Signal Transduction in Ribosome Biogenesis: A Recipe to Avoid Disaster



 and
and
Abstract
:
1. Introduction
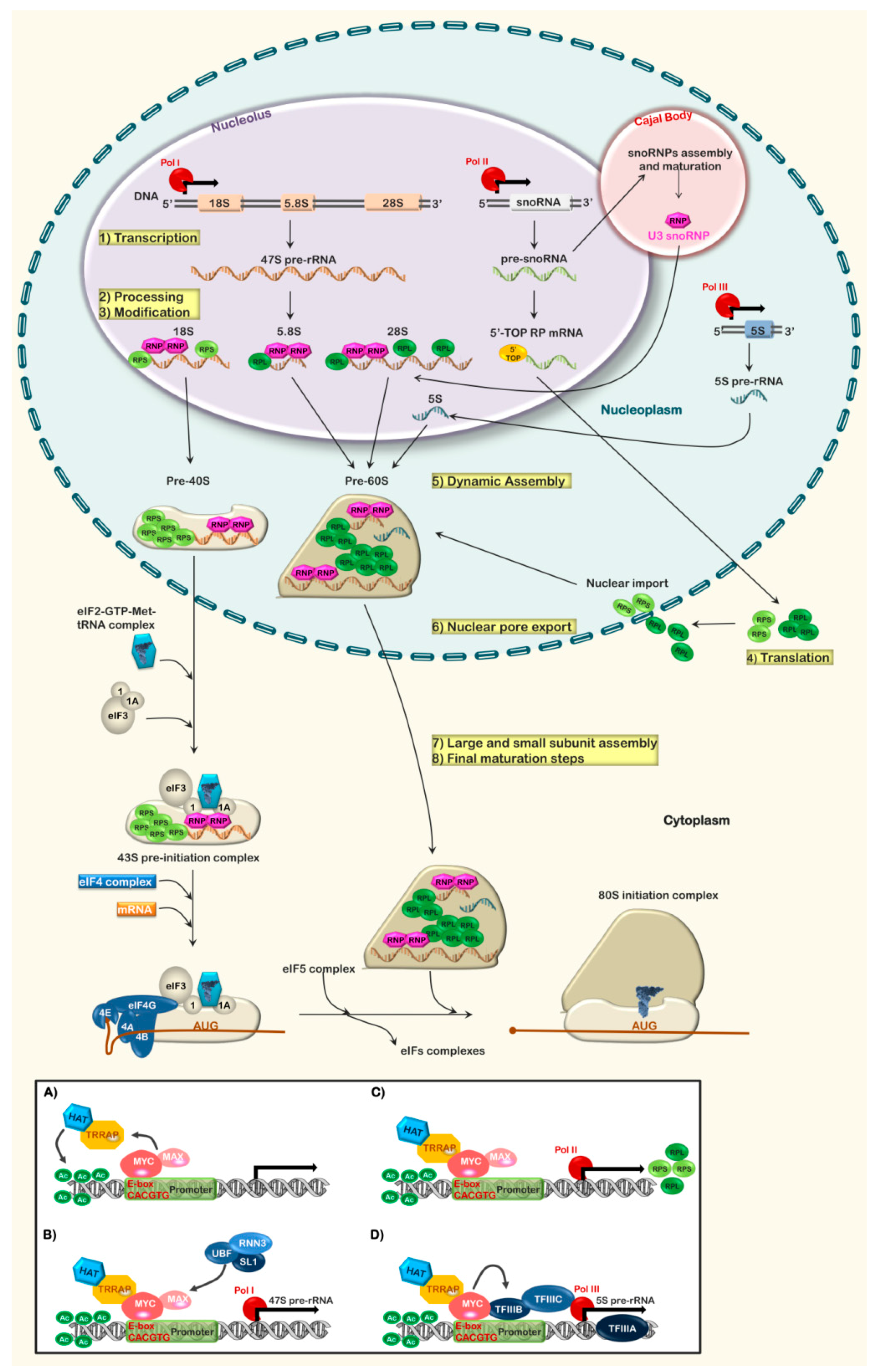
2. Ribosome Biogenesis: An Overview
3. MYC a Global Regulator of Ribosome Biogenesis
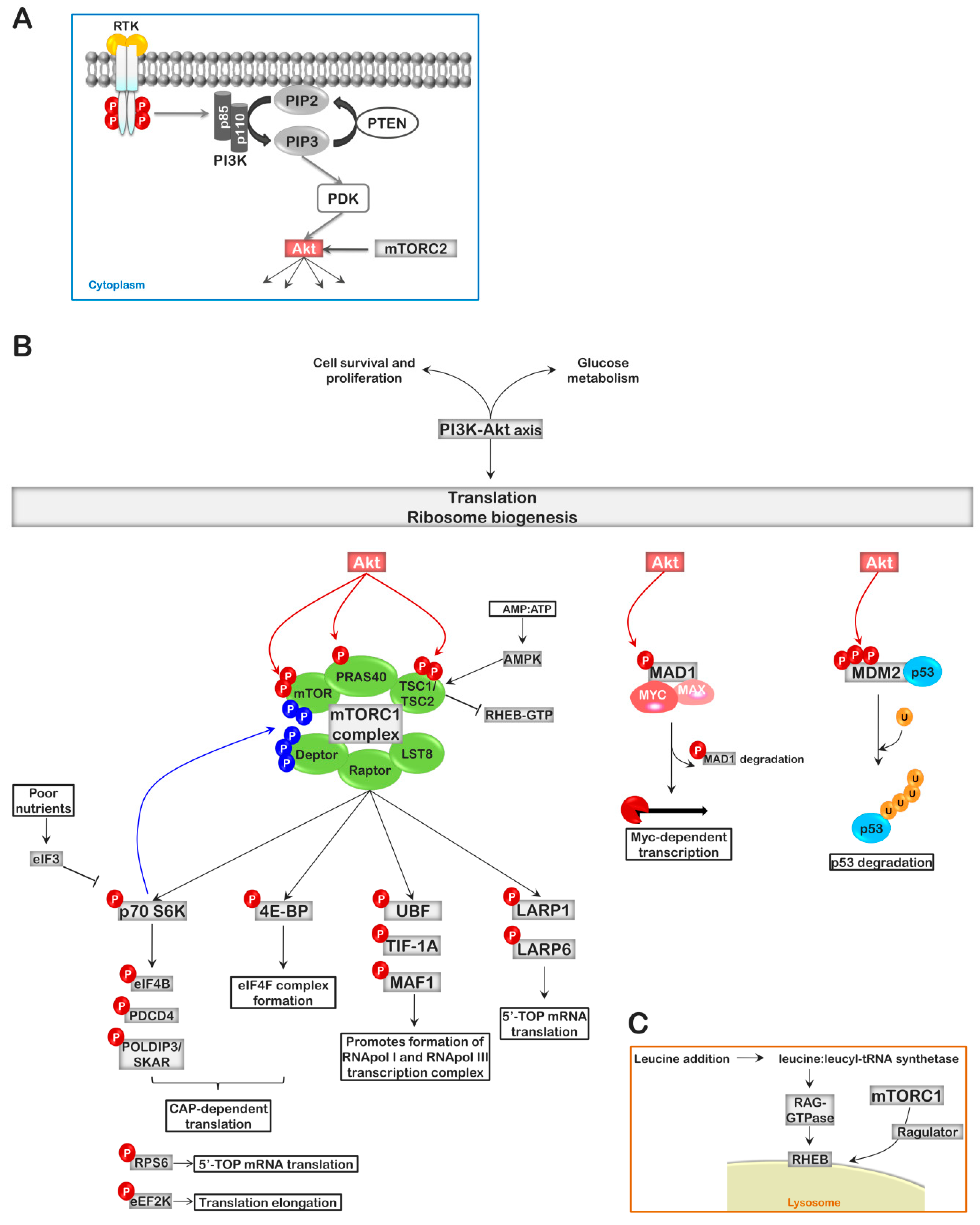
4. The PI3K-AKT-mTOR Pathway, Linking Ribosome Biogenesis to Extracellular Signaling
5. Cell Cycle Regulators and Ribosomal Stress
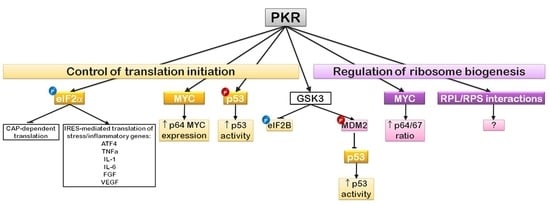
6. EIF2α Regulation and Translation Initiation: The PKR Story
7. RNA Editing/Splicing and Its Potential Role in Ribosome Biogenesis
8. Ribosome Biogenesis and Disease (Ribosomopathies)
8.1. Acquired Ribosomopathies: 5q-Syndrome
8.2. Acquired Ribosomopathies (Potential): T-Cell Acute Lympoblastic Leukemia
8.3. Hereditary Ribosomopathies
8.4. Diamond–Blackfan Anemia (DBA)
8.5. Shwachman–Diamond Syndrome (SDS)
8.6. Dyskeratosis Congenita (DC)
8.7. Treacher–Collins Syndrome (TCS)
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AMP | Adenosine Monophosphate |
| ATF | Activating Transcription Factor |
| ATP | Adenosine Triphosphate |
| BMFD | Bone Marrow Failure Disorder |
| DBA | Diamond-Blackfan Anemia |
| DC | Dyskeratosis Congenita |
| eIF | Eukaryotic Translation Initiation Factor |
| eEF | Eukaryotic Translation Elongation Factor |
| FGF | Fibroblast Growth Factor |
| GDP | Guanosine Diphosphate |
| GTP | Guanosine Triphosphate |
| GTPase | Guanosine Triphosphatase |
| HAT | Histone Acetyltransferase |
| hnRNP | Heterogeneous Nuclear Ribonucleoprotein |
| IFN | Interferon |
| IL | Interleukin |
| IRES | Internal Ribosome Entry Site |
| MDS | Myelodysplastic Syndrome |
| mRNA | Messenger RNA |
| ncRNA | Non-coding RNA |
| PIC | Pre-Initiation Complex |
| PtdIns | Phosphatidylinositol |
| RNP | Ribonucleoprotein |
| RP | Ribosomal Protein |
| RPL | 60S Ribosomal Subunit Protein |
| RPS | 40S Ribosomal Subunit Protein |
| rDNA | Ribosomal DNA |
| rRNA | Ribosomal RNA |
| scaRNA | Small Cajal Body RNA |
| SDS | Shwachman-Diamond Syndrome |
| snRNA | Small Nuclear RNA |
| snRNP | Small Nuclear Ribonucleoprotein |
| snoRNA | Small Nucleolar RNA |
| TCS | Treacher-Collins Syndrome |
| TNFα | Tumor Necrosis Factor-Alpha |
| TRRAP | Transformation/Transcription Domain-Associated Protein |
| tRNA | Transfer RNA |
| uORF | Upstream Open Reading Frame |
| UPC | Ubiquitin Proteosome Complex |
| UPR | Unfolded Protein Response |
| UTR | Untranslated Region |
| VEGF | Vascular Endothelial Growth Factor |
References
- Pelletier, J.; Thomas, G.; Volarević, S. Ribosome biogenesis in cancer: New players and therapeutic avenues. Nat. Rev. Cancer 2018, 18, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Blalock, W.L.; Piazzi, M.; Gallo, A.; Bavelloni, A.; Faenza, I. RNA processing and ribosome biogenesis in bone marrow failure disorders. RNA Dis. 2017, 4, e1531. [Google Scholar] [CrossRef]
- Kjeldgaard, N.O.; Maaloe, O.; Schaechter, M. The transition between different physiological states during balanced growth of Salmonella typhimurium. J. Gen. Microbiol. 1958, 19, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Schaechter, M.; Maaloe, O.; Kjeldgaard, N.O. Dependency on medium and temperature of cell size and chemical composition during balanced grown of Salmonella typhimurium. J. Gen. Microbiol. 1958, 19, 592–606. [Google Scholar] [CrossRef] [PubMed]
- Gamalinda, M.; Woolford, J.L., Jr. Paradigms of ribosome synthesis: Lessons learned from ribosomal proteins. Translation 2015, 3, e975018. [Google Scholar] [CrossRef] [PubMed]
- Henras, A.K.; Plisson-Chastang, C.; O’Donohue, M.F.; Chakraborty, A.; Gleizes, P.E. An overview of pre-ribosomal RNA processing in eukaryotes. Wiley Interdiscip. Rev. RNA 2015, 6, 225–242. [Google Scholar] [CrossRef] [PubMed]
- Kressler, D.; Hurt, E.; Bassler, J. Driving ribosome assembly. Biochim. Biophys. Acta 2010, 1803, 673–683. [Google Scholar] [CrossRef] [Green Version]
- van Riggelen, J.; Yetil, A.; Felsher, D.W. MYC as a regulator of ribosome biogenesis and protein synthesis. Nat. Rev. Cancer 2010, 10, 301–309. [Google Scholar] [CrossRef] [PubMed]
- de Las Heras-Rubio, A.; Perucho, L.; Paciucci, R.; Vilardell, J.; LLeonart, M.E. Ribosomal proteins as novel players in tumorigenesis. Cancer Metastasis Rev. 2014, 33, 115–141. [Google Scholar] [CrossRef]
- Braun, B.R.; Kassavetis, G.A.; Geiduschek, E.P. Bending of the Saccharomyces cerevisiae 5S rRNA gene in transcription factor complexes. J Biol. Chem. 1992, 267, 22562–22569. [Google Scholar]
- Rothfels, K.; Rowland, O.; Segall, J. Zinc fingers 1 and 7 of yeast TFIIIA are essential for assembly of a functional transcription complex on the 5 S RNA gene. Nucleic Acids Res. 2007, 35, 4869–4881. [Google Scholar] [CrossRef] [PubMed]
- Zemp, I.; Kutay, U. Nuclear export and cytoplasmic maturation of ribosomal subunits. FEBS Lett. 2007, 581, 2783–2793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Gerstein, M. Identification and characterization of over 100 mitochondrial ribosomal protein pseudogenes in the human genome. Genomics 2003, 81, 468–480. [Google Scholar] [CrossRef]
- Zhou, Y.; Musalgaonkar, S.; Johnson, A.W.; Taylor, D.W. Tightly-orchestrated rearrangements govern catalytic center assembly of the ribosome. Nat. Commun. 2019, 10, 958. [Google Scholar] [CrossRef]
- Pillet, B.; Mitterer, V.; Kressler, D.; Pertschy, B. Hold on to your friends: Dedicated chaperones of ribosomal proteins: Dedicated chaperones mediate the safe transfer of ribosomal proteins to their site of pre-ribosome incorporation. Bioessays 2017, 39, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Pena, C.; Schutz, S.; Fischer, U.; Chang, Y.; Panse, V.G. Prefabrication of a ribosomal protein subcomplex essential for eukaryotic ribosome formation. Elife 2016, 5, e21755. [Google Scholar] [CrossRef] [PubMed]
- Leidig, C.; Thoms, M.; Holdermann, I.; Bradatsch, B.; Berninghausen, O.; Bange, G.; Sinning, I.; Hurt, E.; Beckmann, R. 60S ribosome biogenesis requires rotation of the 5S ribonucleoprotein particle. Nat. Commun. 2014, 5, 3491. [Google Scholar] [CrossRef]
- Kater, L.; Thoms, M.; Barrio-Garcia, C.; Cheng, J.; Ismail, S.; Ahmed, Y.L.; Bange, G.; Kressler, D.; Berninghausen, O.; Sinning, I.; et al. Visualizing the assembly pathway of nucleolar Pre-60S ribosomes. Cell 2017, 171, 1599–1610. [Google Scholar] [CrossRef]
- Greber, B.J.; Gerhardy, S.; Leitner, A.; Leibundgut, M.; Salem, M.; Boehringer, D.; Leulliot, N.; Aebersold, R.; Panse, V.G.; Ban, N. Insertion of the biogenesis factor rei1 probes the ribosomal tunnel during 60s maturation. Cell 2016, 164, 91–102. [Google Scholar] [CrossRef]
- Turowski, T.W.; Tollervey, D. Cotranscriptional events in eukaryotic ribosome synthesis. Wiley Interdiscip. Rev. RNA 2015, 6, 129–139. [Google Scholar] [CrossRef]
- Fernandez-Pevida, A.; Kressler, D.; de la Cruz, J. Processing of preribosomal RNA in Saccharomyces cerevisiae. Wiley Interdiscip. Rev. RNA 2015, 6, 191–209. [Google Scholar] [CrossRef] [PubMed]
- Mullineux, S.T.; Lafontaine, D.L. Mapping the cleavage sites on mammalian pre-rRNAs: Where do we stand? Biochimie 2012, 94, 1521–1532. [Google Scholar] [CrossRef] [PubMed]
- Burroughs, L.; Woolfrey, A.; Shimamura, A. Shwachman-Diamond syndrome: A review of the clinical presentation, molecular pathogenesis, diagnosis, and treatment. Hematol. Oncol. Clin. N. Am. 2009, 23, 233–248. [Google Scholar] [CrossRef] [PubMed]
- Brina, D.; Miluzio, A.; Ricciardi, S.; Biffo, S. eIF6 anti-association activity is required for ribosome biogenesis, translational control and tumor progression. Biochim. Biophys. Acta 2015, 1849, 830–835. [Google Scholar] [CrossRef] [PubMed]
- Danilova, N.; Gazda, H.T. Ribosomopathies: How a common root can cause a tree of pathologies. Dis. Models Mech. 2015, 8, 1013–1026. [Google Scholar] [CrossRef]
- Bibikova, E.; Youn, M.Y.; Danilova, N.; Ono-Uruga, Y.; Konto-Ghiorghi, Y.; Ochoa, R.; Narla, A.; Glader, B.; Lin, S.; Sakamoto, K.M. TNF-mediated inflammation represses GATA1 and activates p38 MAP kinase in RPS19-deficient hematopoietic progenitors. Blood 2014, 124, 3791–3798. [Google Scholar] [CrossRef]
- Trinkle-Mulcahy, L.; Sleeman, J.E. The Cajal body and the nucleolus: “In a relationship” or “It’s complicated”? RNA Biol. 2017, 14, 739–751. [Google Scholar] [CrossRef]
- Leary, D.J.; Huang, S. Regulation of ribosome biogenesis within the nucleolus. FEBS Lett. 2001, 509, 145–150. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, P.C.; Frank, S.R.; Wang, L.; Schroeder, M.; Liu, S.; Greene, J.; Cocito, A.; Amati, B. Genomic targets of the human c-Myc protein. Genes Dev. 2003, 17, 1115–1129. [Google Scholar] [CrossRef] [Green Version]
- Frank, S.R.; Schroeder, M.; Fernandez, P.; Taubert, S.; Amati, B. Binding of c-Myc to chromatin mediates mitogen-induced acetylation of histone H4 and gene activation. Genes Dev. 2001, 15, 2069–2082. [Google Scholar] [CrossRef] [Green Version]
- McMahon, S.B.; Wood, M.A.; Cole, M.D. The essential cofactor TRRAP recruits the histone acetyltransferase hGCN5 to c-Myc. Mol. Cell. Biol. 2000, 20, 556–562. [Google Scholar] [CrossRef]
- Gomez-Roman, N.; Grandori, C.; Eisenman, R.N.; White, R.J. Direct activation of RNA polymerase III transcription by c-Myc. Nature 2003, 421, 290–294. [Google Scholar] [CrossRef] [PubMed]
- Poortinga, G.; Hannan, K.M.; Snelling, H.; Walkley, C.R.; Jenkins, A.; Sharkey, K.; Wall, M.; Brandenburger, Y.; Palatsides, M.; Pearson, R.B.; et al. MAD1 and c-MYC regulate UBF and rDNA transcription during granulocyte differentiation. EMBO J. 2004, 23, 3325–3335. [Google Scholar] [CrossRef] [Green Version]
- Dai, M.S.; Arnold, H.; Sun, X.X.; Sears, R.; Lu, H. Inhibition of c-Myc activity by ribosomal protein L11. EMBO J. 2007, 26, 3332–3345. [Google Scholar] [CrossRef]
- Ginisty, H.; Amalric, F.; Bouvet, P. Nucleolin functions in the first step of ribosomal RNA processing. EMBO J 1998, 17, 1476–1486. [Google Scholar] [CrossRef] [Green Version]
- Watson, J.D.; Oster, S.K.; Shago, M.; Khosravi, F.; Penn, L.Z. Identifying genes regulated in a Myc-dependent manner. J. Biol. Chem. 2002, 277, 36921–36930. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, E.V. The role of c-myc in regulation of translation initiation. Oncogene 2004, 23, 3217–3221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barna, M.; Pusic, A.; Zollo, O.; Costa, M.; Kondrashov, N.; Rego, E.; Rao, P.H.; Ruggero, D. Suppression of Myc oncogenic activity by ribosomal protein haploinsufficiency. Nature 2008, 456, 971–975. [Google Scholar] [CrossRef] [PubMed]
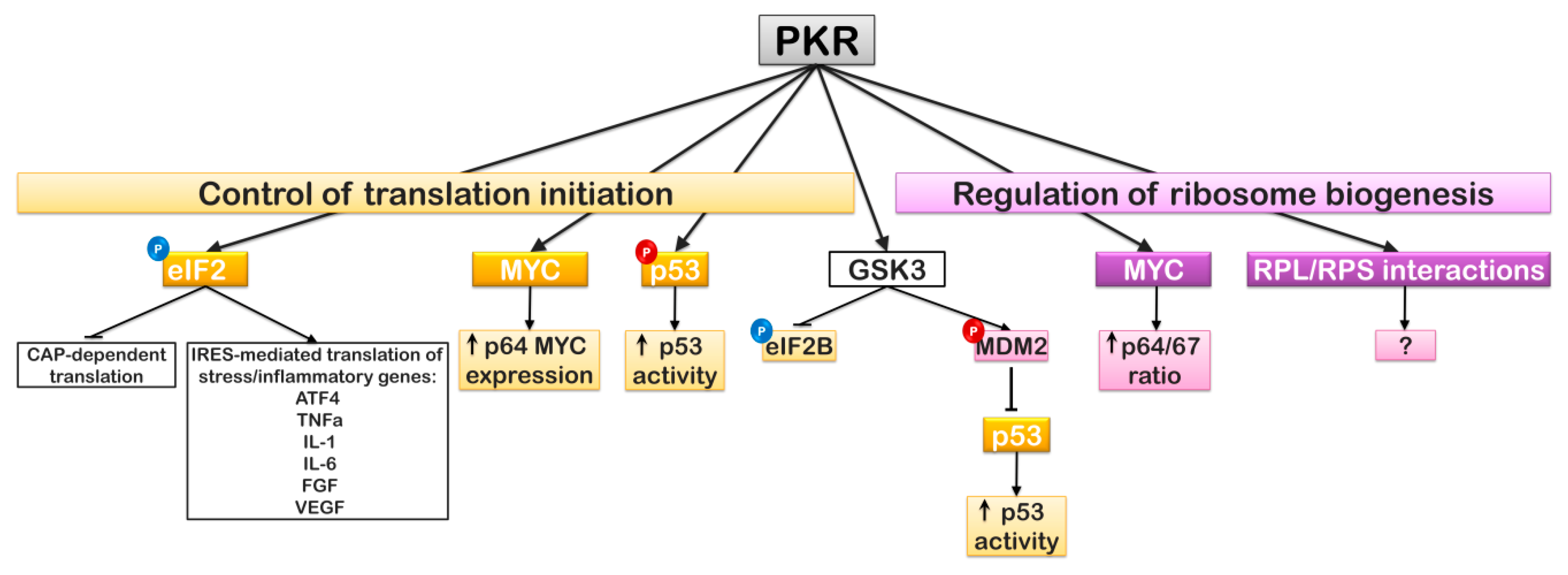
