1. Introduction
The biogenesis of eukaryotic ribosomes is a massive undertaking involving binding of ribosomal proteins (r-proteins) to rRNA and processing of precursor rRNA along pathways that are facilitated by over 250 ribosomal assembly factors [
1,
2]. The process begins in the nucleolus with RNA polymerase I transcribing a long precursor rRNA (“35S” in
Saccharomyces cerevisiae (yeast) and “45–47S” in mammalian cells) that includes sequences for three of the four mature rRNAs (18S rRNA, 5.8S, and 25S) in addition to “Transcribed Spacers” (
Figure 1A) [
3]. The External Transcribed Spacers (5′ and 3′ ETS) make up the 5′ and 3′ parts of the primary transcript, while the Internal Transcribed Spacers (ITS1 and ITS2) are interstitial between the 18S, 5.8S, and 25S rRNA parts. As it is transcribed, the precursor transcript associates with a large number of ribosomal assembly factors to form the “90S” precursor particle, which is subsequently split into subunit-specific entities by cutting the precursor rRNA at the A2 site in ITS1 (
Figure 1A). In rapidly growing yeast cells 70%–80% of nascent precursor rRNA molecules are cleaved while transcription of the rRNAs for the large ribosomal subunit is still ongoing [
4,
5,
6]. Subsequent to the ITS1 cleavage, the 40S and 60S are completed along separate multiple-step pathways [
1,
2]. Ribosomal proteins (r-proteins) are made in the cytoplasm concurrently with the transcription and processing of the rRNA and then chaperoned to the nucleolus or nucleus where they associate with the nascent ribosomal subunits in a hierarchical fashion [
7,
8,
9].
Over the last couple of decades, the ribosome assembly process has been elucidated in very significant detail [
1,
2,
10,
11,
12]. Even though rRNAs for both subunits are co-transcribed, the complexity of the ribosome construction has necessitated separate exploration of the assembly of the two subunits. Incomplete ribosomal precursor particles for each of the subunits have been purified and analyzed for structure and content of rRNA and assembly factors. However, this approach does not clarify the relative timing of association of r-proteins with the ribosomal precursor particles, and cleavage of the primary rRNA transcript in the physiological situation where the assembly of the two subunits occurs simultaneously. Of special interest is the question of whether 60S and 40S proteins are co-assembled before the long