. https://doi.org/10.3390/ijms20102520
Chicago/Turabian StyleBendinelli, Paola, Paola Maroni, Valentina Dall’Olio, Emanuela Matteucci, and Maria Alfonsina Desiderio. 2019. "Bone Metastasis Phenotype and Growth Undergo Regulation by Micro-Environment Stimuli: Efficacy of Early Therapy with HGF or TGFβ1-Type I Receptor Blockade" International Journal of Molecular Sciences 20, no. 10: 2520. https://doi.org/10.3390/ijms20102520
APA StyleBendinelli, P., Maroni, P., Dall’Olio, V., Matteucci, E., & Desiderio, M. A. (2019). Bone Metastasis Phenotype and Growth Undergo Regulation by Micro-Environment Stimuli: Efficacy of Early Therapy with HGF or TGFβ1-Type I Receptor Blockade. International Journal of Molecular Sciences, 20(10), 2520. https://doi.org/10.3390/ijms20102520