- Blalock, W.L.; Piazzi, M.; Bavelloni, A.; Raffini, M.; Faenza, I.; D’Angelo, A.; Cocco, L. Identification of the PKR nuclear interactome reveals roles in ribosome biogenesis, mRNA processing and cell division. J. Cell. Physiol. 2014, 229, 1047–1060. [Google Scholar] [CrossRef] [PubMed]
- Hann, S.R.; Dixit, M.; Sears, R.C.; Sealy, L. The alternatively initiated c-Myc proteins differentially regulate transcription through a noncanonical DNA-binding site. Genes Dev. 1994, 8, 2441–2452. [Google Scholar] [CrossRef] [PubMed]
- Franke, T.F. PI3K/Akt: Getting it right matters. Oncogene 2008, 27, 6473–6488. [Google Scholar] [CrossRef] [PubMed]
- Hannan, K.M.; Sanij, E.; Hein, N.; Hannan, R.D.; Pearson, R.B. Signaling to the ribosome in cancer—It is more than just mTORC1. IUBMB Life 2011, 63, 79–85. [Google Scholar] [CrossRef]
- Papa, A.; Pandolfi, P.P. The PTEN(-)PI3K Axis in Cancer. Biomolecules 2019, 9, 153. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, E.; McGraw, T.E. The Akt kinases: Isoform specificity in metabolism and cancer. Cell Cycle 2009, 8, 2502–2508. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.M., Jr. The AKT genes and their roles in various disorders. Am. J. Med. Genet. A 2013, 161A, 2931–2937. [Google Scholar] [CrossRef]
- Memmott, R.M.; Dennis, P.A. Akt-dependent and -independent mechanisms of mTOR regulation in cancer. Cell. Signal. 2009, 21, 656–664. [Google Scholar] [CrossRef] [Green Version]
- Boultwood, J.; Yip, B.H.; Vuppusetty, C.; Pellagatti, A.; Wainscoat, J.S. Activation of the mTOR pathway by the amino acid (L)-leucine in the 5q- syndrome and other ribosomopathies. Adv. Biol. Regul. 2013, 53, 8–17. [Google Scholar] [CrossRef]
- Kim, E.; Goraksha-Hicks, P.; Li, L.; Neufeld, T.P.; Guan, K.L. Regulation of TORC1 by Rag GTPases in nutrient response. Nat. Cell Biol. 2008, 10, 935–945. [Google Scholar] [CrossRef] [Green Version]
- Sancak, Y.; Bar-Peled, L.; Zoncu, R.; Markhard, A.L.; Nada, S.; Sabatini, D.M. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 2010, 141, 290–303. [Google Scholar] [CrossRef]
- Nicklin, P.; Bergman, P.; Zhang, B.; Triantafellow, E.; Wang, H.; Nyfeler, B.; Yang, H.; Hild, M.; Kung, C.; Wilson, C.; et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 2009, 136, 521–534. [Google Scholar] [CrossRef] [PubMed]
- Han, J.M.; Jeong, S.J.; Park, M.C.; Kim, G.; Kwon, N.H.; Kim, H.K.; Ha, S.H.; Ryu, S.H.; Kim, S. Leucyl-tRNA synthetase is an intracellular leucine sensor for the mTORC1-signaling pathway. Cell 2012, 149, 410–424. [Google Scholar] [CrossRef]
- Inoki, K.; Zhu, T.; Guan, K.L. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Murashige, D.S.; Humphrey, S.J.; James, D.E. A positive feedback loop between Akt and mTORC2 via SIN1 phosphorylation. Cell Rep. 2015, 12, 937–943. [Google Scholar] [CrossRef]
- Blalock, W.L.; Bavelloni, A.; Piazzi, M.; Faenza, I.; Cocco, L. A role for PKR in hematologic malignancies. J. Cell. Physiol. 2010, 223, 572–591. [Google Scholar] [CrossRef]
- Dorrello, N.V.; Peschiaroli, A.; Guardavaccaro, D.; Colburn, N.H.; Sherman, N.E.; Pagano, M. S6K1- and betaTRCP-mediated degradation of PDCD4 promotes protein translation and cell growth. Science 2006, 314, 467–471. [Google Scholar] [CrossRef]
- Galan, J.A.; Geraghty, K.M.; Lavoie, G.; Kanshin, E.; Tcherkezian, J.; Calabrese, V.; Jeschke, G.R.; Turk, B.E.; Ballif, B.A.; Blenis, J.; et al. Phosphoproteomic analysis identifies the tumor suppressor PDCD4 as a RSK substrate negatively regulated by 14-3-3. Proc. Natl. Acad. Sci. USA 2014, 111, E2918–E2927. [Google Scholar] [CrossRef] [Green Version]
- Raught, B.; Peiretti, F.; Gingras, A.C.; Livingstone, M.; Shahbazian, D.; Mayeur, G.L.; Polakiewicz, R.D.; Sonenberg, N.; Hershey, J.W. Phosphorylation of eucaryotic translation initiation factor 4B Ser422 is modulated by S6 kinases. EMBO J. 2004, 23, 1761–1769. [Google Scholar] [CrossRef] [Green Version]
- Richardson, C.J.; Broenstrup, M.; Fingar, D.C.; Julich, K.; Ballif, B.A.; Gygi, S.; Blenis, J. SKAR is a specific target of S6 kinase 1 in cell growth control. Curr. Biol. 2004, 14, 1540–1549. [Google Scholar] [CrossRef]
- Martineau, Y.; Wang, X.; Alain, T.; Petroulakis, E.; Shahbazian, D.; Fabre, B.; Bousquet-Dubouch, M.P.; Monsarrat, B.; Pyronnet, S.; Sonenberg, N. Control of Paip1-eukayrotic translation initiation factor 3 interaction by amino acids through S6 kinase. Mol. Cell. Biol. 2014, 34, 1046–1053. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Blenis, J.; Yuan, J. Activation of PI3K/Akt and MAPK pathways regulates Myc-mediated transcription by phosphorylating and promoting the degradation of Mad1. Proc. Natl. Acad. Sci. USA 2008, 105, 6584–6589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Stefanovic, B. Akt mediated phosphorylation of LARP6; critical step in biosynthesis of type I collagen. Sci. Rep. 2016, 6, 22597. [Google Scholar] [CrossRef] [Green Version]
- Fonseca, B.D.; Lahr, R.M.; Damgaard, C.K.; Alain, T.; Berman, A.J. LARP1 on TOP of ribosome production. Wiley Interdiscip. Rev. RNA 2018, 9, e1480. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Stefanovic, B. mTORC1 phosphorylates LARP6 to stimulate type I collagen expression. Sci. Rep. 2017, 7, 41173. [Google Scholar] [CrossRef] [PubMed]
- Innes, F.; Ramsbottom, B.; White, R.J. A test of the model that RNA polymerase III transcription is regulated by selective induction of the 110 kDa subunit of TFIIIC. Nucleic Acids Res. 2006, 34, 3399–3407. [Google Scholar] [CrossRef] [PubMed]
- Shor, B.; Wu, J.; Shakey, Q.; Toral-Barza, L.; Shi, C.; Follettie, M.; Yu, K. Requirement of the mTOR kinase for the regulation of Maf1 phosphorylation and control of RNA polymerase III-dependent transcription in cancer cells. J. Biol. Chem. 2010, 285, 15380–15392. [Google Scholar] [CrossRef]
- Wang, X.; Li, W.; Williams, M.; Terada, N.; Alessi, D.R.; Proud, C.G. Regulation of elongation factor 2 kinase by p90(RSK1) and p70 S6 kinase. EMBO J. 2001, 20, 4370–4379. [Google Scholar] [CrossRef] [PubMed]
- Ruvinsky, I.; Meyuhas, O. Ribosomal protein S6 phosphorylation: From protein synthesis to cell size. Trends Biochem. Sci. 2006, 31, 342–348. [Google Scholar] [CrossRef]
- Roux, P.P.; Shahbazian, D.; Vu, H.; Holz, M.K.; Cohen, M.S.; Taunton, J.; Sonenberg, N.; Blenis, J. RAS/ERK signaling promotes site-specific ribosomal protein S6 phosphorylation via RSK and stimulates cap-dependent translation. J. Biol. Chem. 2007, 282, 14056–14064. [Google Scholar] [CrossRef]
- Duan, S.; Skaar, J.R.; Kuchay, S.; Toschi, A.; Kanarek, N.; Ben-Neriah, Y.; Pagano, M. mTOR generates an auto-amplification loop by triggering the betaTrCP- and CK1alpha-dependent degradation of DEPTOR. Mol. Cell 2011, 44, 317–324. [Google Scholar] [CrossRef]
- Dibble, C.C.; Asara, J.M.; Manning, B.D. Characterization of Rictor phosphorylation sites reveals direct regulation of mTOR complex 2 by S6K1. Mol. Cell. Biol. 2009, 29, 5657–5670. [Google Scholar] [CrossRef] [PubMed]
- Holz, M.K.; Blenis, J. Identification of S6 kinase 1 as a novel mammalian target of rapamycin (mTOR)-phosphorylating kinase. J. Biol. Chem. 2005, 280, 26089–26093. [Google Scholar] [CrossRef] [PubMed]
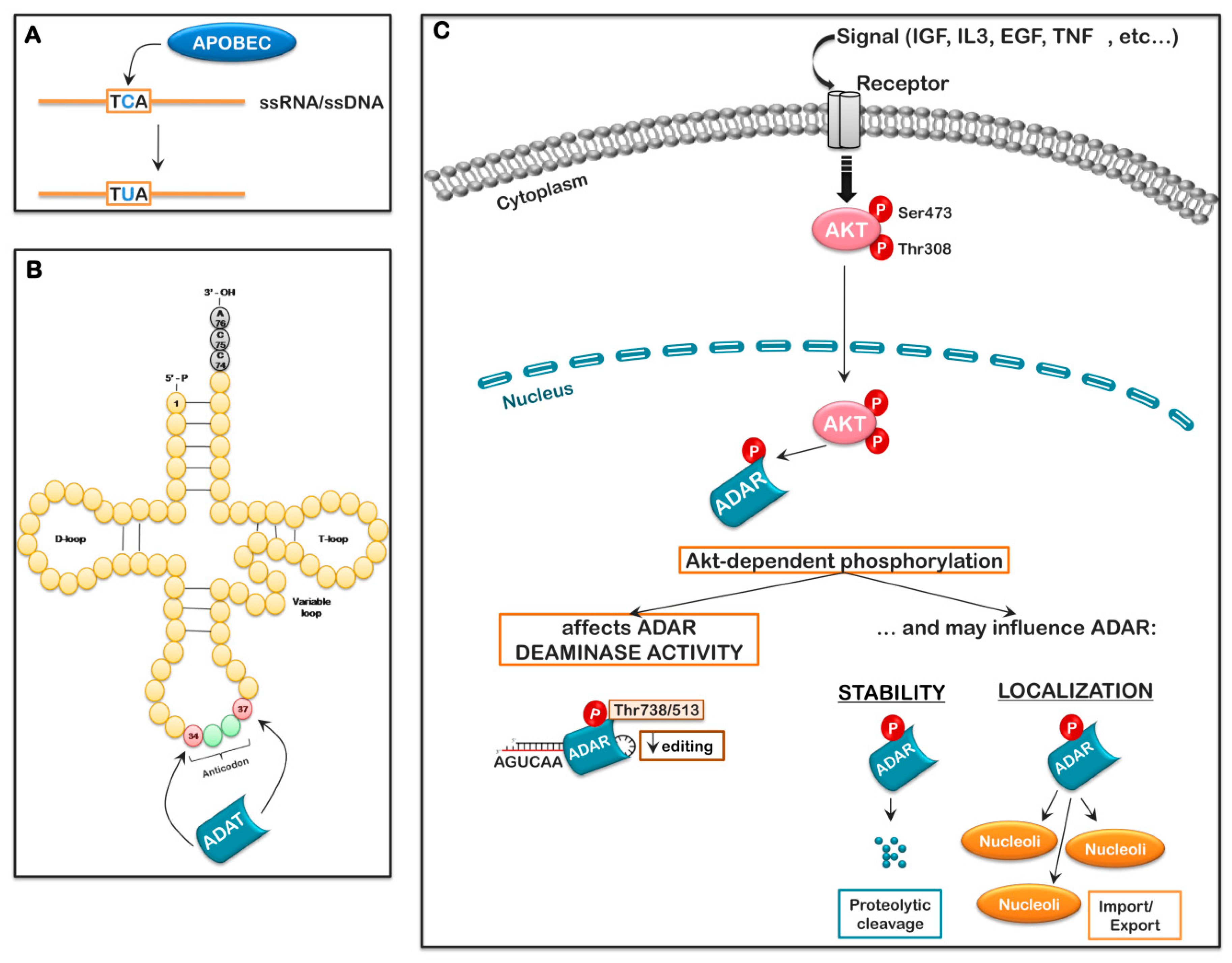
- Bavelloni, A.; Focaccia, E.; Piazzi, M.; Raffini, M.; Cesarini, V.; Tomaselli, S.; Orsini, A.; Ratti, S.; Faenza, I.; Cocco, L.; et al. AKT-dependent phosphorylation of the adenosine deaminases ADAR-1 and -2 inhibits deaminase activity. FASEB J. 2019, fj201800490RR. [Google Scholar] [CrossRef]
- Knudsen, E.S.; Knudsen, K.E. Tailoring to RB: Tumour suppressor status and therapeutic response. Nat. Rev. Cancer 2008, 8, 714–724. [Google Scholar] [CrossRef] [PubMed]
- White, R.J.; Trouche, D.; Martin, K.; Jackson, S.P.; Kouzarides, T. Repression of RNA polymerase III transcription by the retinoblastoma protein. Nature 1996, 382, 88–90. [Google Scholar] [CrossRef]
- Cavanaugh, A.H.; Hempel, W.M.; Taylor, L.J.; Rogalsky, V.; Todorov, G.; Rothblum, L.I. Activity of RNA polymerase I transcription factor UBF blocked by Rb gene product. Nature 1995, 374, 177–180. [Google Scholar] [CrossRef]
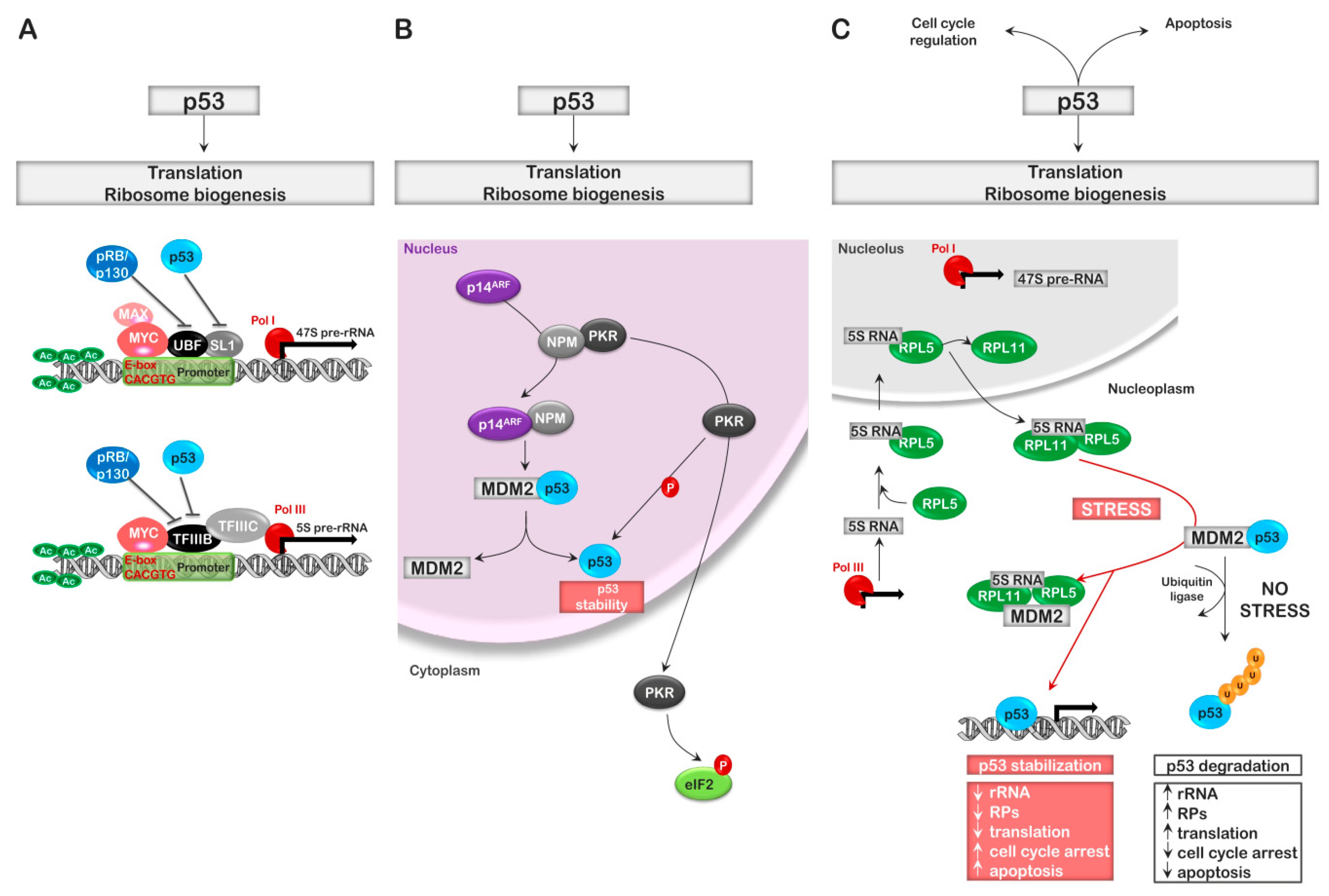
- Lessard, F.; Morin, F.; Ivanchuk, S.; Langlois, F.; Stefanovsky, V.; Rutka, J.; Moss, T. The ARF tumor suppressor controls ribosome biogenesis by regulating the RNA polymerase I transcription factor TTF-I. Mol. Cell 2010, 38, 539–550. [Google Scholar] [CrossRef]
- James, A.; Wang, Y.; Raje, H.; Rosby, R.; DiMario, P. Nucleolar stress with and without p53. Nucleus 2014, 5, 402–426. [Google Scholar] [CrossRef] [Green Version]
- Brady, S.N.; Yu, Y.; Maggi, L.B., Jr.; Weber, J.D. ARF impedes NPM/B23 shuttling in an Mdm2-sensitive tumor suppressor pathway. Mol. Cell. Biol. 2004, 24, 9327–9338. [Google Scholar] [CrossRef]
- Itahana, K.; Bhat, K.P.; Jin, A.; Itahana, Y.; Hawke, D.; Kobayashi, R.; Zhang, Y. Tumor suppressor ARF degrades B23, a nucleolar protein involved in ribosome biogenesis and cell proliferation. Mol. Cell 2003, 12, 1151–1164. [Google Scholar] [CrossRef]
- Llanos, S.; Clark, P.A.; Rowe, J.; Peters, G. Stabilization of p53 by p14ARF without relocation of MDM2 to the nucleolus. Nat. Cell Biol. 2001, 3, 445–452. [Google Scholar] [CrossRef]
- Weber, J.D.; Taylor, L.J.; Roussel, M.F.; Sherr, C.J.; Bar-Sagi, D. Nucleolar Arf sequesters Mdm2 and activates p53. Nat. Cell Biol. 1999, 1, 20–26. [Google Scholar] [CrossRef]
- Garcia, M.A.; Collado, M.; Munoz-Fontela, C.; Matheu, A.; Marcos-Villar, L.; Arroyo, J.; Esteban, M.; Serrano, M.; Rivas, C. Antiviral action of the tumor suppressor ARF. EMBO J. 2006, 25, 4284–4292. [Google Scholar] [CrossRef] [PubMed]
- Itahana, Y.; Itahana, K. Emerging roles of p53 family members in glucose metabolism. Int. J. Mol. Sci. 2018, 19, 776. [Google Scholar] [CrossRef]
- Cairns, C.A.; White, R.J. p53 is a general repressor of RNA polymerase III transcription. EMBO J. 1998, 17, 3112–3123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhai, W.; Comai, L. Repression of RNA polymerase I transcription by the tumor suppressor p53. Mol. Cell. Biol. 2000, 20, 5930–5938. [Google Scholar] [CrossRef]
- Orsolic, I.; Jurada, D.; Pullen, N.; Oren, M.; Eliopoulos, A.G.; Volarevic, S. The relationship between the nucleolus and cancer: Current evidence and emerging paradigms. Semin. Cancer Biol. 2016, 37, 36–50. [Google Scholar] [CrossRef] [PubMed]
- Dai, M.S.; Sun, X.X.; Lu, H. Aberrant expression of nucleostemin activates p53 and induces cell cycle arrest via inhibition of MDM2. Mol. Cell. Biol. 2008, 28, 4365–4376. [Google Scholar] [CrossRef]