rRNA precursor is cleaved and the ribosomal assembly is separated into subunit-specific pathways. Furthermore, determining the content of r-proteins in the precursor subunits is complicated by the fact that the number of mature ribosomes vastly outnumbers assembly intermediates, which may result in transfer of r-proteins from mature ribosomes to assembly intermediates during cell lysis and purification of nascent ribosomes [
12].
Challenged by these questions, we developed an approach to determine the relative timing of addition of r-proteins to, and the cleavage of, the pre-rRNA within ITS1 (
Figure 1A). We focused on the 40S proteins uS4/S9 and eS7/S7, as well as the 60S proteins uL4/L4 and eL13/L13 which are all known to be incorporated early in the assembly before their respective ribosomal subunits migrate from the nucleolus to the nucleus [
7]. Hemagglutinin (HA)-tagged versions of the r-proteins were expressed for a short time and precursor particles isolated using anti-HA. Identification of r-proteins and transcribed spacers in the immuno-precipitate showed that eS7 and uL4 associate with precursor ribosomes prior to the separation of 40S and 60S specific segments, while uS4/S9, uL22, and eL13 bind after, or simultaneously with, ITS1 cleavage
3. Discussion
Electron micrographs of yeast rRNA genes associated with RNA transcripts show that rRNA for both the small and large subunits is folded during transcription [
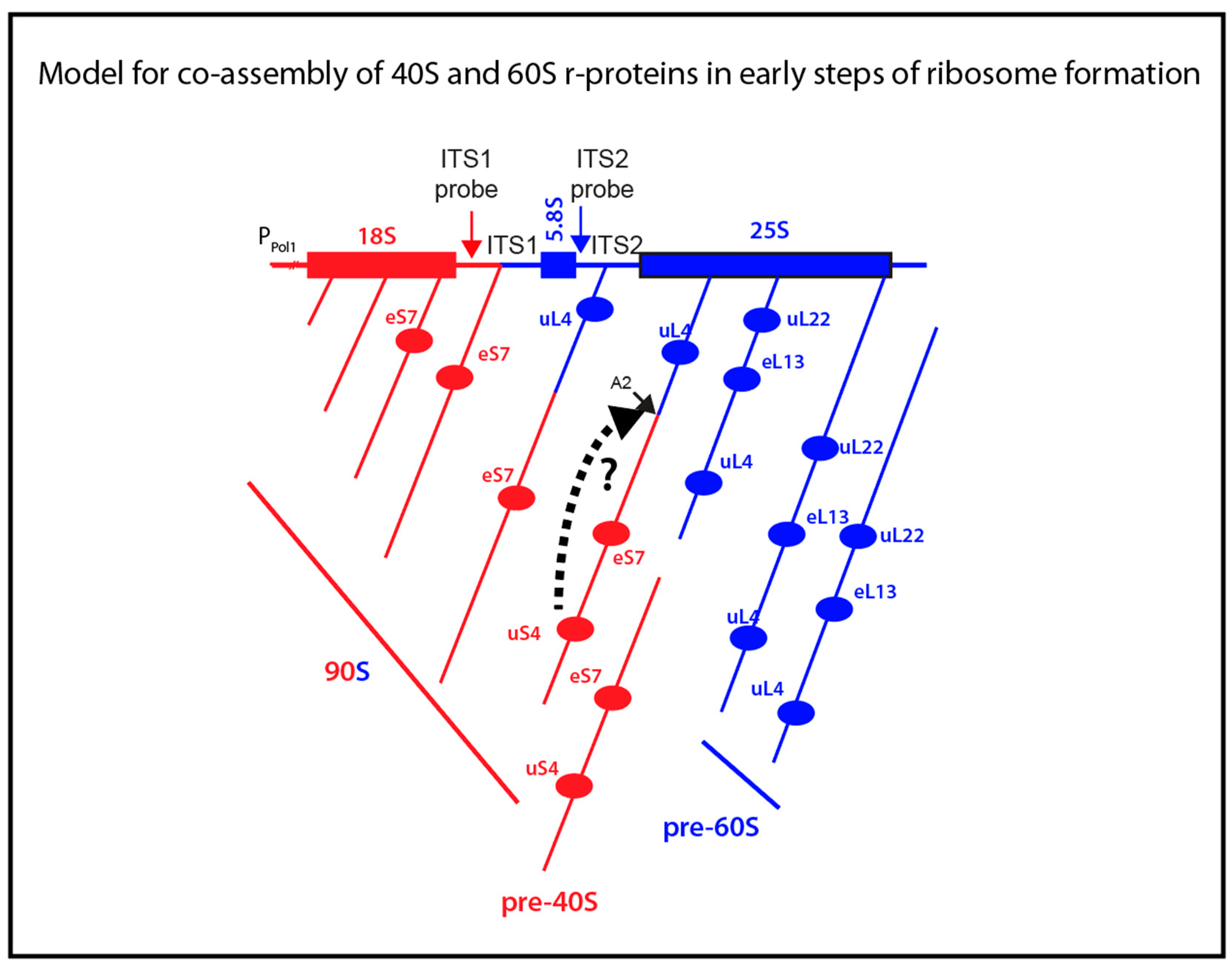
6]. During rapid growth of yeast cells 70%–80% of the transcripts are cleaved at the A2 site in ITS1 (
Figure 1A) into 40S- and 60S-specific segments when RNA I polymerase has transcribed 25% of the 60S-specific segment [
5,
6] (
Figure 6). This raises a central question: Does the assembly of 60S ribosomal proteins begin before or after the rRNA moieties for the two subunits have been separated? The answer to this question is important, because initial co-assembly of 40S and 60S r-proteins raises the possibility that incorporation of proteins from one subunit could affect the assembly pathway and kinetics of the other subunit. Our data suggest that at least a single 60S r-protein, uL4, binds to the nascent rRNA molecule before A2 cleavage separates the 40S and 60S rRNA moieties. The central results justifying this conclusion are that uL4 is associated with precursor particle(s) that also contain a 40S protein (eS7) and both ITS1 and ITS2.