- Bursac, S.; Brdovcak, M.C.; Donati, G.; Volarevic, S. Activation of the tumor suppressor p53 upon impairment of ribosome biogenesis. Biochim. Biophys. Acta 2014, 1842, 817–830. [Google Scholar] [CrossRef] [Green Version]
- Sloan, K.E.; Bohnsack, M.T.; Watkins, N.J. The 5S RNP couples p53 homeostasis to ribosome biogenesis and nucleolar stress. Cell Rep. 2013, 5, 237–247. [Google Scholar] [CrossRef]
- Hinnebusch, A.G. Structural insights into the mechanism of scanning and start codon recognition in eukaryotic translation initiation. Trends Biochem. Sci. 2017, 42, 589–611. [Google Scholar] [CrossRef] [PubMed]
- Hinnebusch, A.G.; Lorsch, J.R. The mechanism of eukaryotic translation initiation: New insights and challenges. Cold Spring Harb. Perspect. Biol. 2012, 4, a011544. [Google Scholar] [CrossRef] [PubMed]
- Sonenberg, N.; Hinnebusch, A.G. Regulation of translation initiation in eukaryotes: Mechanisms and biological targets. Cell 2009, 136, 731–745. [Google Scholar] [CrossRef]
- Wek, R.C.; Jiang, H.Y.; Anthony, T.G. Coping with stress: eIF2 kinases and translational control. Biochem. Soc. Trans. 2006, 34, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Kimball, S.R.; Clemens, M.J.; Tilleray, V.J.; Wek, R.C.; Horetsky, R.L.; Jefferson, L.S. The double-stranded RNA-activated protein kinase PKR is dispensable for regulation of translation initiation in response to either calcium mobilization from the endoplasmic reticulum or essential amino acid starvation. Biochem. Biophys. Res. Commun. 2001, 280, 293–300. [Google Scholar] [CrossRef]
- Shi, Y.; Vattem, K.M.; Sood, R.; An, J.; Liang, J.; Stramm, L.; Wek, R.C. Identification and characterization of pancreatic eukaryotic initiation factor 2 alpha-subunit kinase, PEK, involved in translational control. Mol. Cell. Biol. 1998, 18, 7499–7509. [Google Scholar] [CrossRef]
- Mounir, Z.; Krishnamoorthy, J.L.; Wang, S.; Papadopoulou, B.; Campbell, S.; Muller, W.J.; Hatzoglou, M.; Koromilas, A.E. Akt determines cell fate through inhibition of the PERK-eIF2alpha phosphorylation pathway. Sci. Signal 2011, 4, ra62. [Google Scholar] [CrossRef]
- Berlanga, J.J.; Santoyo, J.; De Haro, C. Characterization of a mammalian homolog of the GCN2 eukaryotic initiation factor 2alpha kinase. Eur. J. Biochem. 1999, 265, 754–762. [Google Scholar] [CrossRef]
- Chen, J.J. Regulation of protein synthesis by the heme-regulated eIF2alpha kinase: Relevance to anemias. Blood 2007, 109, 2693–2699. [Google Scholar] [CrossRef]
- Garcia, M.A.; Gil, J.; Ventoso, I.; Guerra, S.; Domingo, E.; Rivas, C.; Esteban, M. Impact of protein kinase PKR in cell biology: From antiviral to antiproliferative action. Microbiol. Mol. Biol. Rev. 2006, 70, 1032–1060. [Google Scholar] [CrossRef]
- Williams, B.R. PKR; a sentinel kinase for cellular stress. Oncogene 1999, 18, 6112–6120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamanaka, R.B.; Bennett, B.S.; Cullinan, S.B.; Diehl, J.A. PERK and GCN2 contribute to eIF2alpha phosphorylation and cell cycle arrest after activation of the unfolded protein response pathway. Mol. Biol. Cell 2005, 16, 5493–5501. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Warnken, S.P.; May, W.S. Protein synthesis inhibition by flavonoids: Roles of eukaryotic initiation factor 2alpha kinases. Biochem. Biophys. Res. Commun. 1999, 265, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Pervin, S.; Tran, A.H.; Zekavati, S.; Fukuto, J.M.; Singh, R.; Chaudhuri, G. Increased susceptibility of breast cancer cells to stress mediated inhibition of protein synthesis. Cancer Res. 2008, 68, 4862–4874. [Google Scholar] [CrossRef]
- Blalock, W.L.; Bavelloni, A.; Piazzi, M.; Tagliavini, F.; Faenza, I.; Martelli, A.M.; Follo, M.Y.; Cocco, L. Multiple forms of PKR present in the nuclei of acute leukemia cells represent an active kinase that is responsive to stress. Leukemia 2011, 25, 236–245. [Google Scholar] [CrossRef]
- Tafforeau, L. About the ribosomal biogenesis in human. Med. Sci. 2015, 31, 622–628. [Google Scholar] [CrossRef]
- Tomecki, R.; Sikorski, P.J.; Zakrzewska-Placzek, M. Comparison of preribosomal RNA processing pathways in yeast, plant and human cells—Focus on coordinated action of endo- and exoribonucleases. FEBS Lett. 2017, 591, 1801–1850. [Google Scholar] [CrossRef]
- Fernandez, J.; Yaman, I.; Merrick, W.C.; Koromilas, A.; Wek, R.C.; Sood, R.; Hensold, J.; Hatzoglou, M. Regulation of internal ribosome entry site-mediated translation by eukaryotic initiation factor-2alpha phosphorylation and translation of a small upstream open reading frame. J. Biol. Chem. 2002, 277, 2050–2058. [Google Scholar] [CrossRef]
- Yaman, I.; Fernandez, J.; Liu, H.; Caprara, M.; Komar, A.A.; Koromilas, A.E.; Zhou, L.; Snider, M.D.; Scheuner, D.; Kaufman, R.J.; et al. The zipper model of translational control: A small upstream ORF is the switch that controls structural remodeling of an mRNA leader. Cell 2003, 113, 519–531. [Google Scholar] [CrossRef]
- Gerlitz, G.; Jagus, R.; Elroy-Stein, O. Phosphorylation of initiation factor-2 alpha is required for activation of internal translation initiation during cell differentiation. Eur. J. Biochem. 2002, 269, 2810–2819. [Google Scholar] [CrossRef] [PubMed]
- Donze, O.; Deng, J.; Curran, J.; Sladek, R.; Picard, D.; Sonenberg, N. The protein kinase PKR: A molecular clock that sequentially activates survival and death programs. EMBO J. 2004, 23, 564–571. [Google Scholar] [CrossRef] [PubMed]
- Sanz, M.A.; Redondo, N.; Garcia-Moreno, M.; Carrasco, L. Phosphorylation of eIF2alpha is responsible for the failure of the picornavirus internal ribosome entry site to direct translation from Sindbis virus replicons. J. Gen. Virol. 2013, 94, 796–806. [Google Scholar] [CrossRef] [PubMed]
- Stoneley, M.; Willis, A.E. Cellular internal ribosome entry segments: Structures, trans-acting factors and regulation of gene expression. Oncogene 2004, 23, 3200–3207. [Google Scholar] [CrossRef]
- Aggarwal, B.B.; Shishodia, S.; Takada, Y.; Jackson-Bernitsas, D.; Ahn, K.S.; Sethi, G.; Ichikawa, H. TNF blockade: An inflammatory issue. Ernst Schering Res. Found. Workshop 2016, 161–186. [Google Scholar] [CrossRef]
- Aggarwal, B.B.; Shishodia, S.; Sandur, S.K.; Pandey, M.K.; Sethi, G. Inflammation and cancer: How hot is the link? Biochem. Pharmacol. 2006, 72, 1605–1621. [Google Scholar] [CrossRef]
- Koschmieder, S.; D’Alo, F.; Radomska, H.; Schoneich, C.; Chang, J.S.; Konopleva, M.; Kobayashi, S.; Levantini, E.; Suh, N.; Di Ruscio, A.; et al. CDDO induces granulocytic differentiation of myeloid leukemic blasts through translational up-regulation of p42 CCAAT enhancer binding protein alpha. Blood 2007, 110, 3695–3705. [Google Scholar] [CrossRef]
- Lee, Y.Y.; Cevallos, R.C.; Jan, E. An upstream open reading frame regulates translation of GADD34 during cellular stresses that induce eIF2alpha phosphorylation. J. Biol. Chem. 2009, 284, 6661–6673. [Google Scholar] [CrossRef]
- van den Beucken, T.; Magagnin, M.G.; Savelkouls, K.; Lambin, P.; Koritzinsky, M.; Wouters, B.G. Regulation of Cited2 expression provides a functional link between translational and transcriptional responses during hypoxia. Radiother. Oncol. 2007, 83, 346–352. [Google Scholar] [CrossRef]
- Hofmann, W.K.; de Vos, S.; Komor, M.; Hoelzer, D.; Wachsman, W.; Koeffler, H.P. Characterization of gene expression of CD34+ cells from normal and myelodysplastic bone marrow. Blood 2002, 100, 3553–3560. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, K.; Lee, S.H.; Kim, J.S.; Wimalasena, J.; Kitajima, S.; Baek, S.J. Activating transcription factor 3 and early growth response 1 are the novel targets of LY294002 in a phosphatidylinositol 3-kinase-independent pathway. Cancer Res. 2006, 66, 2376–2384. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Arantes, S.; Yan, L.; Kiguchi, K.; McArthur, M.J.; Sahin, A.; Thames, H.D.; Aldaz, C.M.; Macleod, M.C. The transcription factor ATF3 acts as an oncogene in mouse mammary tumorigenesis. BMC Cancer 2008, 8, 268. [Google Scholar] [CrossRef] [PubMed]
- Janz, M.; Hummel, M.; Truss, M.; Wollert-Wulf, B.; Mathas, S.; Johrens, K.; Hagemeier, C.; Bommert, K.; Stein, H.; Dorken, B.; et al. Classical Hodgkin lymphoma is characterized by high constitutive expression of activating transcription factor 3 (ATF3), which promotes viability of Hodgkin/Reed-Sternberg cells. Blood 2006, 107, 2536–2539. [Google Scholar] [CrossRef] [Green Version]
- Calkhoven, C.F.; Muller, C.; Leutz, A. Translational control of C/EBPalpha and C/EBPbeta isoform expression. Genes Dev. 2000, 14, 1920–1932. [Google Scholar] [PubMed]
- Nerlov, C. The C/EBP family of transcription factors: A paradigm for interaction between gene expression and proliferation control. Trends Cell Biol. 2007, 17, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.E.; Zhang, P.; Wang, N.D.; Hetherington, C.J.; Darlington, G.J.; Tenen, D.G. Absence of granulocyte colony-stimulating factor signaling and neutrophil development in CCAAT enhancer binding protein alpha-deficient mice. Proc. Natl. Acad. Sci. USA 1997, 94, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Akasaka, T.; Balasas, T.; Russell, L.J.; Sugimoto, K.J.; Majid, A.; Walewska, R.; Karran, E.L.; Brown, D.G.; Cain, K.; Harder, L.; et al. Five members of the CEBP transcription factor family are targeted by recurrent IGH translocations in B-cell precursor acute lymphoblastic leukemia (BCP-ALL). Blood 2007, 109, 3451–3461. [Google Scholar] [CrossRef] [PubMed]
- Chapiro, E.; Russell, L.; Radford-Weiss, I.; Bastard, C.; Lessard, M.; Struski, S.; Cave, H.; Fert-Ferrer, S.; Barin, C.; Maarek, O.; et al. Overexpression of CEBPA resulting from the translocation t(14;19)(q32;q13) of human precursor B acute lymphoblastic leukemia. Blood 2006, 108, 3560–3563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geletu, M.; Balkhi, M.Y.; Peer Zada, A.A.; Christopeit, M.; Pulikkan, J.A.; Trivedi, A.K.; Tenen, D.G.; Behre, G. Target proteins of C/EBPalphap30 in AML: C/EBPalphap30 enhances sumoylation of C/EBPalphap42 via up-regulation of Ubc9. Blood 2007, 110, 3301–3309. [Google Scholar] [CrossRef]
- Zhu, S.; Yoon, K.; Sterneck, E.; Johnson, P.F.; Smart, R.C. CCAAT/enhancer binding protein-beta is a mediator of keratinocyte survival and skin tumorigenesis involving oncogenic Ras signaling. Proc. Natl. Acad. Sci. USA 2002, 99, 207–212. [Google Scholar] [CrossRef]
- Sterneck, E.; Zhu, S.; Ramirez, A.; Jorcano, J.L.; Smart, R.C. Conditional ablation of C/EBP beta demonstrates its keratinocyte-specific requirement for cell survival and mouse skin tumorigenesis. Oncogene 2006, 25, 1272–1276. [Google Scholar] [CrossRef] [PubMed]
- Ramana, C.V.; Grammatikakis, N.; Chernov, M.; Nguyen, H.; Goh, K.C.; Williams, B.R.; Stark, G.R. Regulation of c-myc expression by IFN-gamma through Stat1-dependent and -independent pathways. EMBO J. 2000, 19, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Takada, Y.; Ichikawa, H.; Pataer, A.; Swisher, S.; Aggarwal, B.B. Genetic deletion of PKR abrogates TNF-induced activation of IkappaBalpha kinase, JNK, Akt and cell proliferation but potentiates p44/p42 MAPK and p38 MAPK activation. Oncogene 2007, 26, 1201–1212. [Google Scholar] [CrossRef] [PubMed]
- Blalock, W.L.; Grimaldi, C.; Fala, F.; Follo, M.; Horn, S.; Basecke, J.; Martinelli, G.; Cocco, L.; Martelli, A.M. PKR activity is required for acute leukemic cell maintenance and growth: A role for PKR-mediated phosphatase activity to regulate GSK-3 phosphorylation. J. Cell. Physiol. 2009, 221, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Bose, A.; Mouton-Liger, F.; Paquet, C.; Mazot, P.; Vigny, M.; Gray, F.; Hugon, J. Modulation of tau phosphorylation by the kinase PKR: Implications in Alzheimer’s disease. Brain Pathol. 2011, 21, 189–200. [Google Scholar] [CrossRef]
- Proud, C.G. eIF2 and the control of cell physiology. Semin. Cell Dev. Biol. 2005, 16, 3–12. [Google Scholar] [CrossRef]
- Wang, X.; Paulin, F.E.; Campbell, L.E.; Gomez, E.; O’Brien, K.; Morrice, N.; Proud, C.G. Eukaryotic initiation factor 2B: Identification of multiple phosphorylation sites in the epsilon-subunit and their functions in vivo. EMBO J. 2001, 20, 4349–4359. [Google Scholar] [CrossRef]
- Kubica, N.; Jefferson, L.S.; Kimball, S.R. Eukaryotic initiation factor 2B and its role in alterations in mRNA translation that occur under a number of pathophysiological and physiological conditions. Prog. Nucleic Acid Res. Mol. Biol. 2006, 81, 271–296. [Google Scholar] [CrossRef]
- Willis, A.E. Translational control of growth factor and proto-oncogene expression. Int. J. Biochem. Cell Biol. 1999, 31, 73–86. [Google Scholar] [CrossRef]
- van den Beucken, T.; Koritzinsky, M.; Wouters, B.G. Translational control of gene expression during hypoxia. Cancer Biol. Ther. 2006, 5, 749–755. [Google Scholar] [CrossRef] [Green Version]
- Spriggs, K.A.; Bushell, M.; Mitchell, S.A.; Willis, A.E. Internal ribosome entry segment-mediated translation during apoptosis: The role of IRES-trans-acting factors. Cell Death Differ. 2005, 12, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Baltzis, D.; Pluquet, O.; Papadakis, A.I.; Kazemi, S.; Qu, L.K.; Koromilas, A.E. The eIF2alpha kinases PERK and PKR activate glycogen synthase kinase 3 to promote the proteasomal degradation of p53. J. Biol. Chem. 2007, 282, 31675–31687. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.R.; He, K.; Landgraf, J.; Pan, X.; Pestka, J.J. Direct activation of ribosome-associated double-stranded RNA-dependent protein kinase (PKR) by deoxynivalenol, anisomycin and ricin: A new model for ribotoxic stress response induction. Toxins 2014, 6, 3406–3425. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, J.; Huang, B.O.; Xu, Y.M.; Li, J.; Huang, L.F.; Lin, J.; Zhang, J.; Min, Q.H.; Yang, W.M.; et al. Mechanism of alternative splicing and its regulation. Biomed. Rep. 2015, 3, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Baralle, D.; Buratti, E. RNA splicing in human disease and in the clinic. Clin. Sci. 2017, 131, 355–368. [Google Scholar] [CrossRef] [PubMed]
- Haferlach, T.; Nagata, Y.; Grossmann, V.; Okuno, Y.; Bacher, U.; Nagae, G.; Schnittger, S.; Sanada, M.; Kon, A.; Alpermann, T.; et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 2014, 28, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Papaemmanuil, E.; Gerstung, M.; Malcovati, L.; Tauro, S.; Gundem, G.; Van Loo, P.; Yoon, C.J.; Ellis, P.; Wedge, D.C.; Pellagatti, A.; et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 2013, 122, 3616–3627. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Sanada, M.; Shiraishi, Y.; Nowak, D.; Nagata, Y.; Yamamoto, R.; Sato, Y.; Sato-Otsubo, A.; Kon, A.; Nagasaki, M.; et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 2011, 478, 64–69. [Google Scholar] [CrossRef]
- Rendleman, J.; Cheng, Z.; Maity, S.; Kastelic, N.; Munschauer, M.; Allgoewer, K.; Teo, G.; Zhang, Y.B.M.; Lei, A.; Parker, B.; et al. New insights into the cellular temporal response to proteostatic stress. Elife 2018, 7, e39054. [Google Scholar] [CrossRef] [PubMed]
- Mei, H.; Wang, Y.; Fan, J.; Lin, Z. Alternative splicing of S6K1 promotes non-small cell lung cancer survival. Tumor Biol. 2016, 37, 13369–13376. [Google Scholar] [CrossRef]
- Mrvova, S.; Frydryskova, K.; Pospisek, M.; Vopalensky, V.; Masek, T. Major splice variants and multiple polyadenylation site utilization in mRNAs encoding human translation initiation factors eIF4E1 and eIF4E3 regulate the translational regulators? Mol. Genet. Genom. 2018, 293, 167–186. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.N.; Yuan, J.H.; Wang, T.T.; Pan, W.; Sun, S.H. An alternative POLDIP3 transcript promotes hepatocellular carcinoma progression. Biomed. Pharmacother. 2017, 89, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.; Warner, J.R. Ribosome-omics of the human ribosome. RNA 2014, 20, 1004–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plocik, A.M.; Guthrie, C. Diverse forms of RPS9 splicing are part of an evolving autoregulatory circuit. PLoS Genet. 2012, 8, e1002620. [Google Scholar] [CrossRef] [PubMed]
- Carlston, C.M.; Afify, Z.A.; Palumbos, J.C.; Bagley, H.; Barbagelata, C.; Wooderchak-Donahue, W.L.; Mao, R.; Carey, J.C. Variable expressivity and incomplete penetrance in a large family with non-classical Diamond-Blackfan anemia associated with ribosomal protein L11 splicing variant. Am. J. Med. Genet. Part A 2017, 173, 2622–2627. [Google Scholar] [CrossRef] [PubMed]
- Aviner, R.; Hofmann, S.; Elman, T.; Shenoy, A.; Geiger, T.; Elkon, R.; Ehrlich, M.; Elroy-Stein, O. Proteomic analysis of polyribosomes identifies splicing factors as potential regulators of translation during mitosis. Nucleic Acids Res. 2017, 45, 5945–5957. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Luo, C.; Luo, Y.; Chen, L.; Liu, Y.; Wang, Y.; Han, J.; Zhang, Y.; Wei, N.; Xie, Z.; et al. MRPL33 and its splicing regulator hnRNPK are required for mitochondria function and implicated in tumor progression. Oncogene 2018, 37, 86–94. [Google Scholar] [CrossRef]
- Ogawa, S. Splicing factor mutations in myelodysplasia. Int. J. Hematol. 2012, 96, 438–442. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.; Li, X.; Lei, L.; Chen, J. APOBEC: From mutator to editor. J. Genet. Genom. 2017, 44, 423–437. [Google Scholar] [CrossRef]
- Nishikura, K. A-to-I editing of coding and non-coding RNAs by ADARs. Nat. Rev. Mol. Cell Biol. 2016, 17, 83–96. [Google Scholar] [CrossRef]
- Eifler, T.; Pokharel, S.; Beal, P.A. RNA-Seq analysis identifies a novel set of editing substrates for human ADAR2 present in Saccharomyces cerevisiae. Biochemistry 2013, 52, 7857–7869. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, E.; Nemzer, S.; Kinar, Y.; Sorek, R.; Rechavi, G.; Levanon, E.Y. Is abundant A-to-I RNA editing primate-specific? Trends Genet. 2005, 21, 77–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keegan, L.P.; Leroy, A.; Sproul, D.; O’Connell, M.A. Adenosine deaminases acting on RNA (ADARs): RNA-editing enzymes. Genome Biol. 2004, 5, 209. [Google Scholar] [CrossRef]
- Torres, A.G.; Pineyro, D.; Filonava, L.; Stracker, T.H.; Batlle, E.; Ribas de Pouplana, L. A-to-I editing on tRNAs: Biochemical, biological and evolutionary implications. FEBS Lett. 2014, 588, 4279–4286. [Google Scholar] [CrossRef] [PubMed]
- Lim, V.I.; Curran, J.F. Analysis of codon: Anticodon interactions within the ribosome provides new insights into codon reading and the genetic code structure. RNA 2001, 7, 942–957. [Google Scholar] [CrossRef] [PubMed]
- Curran, J.F. Decoding with the A: I wobble pair is inefficient. Nucleic Acids Res. 1995, 23, 683–688. [Google Scholar] [CrossRef]
- Keegan, L.; Khan, A.; Vukic, D.; O’Connell, M. ADAR RNA editing below the backbone. RNA 2017, 23, 1317–1328. [Google Scholar] [CrossRef] [PubMed]
- Gallo, A.; Vukic, D.; Michalik, D.; O’Connell, M.A.; Keegan, L.P. ADAR RNA editing in human disease; more to it than meets the I. Hum. Genet. 2017, 136, 1265–1278. [Google Scholar] [CrossRef]
- Patterson, J.B.; Samuel, C.E. Expression and regulation by interferon of a double-stranded-RNA-specific adenosine deaminase from human cells: Evidence for two forms of the deaminase. Mol. Cell. Biol. 1995, 15, 5376–5388. [Google Scholar] [CrossRef] [PubMed]
- George, C.X.; Wagner, M.V.; Samuel, C.E. Expression of interferon-inducible RNA adenosine deaminase ADAR1 during pathogen infection and mouse embryo development involves tissue-selective promoter utilization and alternative splicing. J. Biol. Chem. 2005, 280, 15020–15028. [Google Scholar] [CrossRef] [PubMed]
- Gerber, A.; O’Connell, M.A.; Keller, W. Two forms of human double-stranded RNA-specific editase 1 (hRED1) generated by the insertion of an Alu cassette. RNA 1997, 3, 453–463. [Google Scholar]
- Sansam, C.L.; Wells, K.S.; Emeson, R.B. Modulation of RNA editing by functional nucleolar sequestration of ADAR2. Proc. Natl. Acad. Sci. USA 2003, 100, 14018–14023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakurai, M.; Shiromoto, Y.; Ota, H.; Song, C.; Kossenkov, A.V.; Wickramasinghe, J.; Showe, L.C.; Skordalakes, E.; Tang, H.Y.; Speicher, D.W.; et al. ADAR1 controls apoptosis of stressed cells by inhibiting Staufen1-mediated mRNA decay. Nat. Struct. Mol. Biol. 2017, 24, 534–543. [Google Scholar] [CrossRef] [PubMed]
- Shelton, P.M.; Duran, A.; Nakanishi, Y.; Reina-Campos, M.; Kasashima, H.; Llado, V.; Ma, L.; Campos, A.; Garcia-Olmo, D.; Garcia-Arranz, M.; et al. The secretion of miR-200s by a PKCzeta/ADAR2 signaling axis promotes liver metastasis in colorectal cancer. Cell Rep. 2018, 23, 1178–1191. [Google Scholar] [CrossRef] [PubMed]
- George, C.X.; Ramaswami, G.; Li, J.B.; Samuel, C.E. Editing of cellular Self-RNAs by adenosine deaminase ADAR1 suppresses innate immune stress responses. J. Biol. Chem. 2016, 291, 6158–6168. [Google Scholar] [CrossRef] [PubMed]
- Liddicoat, B.J.; Hartner, J.C.; Piskol, R.; Ramaswami, G.; Chalk, A.M.; Kingsley, P.D.; Sankaran, V.G.; Wall, M.; Purton, L.E.; Seeburg, P.H.; et al. Adenosine-to-inosine RNA editing by ADAR1 is essential for normal murine erythropoiesis. Exp. Hematol. 2016, 44, 947–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aspesi, A.; Ellis, S.R. Rare ribosomopathies: Insights into mechanisms of cancer. Nat. Rev. Cancer 2019, 19, 228–238. [Google Scholar] [CrossRef]