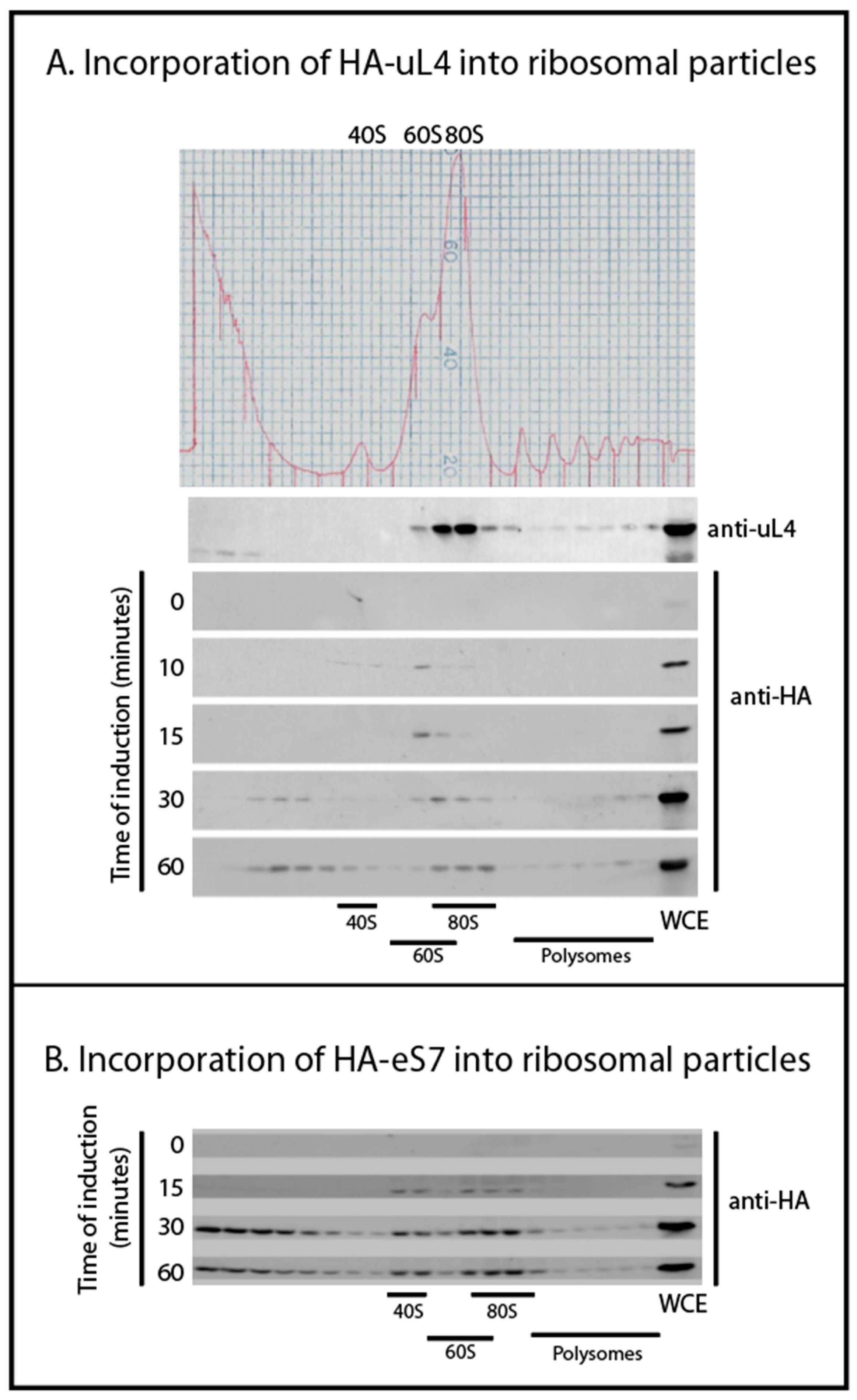
Our experimental strategy was to characterize ribosomal precursor containing specific r-proteins. This was accomplished by inducing the synthesis of specific HA-tagged r-proteins for a short time followed by immune purification on anti-HA coated beads. We observed the incorporation of the HA-tagged protein into ribosomes using a sucrose gradient. As expected, the tagged r-proteins initially entered precursor particles and were not seen in mature subunits competent to combine into 80S ribosomes and polysomes until about 30 min after induction (
Figure 2). Considering that it takes 5–10 min after addition of β-estradiol before accumulation of the tagged proteins raises over the basal level (
Figure 1C), this is in good agreement with the fact that it takes 10–20 min to process primary rRNA transcript into mature 18S and 25S rRNA molecules (see e.g., [
17,
18]). Together these results suggest that the tagged r-proteins go through a normal ribosomal assembly process and, furthermore, that the majority of the tagged proteins initially are in precursor particles, which facilitates purifying precursor ribosomes according to their content of a particular protein. By 15–30 min of induction some tagged protein was also found in complexes sedimenting close to the top of the gradient. We suggest that tagged r-proteins synthesized in excess over other ribosomal form artificial complexes, e.g. because unstructured regions in the free proteins could cause a propensity for aggregation. Alternatively, the slowly sedimenting complexes of r-proteins with their chaperones might accumulate, because the chaperone cannot deliver their freight to ribosomal assembly complexes.