- Yu, Q.H.; Wang, S.Y.; Wu, Z. Advances in genetic studies of inherited bone marrow failure syndromes and their associated malignancies. Transl. Pediatr. 2014, 3, 305–309. [Google Scholar] [CrossRef]
- Bagby, G.C.; Meyers, G. Bone marrow failure as a risk factor for clonal evolution: Prospects for leukemia prevention. Hematol. Am. Soc. Hematol. Educ. Progr. 2007, 40–46. [Google Scholar] [CrossRef]
- Cazzola, M.; Della Porta, M.G.; Malcovati, L. The genetic basis of myelodysplasia and its clinical relevance. Blood 2013, 122, 4021–4034. [Google Scholar] [CrossRef] [Green Version]
- Malcovati, L.; Ambaglio, I.; Elena, C. The genomic landscape of myeloid neoplasms with myelodysplasia and its clinical implications. Curr. Opin. Oncol. 2015, 27, 551–559. [Google Scholar] [CrossRef] [PubMed]
- Follo, M.Y.; Finelli, C.; Mongiorgi, S.; Clissa, C.; Bosi, C.; Martinelli, G.; Blalock, W.L.; Cocco, L.; Martelli, A.M. PKR is activated in MDS patients and its subcellular localization depends on disease severity. Leukemia 2008, 22, 2267–2269. [Google Scholar] [CrossRef] [PubMed]
- Papaemmanuil, E.; Cazzola, M.; Boultwood, J.; Malcovati, L.; Vyas, P.; Bowen, D.; Pellagatti, A.; Wainscoat, J.S.; Hellstrom-Lindberg, E.; Gambacorti-Passerini, C.; et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N. Engl. J. Med. 2011, 365, 1384–1395. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Sashida, G.; Saraya, A.; Ishiga, R.; Koide, S.; Oshima, M.; Isono, K.; Koseki, H.; Iwama, A. Depletion of Sf3b1 impairs proliferative capacity of hematopoietic stem cells but is not sufficient to induce myelodysplasia. Blood 2014, 123, 3336–3343. [Google Scholar] [CrossRef] [Green Version]
- Pellagatti, A.; Boultwood, J. Recent Advances in the 5q- Syndrome. Mediterr. J. Hematol. Infect. Dis. 2015, 7, e2015037. [Google Scholar] [CrossRef]
- Goudarzi, K.M.; Lindstrom, M.S. Role of ribosomal protein mutations in tumor development (Review). Int. J. Oncol. 2016, 48, 1313–1324. [Google Scholar] [CrossRef] [Green Version]
- De Keersmaecker, K.; Atak, Z.K.; Li, N.; Vicente, C.; Patchett, S.; Girardi, T.; Gianfelici, V.; Geerdens, E.; Clappier, E.; Porcu, M.; et al. Exome sequencing identifies mutation in CNOT3 and ribosomal genes RPL5 and RPL10 in T-cell acute lymphoblastic leukemia. Nat. Genet. 2013, 45, 186–190. [Google Scholar] [CrossRef]
- Palomero, T.; Sulis, M.L.; Cortina, M.; Real, P.J.; Barnes, K.; Ciofani, M.; Caparros, E.; Buteau, J.; Brown, K.; Perkins, S.L.; et al. Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nat. Med. 2007, 13, 1203–1210. [Google Scholar] [CrossRef] [Green Version]
- Martelli, A.M.; Paganelli, F.; Fazio, A.; Bazzichetto, C.; Conciatori, F.; McCubrey, J.A. The key roles of PTEN in T-Cell acute lymphoblastic leukemia development, progression, and therapeutic response. Cancers 2019, 11, 629. [Google Scholar] [CrossRef]
- Villegas, S.N.; Gombos, R.; Garcia-Lopez, L.; Gutierrez-Perez, I.; Garcia-Castillo, J.; Vallejo, D.M.; Da Ros, V.G.; Ballesta-Illan, E.; Mihaly, J.; Dominguez, M. PI3K/Akt cooperates with oncogenic notch by inducing nitric oxide-dependent inflammation. Cell Rep. 2018, 22, 2541–2549. [Google Scholar] [CrossRef]
- Grzes, K.M.; Swamy, M.; Hukelmann, J.L.; Emslie, E.; Sinclair, L.V.; Cantrell, D.A. Control of amino acid transport coordinates metabolic reprogramming in T-cell malignancy. Leukemia 2017, 31, 2771–2779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sulima, S.O.; Patchett, S.; Advani, V.M.; De Keersmaecker, K.; Johnson, A.W.; Dinman, J.D. Bypass of the pre-60S ribosomal quality control as a pathway to oncogenesis. Proc. Natl. Acad. Sci. USA 2014, 111, 5640–5645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipton, J.M.; Ellis, S.R. Diamond Blackfan anemia 2008–2009: Broadening the scope of ribosome biogenesis disorders. Curr. Opin. Pediatr. 2010, 22, 12–19. [Google Scholar] [CrossRef]
- Wang, R.; Yoshida, K.; Toki, T.; Sawada, T.; Uechi, T.; Okuno, Y.; Sato-Otsubo, A.; Kudo, K.; Kamimaki, I.; Kanezaki, R.; et al. Loss of function mutations in RPL27 and RPS27 identified by whole-exome sequencing in Diamond-Blackfan anaemia. Br. J. Haematol. 2015, 168, 854–864. [Google Scholar] [CrossRef] [PubMed]
- Kirwan, M.; Dokal, I. Dyskeratosis congenita: A genetic disorder of many faces. Clin. Genet. 2008, 73, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Morimoto, K.; Danilova, N.; Zhang, B.; Lin, S. Zebrafish models for dyskeratosis congenita reveal critical roles of p53 activation contributing to hematopoietic defects through RNA processing. PLoS ONE 2012, 7, e30188. [Google Scholar] [CrossRef] [PubMed]
- Pereboom, T.C.; van Weele, L.J.; Bondt, A.; MacInnes, A.W. A zebrafish model of dyskeratosis congenita reveals hematopoietic stem cell formation failure resulting from ribosomal protein-mediated p53 stabilization. Blood 2011, 118, 5458–5465. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Acc # | Gene Name | Name | Function | AKT pSites | Known Sites | AKT/p70 S6K Sub |
|---|---|---|---|---|---|---|
| P55265 | ADAR | Double-stranded RNA-specific adenosine deaminase (NB4) | Catalyzes the hydrolytic deamination of multiple adenosines to inosines in RNA. This can result in diverse effects as a consequence of RNA modification. | (5/7) | Y (1/1) | X |
| Q86V81 | ALYREF | THO complex subunit 4 | Export adapter protein; functions in the export of spliced and unspliced mRNAs from the nucleus and mRNA processing. | (2/2) | Y (2/2) | X |
| Q9UIG0 | BAZ1B | Tyrosine-protein kinase BAZ1B | Chromatin remodeling factor; involved in the promoting RNA polymerase (pol I, pol II, and pol III) activity. | (11/12) | Y (3/4) | |
| P17844 | DDX5 | Probable ATP-dependent RNA helicase DDX5 | RNA helicase; involved in alternate pre-mRNA splicing; | (1/4) | Y (1/3) | |
| Q92841 | DDX17 | Probable ATP-dependent RNA helicase DDX17 | RNA helicase; involved in RNA splicing, alternative RNA splicing, alteration of RNA secondary structure; involved in rRNA and miRNA processing; a transcriptional coactivator. | (6/7) | Y (3/3) | |
| Q9NR30 | DDX21 | Nucleolar RNA helicase 2 | RNA helicase that senses the status of RNA pol I and RNA pol II activity; binds rRNAs, snoRNAs, and mRNAs; influences RNA pol II transcription; binds dsRNA and acts as a sensor for cytoplasmic dsRNA; activates inflammatory cascade. | (3/4) | N | |
| Q08211 | DHX9 | ATP-dependent RNA helicase A | RNA-DNA helicase with role in DNA replication, RNA transcription, translation, and RNA silencing; hnRNP actin binding. Transcriptional activator; mediates MYC mRNA stability: Interacts with RELA, IGFBP1, CREB-BP. Involved in viral infection and inflammasome activation; known substrate for EIF2AK2 (PKR). | (7/9) | N | |
| O43143 | DHX15 | Pre-mRNA-splicing factor ATP-dependent RNA helicase DHX15 | RNA helicase; pre-mRNA splicing factor involved in the disassembly of the spliceosome. | (3/4) | Y (1/1) | |
| Q99848 | EBNA1BP2 | Probable rRNA-processing protein EBP2 | Required for the processing of the 27S pre-rRNA; interacts with Ebstein–Barr virus (EBV) EBNA1 protein; required for stable EBV episome segregation | (1/1) | N | |
| P68104 | EEF1A1 | Elongation factor 1-alpha 1 | Promotes the GTP-dependent binding of the aminoacetyl-tRNA to the A site of the ribosome. | (1/1) | N | |
| P05198 | EIF2S1 | Eukaryotic translation initiation factor 2 subunit 1 | Alpha subunit of the eIF2 translation initiation factor; forms the ternary complex with GTP and the initiating tRNA. GTP hydrolysis catalyzes the formation of the 80S initiation complex. | (1/1) | Y (1/1) | |
| P20042 | EIF2S2 | Eukaryotic translation initiation factor 2 subunit 2 | Beta subunit of the eIF2 translation initiation factor; forms the ternary complex with GTP and the initiating tRNA. GTP hydrolysis catalyzes the formation of the 80S initiation complex. | (2/2) | Y (1/1) | |
| O15371 | EIF3D | Eukaryotic translation initiation factor 3 subunit D | mRNA CAP-binding component of the eIF3 complex; eIF3 is responsible for the recruitment of other initiation factors to form the 43S PIC; stimulates recruitment of mRNA to the 43S PIC and codon scanning to localize the initiator AUG. | (2/2) | Y (1/1) | |
| Q99613 | EIF3C | Eukaryotic translation initiation factor 3 subunit C | Component of the eIF3 complex; eIF3 is responsible for the recruitment of other initiation factors to form the 43S PIC; stimulates recruitment of mRNA to the 43S PIC and codon scanning to localize the initiator AUG. | (1/2) | N | |
| O60841 | EIF5B | Eukaryotic translation initiation factor 5B | Translation GTPase that catalyzes the assembly of the 80S translation initiation complex. | (4/4) | Y (2/2) | |
| Q15717 | ELAVL1 | ELAV-like protein 1 | Ribonucleoprotein complex; involved in 3′ UTR AU-rich element (ARE) dependent MYC, FOS, and IL3 stabilization; binds p53 mRNA to facilitate its export from the nucleus. | (1/2) | Y (1/1) | |
| Q8IY81 | FTSJ3 | Pre-rRNA 2’-O-ribose RNA methyltransferase FTSJ3 | RNA 2’-O-methyltransferase involved in early processing of 18S rRNA and formation of the 40S ribosomal subunit; maturation of the 5.8S rRNA. | (4/4) | Y (1/1) | |
| P35637 | FUS | RNA-binding protein FUS | DNA/RNA -binding protein that influences transcription, RNA splicing, RNA transport and DNA damage repair. | (2/2) | Y (2/2) | |
| P09651 | HNRNPA1 | Heterogeneous nuclear ribonucleoprotein A1 (NB4) | Involved in pre-mRNA packaging into hnRNP; affects nuclear-cytoplasmic transport of polyA RNA; affects splicing. | (2/2) | Y (2/2) | X |
| P52597 | HNRNPF | Heterogeneous nuclear ribonucleoprotein F | A component of the hnRNP complexes; involved in pre-mRNA processing. | (3/4) | Y (3/3) | |
| Q00839 | HNRNPU | Heterogeneous nuclear ribonucleoprotein U | DNA/RNA-binding protein involved in RNA splicing, alternative splicing, and stability; influences chromatin structure and suppresses RNA pol II-dependent transcription. | (2/4) (m) | Y (2/2) (m) | |
| Q12905 | ILF2 | lnterleukin enhancer-binding factor 2 | Functions as a heterodimer with ILF3 to regulate transcription of IL-2. | (2/2) | Y (1/1) | |
| Q07666 | KHDRBS1 | KH domain-containing, RNA-binding, signal transduction-associated protein 1 | RNA-binding protein that regulates nuclear-cytoplasmic export and alternative splicing of mRNA. | (3/3) | Y (1/1) | |
| Q9NX58 | LYAR | Cell growth-regulating nucleolar protein | Acts as a transcriptional regulator; functions in the processing of 47S rRNA to 18S and 28S rRNAs; part of the 90S, 60S, and 40S RNP complexes, but not polysomes; prevents nucleolin self cleavage. | (2/2) | Y (2/2) | |
| P43243 | MATR3 | Matrin-3 | May function in the nuclear retention of defective RNAs; involved in the activation of the innate immune response. | (8/9) | Y (8/8) | |
| Q9BQG0 | MYBBP1A | Myb-binding protein 1A | DNA-binding protein that may activate or repress transcription; has a role in ribosome biogenesis. | (7/10) | Y (3/4) | |
| Q9H0A0 | NAT10 | RNA cytidine acetyltransferase | RNA cytidine acetyltransferase; modifies mRNA, 18S rRNA, and tRNA; enhances translation efficiency; may acetylate lysine in some proteins such as p53. | (4/6) | N | |
| P19338 | NCL | Nucleolin (NB4) | RNA-binding protein that influence RNA pol I and pol II transcription; plays a role in ribosome assembly | (2/2) | Y (2/2) | |
| Q15233 | NONO | Non-POU domain-containing octamer-binding protein | DNA/RNA-binding protein involved in pre-mRNA splicing; plays a role in nuclear retention of defective RNAs; involved in DNA double-strand break repair; serve a role in ILF3 phosphorylation and innate immune response activation. | (1/2) | N | |
| P46087 | NOP2 | Probable 28S rRNA (cytosine (4447)-C(5))-methyltransferase NOP2 | S-adenosyl-L-methionine-dependent methyltransferase that specifically methylates the cytosine 4447 in 28S rRNA; involved in the assembly of the 60S ribosomal subunit. | (6/7) | Y (4/5) | |
| O00567 | NOP56 | Nucleolar protein 56 | Core component of box C/D small nucleolar ribonucleoprotein (snoRNP) particles. Required for the biogenesis of box C/D snoRNAs; involved in the processing and maturation of the 60S ribosomal subunit. | (4/5) | Y (3/4) | |
| P06748 | NPM1 | Nucleophosmin (NB4) | Involved in cellular division, ribosome biogenesis, and ribosomal export; regulates p53 and p14ARF; enhances MYC transcriptional activity; involved in assembly and export of the 40S and 60S ribosomal subunits; negatively regulates EIF2AK2 (PKR). | (1/1) | Y (1/1) | |
| P09874 | PARP1 | Poly [ADP-ribose] polymerase 1 | DNA ribosyltransferase; promotes RNA pol II-dependent transcription; involved in DNA repair. | (2/2) | N | |
| Q6P2Q9 | PRPF8 | Pre-mRNA-processing-splicing factor 8 | RNA-binding protein that associates with both 5’ and 3’ splice sites to position the U2, U5, and U6 for spliceosome formation. | (5/5) | Y (2/2) | |
| Q9UMS4 | PRPF19 | Pre-mRNA-processing factor 19 | Ubiquitin protein ligase involved in pre-mRNA spliceosome assembly and DNA repair. | (3/3) | Y (1/1) | X |
| Q09028 | RBBP4 | Histone-binding protein RBBP4 | Component of the chromatin assembly factor 1 (CAF-1) complex, which is required for chromatin assembly following DNA replication and DNA repair; the core histone deacetylase (HDAC) complex, which promotes histone deacetylation and consequent transcriptional repression; the nucleosome remodeling and histone deacetylase complex (the NuRD complex), which promotes transcriptional repression by histone deacetylation and nucleosome remodeling; the PRC2/EED-EZH2 complex, which promotes repression of homeotic genes during development; and the NURF (nucleosome remodeling factor) complex. | (1/10) (m) | N (m) | |
| Q96PK6 | RBM14 | RNA-binding protein 14 | Acts a transcriptional coactivator (isoform 1) or repressor (isoform 2); aids in the activation of the innate immune response through ILF3 activation. | (2/2) | Y (2/2) | |
| Q14498 | RBM39 | RNA-binding protein 39 | Transcriptional coactivator involved in RNA processing and splicing. | (16/18) | Y (7/8) | |
| P38159 | RBMX | RNA-binding motif protein, X chromosome | RNA-binding protein that regulates pre- and post-transcriptional processes; involved in RNA pol II transcription; involved in mRNA splicing and alternative splice site selection. | (5/9) | Y (5/9) | |
| P39023 | RPL3 | 60S ribosomal protein L3 (NB4) | Component of the large ribosomal subunit; binds 5S rRNA. | (3/3) | Y (1/3) | |
| P36578 | RPL4 | 60S ribosomal protein L4 (NB4) | Structural component of the 60S ribosomal subunit. | (1/1) | Y (1/1) | |
| P62917 | RPL8 | 60S ribosomal protein L8 | Structural component of the 60S ribosomal subunit; binds rRNA. | (1/2) (m) | Y (0/1) (m) | |
| P26373 | RPL13 | 60S ribosomal protein L13 (NB4) | Structural component of the 60S ribosomal subunit. | (4/5) | Y (3/4) | |
| P40429 | RPL13A | 60S ribosomal protein L13a (NB4) | Associated with the ribosome but is not a required component; associates with 3’-UTR inflammatory mRNAs; interacts with eIF4G near the eIF3 binding site to prevent 43S ribosomal complex assembly. | (1/1) | N | |
| P84098 | RPL19 | 60S ribosomal protein L19 | Structural component of the 60S ribosomal subunit; 5.8S rRNA binding. | (2/2) | Y (2/2) | |
| P23396 | RPS3 | 40S ribosomal protein S3 (NB4) | Structural component of the 40S ribosomal subunit; has endonuclease activity; involved in DNA damage repair; interacts with MDM2 resulting in p53 stability. | (1/1) | Y (1/1) | X |
| P62753 | RPS6 | 40S ribosomal protein S6 | Structural component of the 40S ribosomal subunit; involved in rRNA processing; involved in the selective translation of a certain class of mRNAs. | (1/1) | Y (1/1) | |
| P62081 | RPS7 | 40S ribosomal protein S7 | Structural component of the 40S ribosomal subunit; involved in rRNA processing/maturation; binds 3’-UTR and 5’-UTR of mRNA; involved in translation initiation. | (2/2) | Y (2/2) | |
| P62241 | RPS8 | 40S ribosomal protein S8 (NB4) | Structural component of the 40S ribosomal subunit; involved in the maturation of the 18S rRNA. | (1/1) | N | |
| Q14684 | RRP1B | Ribosomal RNA processing protein 1 homolog B | Acts as a transcriptional coactivator; involved in mRNA splicing; promotes RNA pol II transcription; involved in rRNA processing. | (3/5) | Y (1/1) | |
| O76021 | RSL1D1 | Ribosomal L1 domain-containing protein 1 | Involved in large subunit rRNA processing/maturation; inhibits PTEN translation. | (4/8) (m) | Y (2/4) (m) | |
| Q9Y265 | RUVBL1 | RuvB-like 1 | ATP-dependent DNA helicase; component of the NuA4 histone acetyltransferase complex; binds to the TF-IID transcription complex; involved in H2A and H4 acetylation and RNA pol II transcriptional activation; involved in C/D snoRNP assembly; has a role in DNA repair; required for MYC oncogenesis. | (3/6) (m) | N (m) | |
| Q9Y230 | RUVBL2 | RuvB-like 2 | ATP-dependent DNA helicase; component of the NuA4 histone acetyltransferase complex; binds to the TF-IID transcription complex; involved in H2A and H4 acetylation and RNA pol II transcriptional activation; involved in C/D snoRNP assembly; has a role in DNA repair; binds β-catenin; required for MYC oncogenesis; suppresses expression of ATF2 and endoplasmic reticulum stress response genes. | (4/4) | Y (2/2) | |
| Q13435 | SF3B2 | Splicing factor 3B subunit 2 | Part of the SF3B complex; involved in pre-mRNA splicing. | (4/4) | Y (1/1) | |
| Q15393 | SF3B3 | Splicing factor 3B subunit 3 (NB4) | Part of the SF3B complex; involved in pre-mRNA splicing. | (2/2) | N | |
| P23246 | SFPQ | Splicing factor, proline- and glutamine-rich (NB4) | DNA/RNA-binding protein; essential for spliceosome complex formation; enhances RNA pol II transcription; involved in alternative splicing. | (4/4) | Y (3/3) | |
| O60264 | SMARCA5 | SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily A member 5 | DNA-binding helicase; represses rDNA transcription. | (4/4) | N | |
| Q7KZF4 | SND1 | Staphylococcal nuclease domain-containing protein 1 (NB4) | Transcriptional coactivator of STAT5 and STAT6; mediates miRNA decay. | (2/3) | Y (1/1) | |
| P08579 | SNRPB2 | U2 small nuclear ribonucleoprotein B | Associated with the U2 snRNP involved in pre-mRNA splicing. | (1/1) (m) | Y (1/1) (m) | |
| Q07955 | SRSF1 | Serine/arginine-rich splicing factor 1 | Involved in regulating the accuracy of splicing and alternative splicing by preventing exon skipping; associates with U1 snRNP and U2AF; involved with mRNA nuclear-cytoplasmic export. | (13/14) | Y (13/14) | |
| Q01130 | SRSF2 | Serine/arginine-rich splicing factor 2 | Required for pre-mRNA splicing; facilitates U1 and U2 snRNP association with pre-mRNA; links 5’ and 3’ splice site components U1 snRNP and U2AF, respectively; regulates alternative splicing; facilitates mRNA export from the nucleus; acts as a transcriptional corepressor. | (27/27) | Y (12/12) | |
| P12270 | TPR | Nucleoprotein TPR | Component of the nuclear pore; involved in protein and RNA export/import. | (9/9) | Y (2/2) | |
| P08670 | VIM | Vimentin | Involved with LARP6 to stabilize certain mRNAs. | (1/3) | Y (1/3) | X |
| O95218 | ZRANB2 | Zinc finger Ran-binding domain-containing protein 2 | Involved in alternative splicing by modifying 5’-splice site selection. | (29/33) | Y (10/10) |
| Acc # | Gene Name | Description | Function | Involvement in Disease |
|---|---|---|---|---|
| P55265 | ADAR | Double-stranded RNA-specific adenosine deaminase (1) | Catalyzes the hydrolytic deamination of multiple adenosines to inosines in RNA. This can result in diverse effects as a consequence of RNA modification. | Dyschromatosis hereditaria, Aicardi-Goutieres syndrome 6 |
| Q8WYP5 | AHCTF1 | Protein ELYS | Required for the assembly of a functional nuclear pore complex (NPC). The NPC is required for nuclear-cytoplasmic transport of RNA species and ribonucleoproteins (RNP) complexes and vice versa for the transport of ribosomal proteins (RPs); has effects on RNA pol II activity. | |
| Q13838 | BAT1 | Spliceosome RNA helicase DDX39B | Component of the THO subcomplex of the TREX complex that specifically associates with spliced mRNA; has a role in the nuclear export of unspliced mRNAs. Weak RNA helicase activity that catalyzes the first step in spliceosome assembly for the subsequent binding of the U2 snRNP. | |
| Q14692 | BMS1 | Ribosome biogenesis protein BMS1 homolog | Maturation of the 40S ribosomal subunit in the nucleolus; binds U3 snoRNA and may be required for the maturation of rRNA. | Aplasia cutis congenita (ACC) |
| Q9Y3Y2 | C1orf77 | Chromatin target of PRMT1 protein | Associates with the methylsome complex to induce gene transcription; is a component of the TREX complex; associates upstream of the exon junction complex (EJC) on spliced mRNAs and facilitates their nuclear export. | |
| Q9BRJ6 | C7orf50 | Uncharacterized protein C7orf50 | Not reported. Has RNA binding ability; associates with multiple proteins involved in processing of 27S and 18S rRNAs. | |