To investigate if some 60S r-proteins are incorporated into the emerging ribosomal assembly intermediates prior to the separation of the precursors for 40S and 60S subunits, we first searched for ribosomal precursor particles containing both 40S and 60S proteins. Particles purified on anti-HA coated beads after induction of HA-eS7 synthesis were found to also contain uL4 (
Figure 3B), but only trace amounts of uL22, even though uL22 was efficiently co-precipitated with HA-uL4 in the control experiment (
Figure 3A). Inspection of structure models of 90S precursor ribosomes and mature 80S suggests that the failure to precipitate 80S containing HA-eS7 on anti-HA beads is due to obstruction of the access to the N-terminal end of HA-eS7 in 80S ribosomes, but not in the 90S precursor ribosomes (
Figure 4). Furthermore, the N-terminal end of uL4 is ready accessed 80S ribosomes explaining the pull down of uL22 after induction of HA-uL4 synthesis. We therefore interpret the results from co-precipitation of r-proteins to indicate that eS7 and uL4 both bind to the nascent rRNA before it is cleaved in ITS1, while uL22 binds after the cleavage.
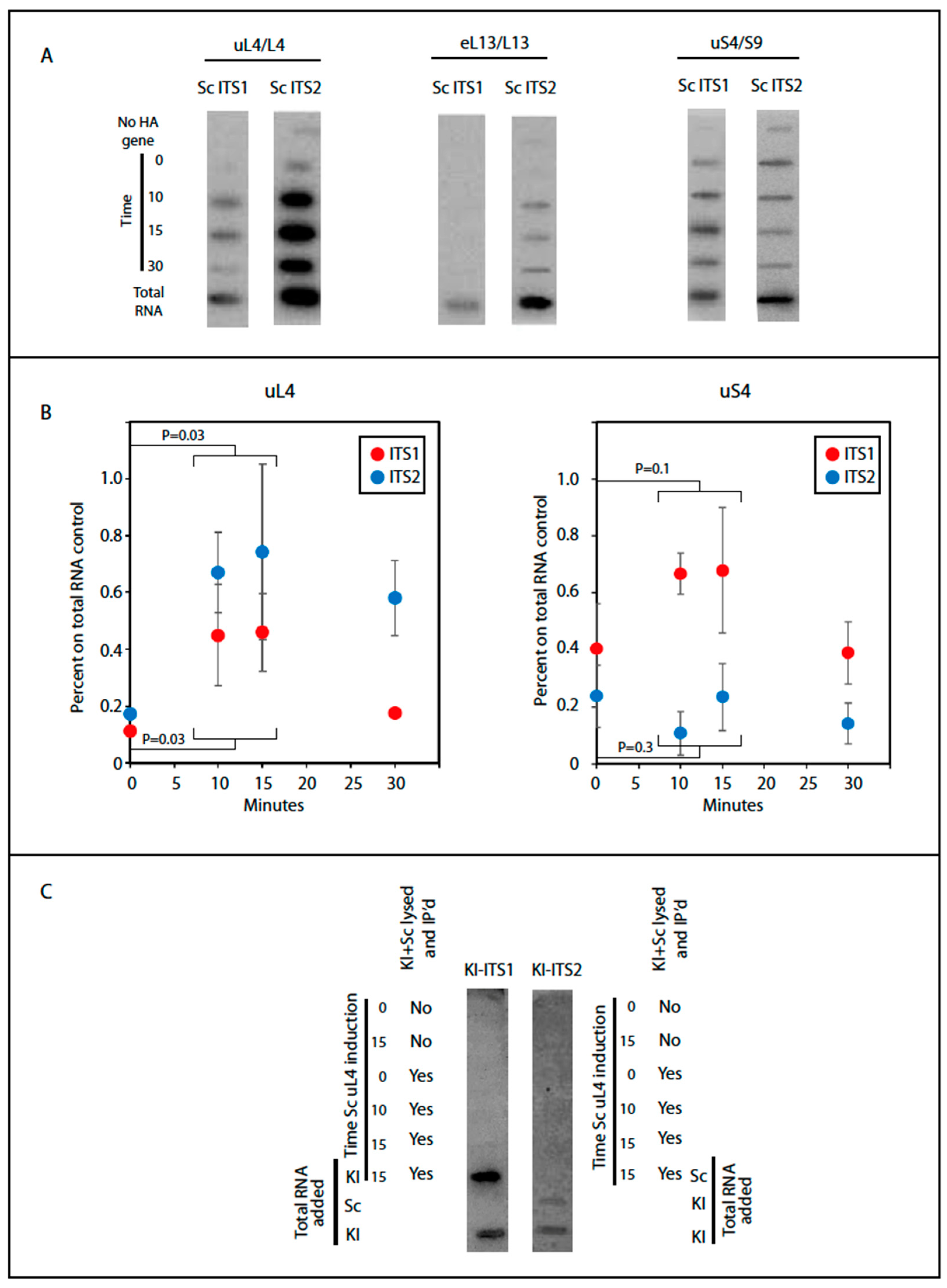
To interrogate the notion of incorporation of uL4 prior to A2 cleavage, we determined the content of the rRNA internal transcribed spacers (ITS1 and ITS2) in particles pulled down by anti-HA purification. These sequences are markers for precursor ribosomes, because they are degraded during ribosome assembly. Furthermore, ITS1 and ITS2 coexist in early ribosomal precursor particles, but are separated into different particles after A2 cleavage (
Figure 6). Accordingly, we determined if ITS1 and ITS2 co-precipitate with HA-uL4. Indeed, we found that both ITS1 and ITS2 co-precipitated with HA-uL4 (
Figure 5). The co-precipitation of ITS2 is expected, since the ITS2 probe maps upstream of the C2 cleavage site and the exosome-mediated removal of ITS2 RNA from the 3′ end of 5.8S rRNA occurs late in the nuclear maturation process [