| O14646 | CHD1 | Chromodomain-helicase-DNA-binding protein 1 | Involved in chromatin remodeling; substrate recognition component of the transcription regulatory histone acetylation (HAT) complex SAGA; regulates RNA polymerase I and II transcription. Associated with diverse mRNA splicing complexes (FACT, PAF and U2 snRNP); blocks DNA replication. | Pilarowski-Bjornsson syndrome (PILBOS) |
| P38432 | COIL | Coilin | Major component of Cajal Bodies; involved in the function or assembly/disassembly of nucleoplasmic snRNPs. | |
| Q92499 | DDX1 | ATP-dependent RNA helicase DDX1 | RNA helicase with activity toward RNA-RNA and RNA-DNA helices. Binds poly A mRNA and may be involved in the processing and polyadenylation of the 3’-end of mRNA; involved in tRNA splicing. Acts as a sensor of dsRNA and is involved in the induction of inflammatory cytokines. | |
| Q8TDD1 | DDX54 | ATP-dependent RNA helicase DDX54 | Represses the transcriptional activity of nuclear receptors. Involved in RNA processing. | |
| Q08211 | DHX9 | ATP-dependent RNA helicase A (1) | RNA-DNA helicase with role in DNA replication, RNA transcription, translation, and RNA silencing; hnRNP actin binding. Transcriptional activator; mediates MYC mRNA stability: Interacts with RELA, IGFBP1, CREB-BP. Involved in viral infection and inflammasome activation; known substrate for EIF2AK2 (PKR). | |
| Q8IY37 | DHX37 | Probable ATP-dependent RNA helicase DHX37 | Has a role in rRNA processing. | |
| O60832 | DKC1 | H/ACA ribonucleoprotein complex subunit 4 | Required for ribosome biogenesis and telomere maintenance; promotes cell to cell and cell to substratum adhesion, increases the cell proliferation rate; catalytic unit of the H/ACA snoRNP complex which is required for pseudouridylation of rRNA; required for correct processing/trafficking of TERC | X-linked Dyskeratosis Congenita (XDKC), Hoyeraal-Hreidarsson syndrome (HHS) |
| Q99848 | EBNA1BP2 | Probable rRNA-processing protein EBP2 (1) | Required for the processing of the 27S pre-rRNA; interacts with Ebstein-Barr virus (EBV) EBNA1 protein; required for stable EBV episome segregation | |
| P19525 | EIF2AK2 | Interferon-induced, dsRNA-activated protein kinase | dsRNA-binding kinase activated in response to diverse stresses; phosphorylates eIF2α leading to inhibition of general translation; activation may favor IRES-mediated translation; phosphorylates p53 to stabilize it; alters MYC isoform expression. | Elevated constitutive activity associated with diverse diseases. |
| P38919 | EIF4A3 | Eukaryotic initiation factor 4A-III | ATP-dependent RNA helicase; component of a splicing-dependent multiprotein exon junction complex (EJC) deposited at splice junction on mRNAs; affects nuclear-cytoplasmic transport of mRNAs; enhances translation of spliced mRNA. | Richieri-Costa-Pereira syndrome (RCPS) |
| P56537 | EIF6 | Eukaryotic translation initiation factor 6 (1) | Binds to the 60S ribosomal subunit and prevents its premature association with the 40S ribosomal subunit to form the 80S initiation complex in the cytoplasm; affects 60S ribosomal subunit export from the nucleus; enhances the translation of certain transcription factor mRNAs (CEBP, ATF4); affects miRNA silencing of mRNAs; controls the expression mitochondrial respiratory chain genes. | High expression in colon carcinoma |
| Q15717 | ELAVL1 | ELAV-like protein 1 | Ribonucleoprotein complex; involved in 3′ UTR AU-rich element (ARE) dependent MYC, FOS, and IL3 stabilization; binds p53 mRNA to facilitate its export from the nucleus. | |
| Q9BVP2 | GNL3 | Guanine nucleotide-binding protein-like 3 (1) | Stabilizes MDM2 by preventing its ubiquitination, and proteasomal degradation. | |
| O60812 | HNRNPCL1 | Heterogeneous nuclear ribonucleoprotein C-like 1 (1) | Ribonucleosome component affecting hnRNPs. | |
| Q14103 | HNRNPD | Heterogeneous nuclear ribonucleoprotein D0 | Component of the ribonucleosomes; binds 3′ AU-rich elements (AREs) of mRNA to destabilize transcripts, binds ssDNA, and can act as a transcription factor; involved in coupled mRNA translation and turn-over. | |
| P52272 | HNRNPM | Heterogeneous nuclear ribonucleoprotein M | Pre-mRNA binding protein; part of the spliceosome C complex; binds poly (G) and poly (U) stretches; may affect signaling events leading to TNFα, IL-1α, IL6, and IL10. | |
| P07910 | HNRPC | Heterogeneous nuclear ribonucleoproteins C1/C2 (1) | Mediates 40S hnRNP particles assembly; binds 5’ and 3’ poly (U) tracks of mRNA affecting their stability and translation; may play a role in spliceosome assembly and influence splicing of mRNAs through early association with pre-mRNA. | |
| O14979 | HNRPDL | Heterogeneous nuclear ribonucleoprotein D-like | Transcriptional regulator of DNA; promotes transcriptional activation in differentiated myotubes; binds 3′ UTR AU-rich elements (AREs) in mRNAs; RNA processing. | Muscular dystrophy, limb-girdle, autosomal dominant 3 (LGMDD3) |
| Q12906 | ILF3 | Interleukin enhancer-binding factor 3 | Involved in biogenesis of circular RNAs from back splicing by binding regulatory elements flanking introns; binds AU-rich element of target RNAs; participates in diverse transcriptional and post-transcriptional event; is an EIF2AK2 (PKR) substrate; phosphorylation results in ILF3 release of circular RNAs. | |
| P52292 | KPNA2 | Importin subunit alpha-2 | Functions in nuclear protein import as an adapter protein for nuclear receptor KPNB1; Ran-dependent. | |
| O00629 | KPNA4 | Importin subunit alpha-4 | Functions in nuclear protein import as an adapter protein for nuclear receptor KPNB1; Ran-dependent. | |
| Q13601 | KRR1 | KRR1 small subunit processome component homolog | Involved in nucleolar processing of pre-18S ribosomal RNA and 40S ribosome biogenesis. | |
| Q6PKG0 | LARP1 | La-related protein 1 (1) | Regulates the translation of specific mRNAs downstream of mTORC1 signaling; when unphosphorylated associates with 5’ UTRs of 5’TOP mRNAs blocking translation by inhibiting eIF4G binding; phoshorylation by mTORC1 results in dissociation from 5’TOP mRNAs favoring their translation; under favorable growth conditions, association with 3’UTR of most mRNAs favors their translation. | |
| Q9NX58 | LYAR | Cell growth-regulating nucleolar protein (1) | Acts as a transcriptional regulator; functions in the processing of 47S rRNA to 18S and 28S rRNAs; part of the 90S, 60S, and 40S RNP complexes, but not polysomes; prevents nucleolin self cleavage. | |
| O95251 | MYST2 | Histone acetyltransferase KAT7 | HBO1 (HAT) complex which has H4-specific acetyltransferase activity, reduced activity toward H3; positive regulator of RNA pol II; promotes p53 transcription. | |
| Q8NEJ9 | NGND | Neuroguidin | Translational repression of cytoplasmic polyA element containing transcripts; involved in the maturation of 40S subunits rRNA. | |
| Q8WTT2 | NOC3L | Nucleolar complex protein 3 homolog (1) | Specifically influences RNA pol II transcriptional activity. | |
| Q9UGY1 | NOL12 | Nucleolar protein 12 | Binds 28S rRNA; involved in rRNA processing; stabilizes the nucleus; inhibits apoptosis. | |
| P46087 | NOP2 | Putative ribosomal RNA methyltransferase NOP2 (1) | S-adenosyl-L-methionine dependent methyltransferase that catalyzes the methylation of cytosine 4447 in 28S rRNA; affects 60S subunit assembly; regulates RNA pol II-mediated transcription; associated with cell proliferation. | |
| P06748 | NPM1 | Nucleophosmin (1) | Involved in cellular division, ribosome biogenesis and ribosomal export; regulates p53 and p14ARF; enhances MYC transcriptional activity; involved in assembly and export of the 40S and 60S ribosomal subunits; negatively regulates EIF2AK2 (PKR). | Myelodysplastic syndromes (MDS) Leukemia, non-Hodgkin’s lymphoma |
| Q9UQ80 | PA2G4 | Proliferation-associated protein 2G4 | Inhibits transcription of some E2F1-regulated promoters by sequestering the HAT complex; associates with 28S 18S and 5.8S rRNAs and U3 snRNAs; involved in the intermediate and late stages of rRNA maturation; mediates cap-independent translation of specific viral IRES containing mRNAs. | |
| Q9BY77 | POLDIP3 | Polymerase delta-interacting protein 3 | Positive regulation of translation; recruits p70 S6 kinase to the ribosome; involved in mRNA export; associates with spliced RNA-protein complexes favoring translation of spliced mRNAs. | |
| P62136 | PPP1CA | Serine/threonine-protein phosphatase PP1-alpha catalytic subunit | Protein phosphatase 1 (PP1) is essential for cell division, and participates in the regulation of glycogen metabolism, muscle contractility, and protein synthesis, cell migration; dephosphorylates a variety of substrates including eIF2α. | |
| P63244 | RACK1 | Receptor of activated protein kinase C1 | Scaffolding protein; binds to and stabilizes activated protein kinase C (PKC), increasing PKC-mediated phosphorylation of EIF6 causing its dissociation from the 60S ribosomal subunit; inhibits Src kinases, prolongs G1/G0, inhibits Wnt signaling, promotes BAX oligomerization; binds HIV NEF1. | Elevated expression in hepatocellular carcinoma |
| Q09028 | RBBP4 | Histone-binding protein RBBP4 | Component of the chromatin assembly factor 1 (CAF-1) complex, which is required for chromatin assembly following DNA replication and DNA repair; the core histone deacetylase (HDAC) complex, which promotes histone deacetylation and consequent transcriptional repression; the nucleosome remodeling and histone deacetylase complex (the NuRD complex), which promotes transcriptional repression by histone deacetylation and nucleosome remodeling; the PRC2/EED-EZH2 complex, which promotes repression of homeotic genes during development; and the NURF (nucleosome remodeling factor) complex. | |
| P39023 | RPL3 | 60S ribosomal protein L3 | Component of the large ribosomal subunit; binds 5S rRNA. | |
| P27635 | RPL10 | 60S ribosomal protein L10 | Component of the 60S ribosomal subunit; may have an active role in translation initiation; has a role in the negative regulation of RNA pol II. | Autism, X-linked 5 (AUTSX5) |
| Q96L21 | RPL10L | 60S ribosomal protein L10-like (1) | Component of the 60S ribosomal subunit; may play a role in compensating for the inactivated X-linked gene during spermatogenesis. | |
| P35268 | RPL22 | 60S ribosomal protein L22 | Component of the 60S ribosomal subunit; may have a role in translation initiation; binds Ebstein–Barr virus (EBV) EBER transcripts and heparin. | |
| P62829 | RPL23 | 60S ribosomal protein L23 | Component of the 60S ribosomal subunit; associates with rRNA; negatively regulates RNA pol II transcription; negative regulation of ubiquitin protein ligase activity; | |