19]. The co-precipitation of ITS1 with HA-uL4 confirms that uL4 binds to precursor ribosomes prior to A2 cleavage, because the probe for ITS1 maps upstream of A2 in the part of ITS1 that is associated with the rRNA in precursor 40S subunits (
Figure 6). The co-precipitation of both ITS1 and ITS2 with HA-uL4 thus bears out the hypothesis formed from the co-precipitation of uL4 with HA-eS7 (
Figure 3).
We also determined the ITS1 and ITS2 content in particles pulled down with HA-uS4. In fact, the autoradiogram in
Figure 5A shows bands for both internal spacers. However, only the content of ITS1 increased after induction of HA-uS4, indicating that ITS2 in the immunoprecipitated is due to unspecific association. The presence of ITS1 in the HA-uS4 pull down agrees with the fact that it is not removed by cleavage at site D (
Figure 1A) until the 40S precursor has been exported to the cytoplasm [
3]. The analysis of internal transcribed spacers in the uS4 pull-down therefore suggests that uS4 is incorporated into the 40S precursor particle after A2 cleavage. However, depletion of uS4 inhibits A2 cleavage and formation of 20S precursor rRNA, suggesting that uS4 binding is a prerequisite for cleavage in ITS1 [
20]. One potential explanation reconciling these observations is that uS4 is a trigger for ITS1 cleavage as is illustrated by the broken-line arrow between uS4 and the A2 site in
Figure 6. If A2 cleavage therefore follows immediately after uS4 binding, particles harboring both uS4 and ITS2 would not accumulate to detectable levels. Ribosomal particles isolated by pull-down with HA-eL13 also contained only ITS2 and not ITS1, suggesting that this protein also binds after A2 cleavage (
Figure 6). Finally, the absence of uL22 in the eS7 pull-down suggests that uL22 is also added after A2 cleavage (
Figure 6).
Our contention that uL4 is incorporated before separation of the 40S and 60S assembly pathways makes sense because (i) uL4 binds to the 60S precursor early in its assembly and is present in high amounts in the 90S particle pulled down with antibodies against Rrp5, an assembly factor with function in the earliest steps of ribosome formation [
21], (ii) uL4 initially binds the Domain I of the large subunit rRNA, even though it contacts five different rRNA domains of the large subunit rRNA in the mature subunit [
22,
23], and (iii) the uL4 target in Domain I is within the 25% of the 60S rRNA that is transcribed by the time the rRNA is cleaved at A2.
Our discussion of the results is based on the model of co-transcriptional ITS1 cleavage. This is justified since we did not see ITS2 in the HA-uS4 precipitate and uL22 was absent in the HA-eS7 precipitate. Furthermore, 70–80% of the rRNA transcripts are cleaved co-transcriptionally in ITS1 [
5,
6]. Any contribution from post-transcriptional rRNA processing would therefore be modest. We also speculate post-transcriptional processing might occur if uS4 for some reason is not added at the time the RNA polymerase has progressed to the point of cleavage in co-transcriptional processing. If the cleavage was missed, because uS4 did not bind, transcription may have to be completed before the next chance of cleavage appears. However, it is also clear that uS4 binding is not the only parameter affecting the co-/post-transcriptional balance, because poor growth conditions, depletion of at least two different 60S proteins (uL1 and uL4), and inhibition of exonuclease Rat1 also favor post transcriptional processing [
4,
5,
6].
The fact that we only found two proteins that bind before A2 cleavage, and three that do not, could be accidental (
Figure 6). However, it is worth noting that while it takes about 20 min to fully process rRNA, the molecule is cleaved at A2 when about half of the precursor 35S precursor (about 3000 nucleotides) has been transcribed. Since rRNA is polymerized at a rate of about 50 nucleotides per second, A2 cleavage occurs already about a minute after transcription start, i.e. very early in the total ribosome assembly process.
In summary, we presented evidence that the 40S protein eS7 and 60S protein uL4 bind to the nascent rRNA prior to separation of the 40S and 60S precursor ribosomes. The significance of this is that 40S protein(s) could affect the folding and assembly of the 5′ 60S rRNA, and, vice versa, 60S protein(s) might affect assembly of the 40S. Therefore, we propose that even though r-proteins are essential only for the assembly of their “own subunit”, binding of r-proteins prior to separation of the subunit precursors could affect pathways and kinetics of the assembly of the ribosomal subunit to which they do not belong. This notion is commensurate with the finding that a deletion in the long loop of the Escherichia coli 50S protein uL4 changes the abundance of pre-16S rRNA (17S), but does not block 30S subunit formation. The uL4 mutation also changes the kinetics of 50S assembly, but does not block 50S formation [
24]. Thus,
E. coli uL4 mutant supports the notion of kinetic interactions between the assembly of the subunits.
Finally, we note that in slower growing cells, cleavage of the long precursor rRNA into subunit specific moieties occurs more frequently after completion of the transcription [
4]. Therefore, more of the r-proteins must bind to the rRNA prior to cleavage in ITS1. Our model thus suggests that in slow growing cells, perhaps including mammalian cells, kinetic interaction of the assembly of the two subunits is more extensive than in fast growing yeast.
4. Materials and Methods
Nomenclature. We used the universal nomenclature for r-proteins [
25] with the classic nomenclature added after a slash at the first appearance of the name of a given r-protein.