| P61254 | RPL26 | 60S ribosomal protein L26 | Component of the 60S ribosomal subunit; involved in rRNA processing; involved in translation initiation; involved in DNA damage response favoring p53-dependent transcription; associates with the 5’ UTR of mRNAs. | Diamond–Blackfan anemia, type 11 |
| P46776 | RPL27A | 60S ribosomal protein L27a | Component of the 60S ribosomal subunit; involved in translation initiation; binds RNA. | |
| P62910 | RPL32 | 60S ribosomal protein L32 | Component of the 60S ribosomal subunit; involved in translation initiation; binds RNA. | |
| P49207 | RPL34 | 60S ribosomal protein L34 | Component of the 60S ribosomal subunit; involved in translation initiation; binds RNA; binds cadherin. | |
| P42766 | RPL35 | 60S ribosomal protein L35 | Component of the 60S ribosomal subunit; involved in the maturation of the 60 S subunit rRNA; involved in translation initiation; binds mRNA. | |
| Q9Y3U8 | RPL36 | 60S ribosomal protein L36 | Component of the 60S ribosomal subunit; involved in translation initiation; binds RNA. | |
| P46783 | RPS10 | 40S ribosomal protein S10 | Component of the 40S ribosomal subunit; involved in translation initiation; binds RNA. | Diamond–Blackfan anemia type 9 |
| Q9NQ39 | RPS10L | Putative 40S ribosomal protein S10-like | Component of the 40S ribosomal subunit; localized to the cytosol only; may result from a pseudogene. | |
| P39019 | RPS19 | 40S ribosomal protein S19 | Component of the 40S ribosomal subunit; required for pre-rRNA processing and maturation of 40S ribosomal subunits; involved in translation initiation; binds RNA; protein kinases and fibroblast growth factor (FGF); involved in NOTCH signaling. | Diamond–Blackfan anemia, type 1; highly expressed in colon carcinoma. |
| P62854 | RPS26 | 40S ribosomal protein S26 | Component of the 40S ribosomal subunit; involved in translation initiation; binds RNA and mRNA; binds cadherin; negatively regulates pre-mRNA splicing; | Diamond–Blackfan anemia, type 10 |
| P56182 | RRP1 | Ribosomal RNA processing protein 1 homolog A | RNA binding protein critical to the generation of 28S rRNA. | |
| Q9Y3B9 | RRP15 | RRP15-like protein | Involved in rRNA processing. | |
| Q96EU6 | RRP36 | Ribosomal RNA processing protein 36 homolog | Involved in the early processing steps of the pre-rRNA in maturation pathway leading to the 18S rRNA; involved in the cleavage to liberate 18S rRNA. | |
| Q9UHA3 | RSL24D1 | Probable ribosome biogenesis protein RLP24 | Involved in the biogenesis of the 60S ribosomal subunit; insures NOG1 docking to 60S ribosomal subunit; structural component of the ribosome; involved in translation. | |
| Q9Y265 | RUVBL1 | RuvB-like 1 | ATP-dependent DNA helicase; component of the NuA4 histone acetyltransferase complex; binds to the TF-IID transcription complex; involved in H2A and H4 acetylation and RNA pol II transcriptional activation; involved in C/D snoRNP assembly; has a role in DNA repair; required for MYC oncogenesis. | |
| Q9Y230 | RUVBL2 | RuvB-like 2 | ATP-dependent DNA helicase; component of the NuA4 histone acetyltransferase complex; binds to the TF-IID transcription complex; involved in H2A and H4 acetylation and RNA pol II transcriptional activation; involved in C/D snoRNP assembly; has a role in DNA repair; binds β-catenin; required for MYC oncogenesis; suppresses expression of ATF2 and endoplasmic reticulum stress response genes. | |
| P28370 | SMARCA1 | Probable global transcription activator SNF2L1 (1) | Chromatin remodeling DNA-binding protein; positively regulates RNA pol II transcription. | |
| P51532 | SMARCA4 | Transcription activator BRG1 | Chromatin remodeling DNA-binding protein; associates with p53; associates with long non-coding RNAs; acts as a transcriptional coactivator or corepressor; positive regulation of RNA pol II pre-miRNA transcription. | Rhab doid tumors and Coffin-Siris syndrome 4 (CSS4) |
| O60264 | SMARCA5 | SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily A member 5 | Chromatin remodeling DNA-binding protein; represses RNA pol-dependent transcription of rDNA; regulates chromatin silencing by recruiting DNA methyltransferases to stretches of rDNA. | |
| Q92922 | SMARCC1 | SWI/SNF complex subunit SMARCC1 | Chromatin remodeling DNA-binding protein; component of the SWI/SNF chromatin remodeling complex; acts as a positive regulator of RNA pol II-dependent transcription; may repress certain genes. | |
| Q8TAQ2 | SMARCC2 | SWI/SNF complex subunit SMARCC2 | Chromatin remodeling DNA-binding protein; component of the SWI/SNF chromatin remodeling complex; acts as a positive regulator of RNA pol II-dependent transcription; may repress certain genes. | |
| P09661 | SNRPA1 | U2 small nuclear ribonucleoprotein A’ | Part of the spliceosome complex involved in pre-mRNA splicing; part of the U2 snRNP; associates with U2 snRNA. | |
| P62316 | SNRPD2 | Small nuclear ribonucleoprotein Sm D2 | Core component of the SMN-Sm complex that mediates snRNP assembly, which occurs in the cytoplasm; facilitates nuclear import of snRNPs; is a component of the U1, U2, U4, and U5 snRNPs; binds RNA; required for pre-mRNA splicing; auto-antibodies found in lupus. | |
| P62318 | SNRPD3 | Small nuclear ribonucleoprotein Sm D3 | Core component of the SMN-Sm complex that mediates snRNP assembly, which occurs in the cytoplasm; facilitates nuclear import of snRNPs; is a component of the U1, U2, U4, and U5 snRNPs; binds RNA; required for pre-mRNA splicing; binds to the downstream cleavage product (DCP) of histone pre-mRNA in a U7 snRNP dependent manner; auto-antibodies found in lupus. | |
| P18583 | SON | Protein SON | mRNA splicing cofactor; enhances splicing of mRNAs with weak splice sites, including many cell cycle and DNA repair genes, through the interaction with SRSF2 and RNA pol II. | ZTTK syndrome |
| O15042 | SR140 | U2 snRNP-associated SURP motif-containing protein | RNA binding; involved in mRNA splicing. | |
| Q8IYB3 | SRRM1 | Serine/arginine repetitive matrix protein 1 | Part of pre- and post-splicing multiprotein mRNP complexes; links sequence-specific splicing factors with snRNP factors of the splicesome; involved in mRNA nuclear export. | |
| Q01130 | SRSF2 | Serine/arginine-rich splicing factor 2 | Required for pre-mRNA splicing; facilitates U1 and U2 snRNP association with pre-mRNA; links 5’ and 3’ splice site components U1 snRNP and U2AF, respectively; regulates alternative splicing; facilitates mRNA export from the nucleus; acts as a transcriptional corepressor. | |
| Q13247 | SRSF6 | Serine/arginine-rich splicing factor 6 | Plays a role in constitutive splicing and can modulate the selection of alternative splice sites; involved in mRNA export from the nucleus. | |
| Q08945 | SSRP1 | FACT complex subunit SSRP1 | DNA-binding component of the FACT complex; involved in RNA pol II mRNA elongation, DNA replication, and DNA repair; may influence p53 stability; auto-antibodies found in lupus. | |
| P53999 | SUB1 | Activated RNA polymerase II transcriptional coactivator p15 | RNA and DNA binding protein that acts as a coactivator of TAF complex-mediated RNA polymerase II transcription. | |
| O75683 | SURF6 | Surfeit locus protein 6 | RNA and DNA binding protein likely involved in ribosome biogenesis and assembly. | |
| O60506 | SYNCRIP | Heterogeneous nuclear ribonucleoprotein Q | An hnRNP implicated mRNA processing; three isoforms function diversely; involved in splicing and mRNA turnover; inhibits translation; promotes MYC mRNA stability; part of APOB editsome; may facilitate cytoplasmic vesicle transport of RNAs. | |
| Q92804 | TAF15 | TATA-binding protein-associated factor 2N | An RNA and ssDNA binding protein with RNA pol II-mediated transcription promoting characteristics. | Extra skeletal myxoid chondrosarcoma |
| Q8NI27 | THOC2 | THO complex subunit 2 | A component of the THO complex that is a sub-component of the TREX complex; associates with spliced and polyA mRNA to export then from the nucleus; required for the release of mRNA from nuclear speckles. | Mental retardation, X-linked 12 (MRX12) |
| P62995 | TRA2B | Transformer-2 protein homolog beta | Sequence-specific RNA-binding protein that participates in the control of pre-mRNA splicing; can promote or inhibit exon inclusion. | |
| Q01081 | U2AF1 | Splicing factor U2AF 35 kDa subunit | Constitutive and enhancer-dependent splicing by mediating protein–protein interactions and protein–RNA interactions required for accurate 3’-splice site selection; facilitates mRNA nuclear export. | Myelodysplastic syndromes (MDS) |
| P26368 | U2AF2 | Splicing factor U2AF 65 kDa subunit | Has a role in splicing of and 3’ processing of pre-mRNA; required for mRNA export; may link the processes of transcription termination, polyadenylation and export. | |
| Q9NQZ2 | UTP3 | Something about silencing protein 10 | Role in the structure of silenced chromatin; has a role in 40S subunit rRNA processing. | |
| Q15061 | WDR43 | WD repeat-containing protein 43 | Ribosome biogenesis factor; required for RNA pol I-mediated transcription; involved in pre-18S rRNA processing. | |
| P67809 | YBX1 | Nuclease-sensitive element-binding protein 1 | pre-mRNA alternative splicing regulation; stabilizes cytoplasmic mRNAs; promotes the interaction of mRNA with translation initiation factors; acts a transcription factor influencing the expression of numerous genes through RNA pol II-specific means; possesses endonuclease activity; promotes MYC mRNA stability. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piazzi, M.; Bavelloni, A.; Gallo, A.; Faenza, I.; Blalock, W.L. Signal Transduction in Ribosome Biogenesis: A Recipe to Avoid Disaster. Int. J. Mol. Sci. 2019, 20, 2718. https://doi.org/10.3390/ijms20112718
Piazzi M, Bavelloni A, Gallo A, Faenza I, Blalock WL. Signal Transduction in Ribosome Biogenesis: A Recipe to Avoid Disaster. International Journal of Molecular Sciences. 2019; 20(11):2718. https://doi.org/10.3390/ijms20112718
Chicago/Turabian StylePiazzi, Manuela, Alberto Bavelloni, Angela Gallo, Irene Faenza, and William L. Blalock. 2019. "Signal Transduction in Ribosome Biogenesis: A Recipe to Avoid Disaster" International Journal of Molecular Sciences 20, no. 11: 2718. https://doi.org/10.3390/ijms20112718
APA StylePiazzi, M., Bavelloni, A., Gallo, A., Faenza, I., & Blalock, W. L. (2019). Signal Transduction in Ribosome Biogenesis: A Recipe to Avoid Disaster. International Journal of Molecular Sciences, 20(11), 2718. https://doi.org/10.3390/ijms20112718