Strain Construction and Culture Growth. To construct plasmids harboring genes for r-proteins tagged at the N-terminal with hemagglutinin (HA) that can be induced by β-estradiol, we replaced the
lacZ gene on pLGSD5 [
13] with the following DNA segments listed in the direction of transcription: a BamHI restriction enzyme site, an ATG start codon, the HA-tag coding sequence, a SalI restriction enzyme site, the
RPL4A 5′ UTR sequence lacking the start codon, the
RPL4A coding sequence, a RsrII restriction enzyme site, the 3′ UTR of the
RPL4A gene sequence including the stop codon. We then utilized the SalI and RsrII restriction sites to sub-clone other r-protein genes (without the ATG and termination codons) into the plasmid. These plasmids were transformed into DBY12021, which contains a gene for a hybrid transcription factor (GEV) a fusion protein consisting of the galactose DNA binding domain, an estrogen response domain, and the VP16 transactivation domain [
15]. Cultures were grown in YPD medium to OD
600 = 0.8–1 (approximately 2 × 10
7 cells per mL) and induced by adding β-estradiol to a final concentration of 100 nM. Aliquots of the cultures were placed on ice at the indicated times, harvested by centrifugation and frozen at −80 °C until used for preparation of whole cell lysates.
Sucrose Gradient Sedimentation. Cell pellets from 250 ml cultures were resuspended in 1.25 mL lysis buffer (50 mM Tris-HCl pH 7.5, 30 mM MgCl2, 100 mM NaCl, 50 µg/mL cycloheximide, and 200 µg/mL heparin) and lysed by vortexing with glass beads six times for 30 s interrupted with about 3 min incubation periods on ice. An additional 1.25 mL of lysis buffer or gradient buffer (50 mM Tris acetate (pH 7), 50 mM NH4Cl, 12 mM MgCl2, and 1 mM dithiothreitol was added after lysis. Finally, the lysate was clarified by centrifuging at 10,000 RPM for 20 min, and 10 A260 units of lysate were layered on top of a 10–50% sucrose gradient in gradient buffer and centrifuged at 40,000 RPM for 4 h in an SW40Ti Beckman rotor. The gradients where then pumped through a colorimeter with a flow cell and a 254 nm light source while fractions were collected in an ISCO Foxy Jr gradient collector.
Immunoprecipitations. Cell pellets from 250 mL cultures were resuspended in 1.25 mL Pull-Down Lysis buffer (25 mM Tris-HCl (pH 7.5), 0.15 M NaCl, 0.05% Tween 20, 30 mM MgCl2, complete protease and phosphatase inhibitor cocktail (Roche), 0.25% Triton 100, and anti-foam B (Sigma)) and vortexed for 15 min at top speed at 4 °C. The samples were then clarified by centrifuging at 10,000 RPM for 15 min, and stored at −80 °C in 300 µL aliquots until used. The samples were then thawed on ice and 20 µL of anti-HA agarose resin (Thermo Scientific PI88836 or 26181) was added then rotated end over end at 4 °C for at least 2 h. The resin was pelleted by centrifuging at 3000 RPM for 5 min at 4 °C and the supernatant was removed. Alternatively, magnetic anti-HA coated beads were used. A sample was taken (FT) and the resin was then resuspended in wash buffer (25 mM Tris-HCl (pH 7.5), 0.15 M NaCl, 0.05% Tween 20, 30 mM MgCl2) and pelleted three times. Samples from the first supernatant were taken (Wash). Finally, protein was eluted from the resin by incubating with 1.5× sample buffer for standard sodium dodecyl sulfate poly-acrylamide gel at 95 °C for 5 min. For analysis of RNA co-precipitating with the HA-r-protein, the salt concentration for the wash buffer was optimized for each HA-r-protein by washing with wash buffer containing 0.25, 0.4, 0.5, 0.75, or 0.9 M NaCl. RNA was eluted with 8 M urea at 42 °C for 5 min and fractionated on 1% agarose gels in 90 mM Tris-borate (pH 8.3) buffer. The salt concentration for which most rRNA was eluted from the resin was subsequently used for preparing samples for slot blotting.
Western blots were performed as described previously [
25]. Anti-uL4 serum was prepared for our lab by Covance (Princeton, NJ, USA), anti-HA was purchased from Sigma (H9658), and anti-uL22 was kindly provided by Sabine Rospert (University of Freiburg, Germany).
Slot blotting. RNA samples eluted from the anti-HA beads were deposited onto an Amersham Hybond-N nylon membrane (GE Healthcare Life Sciences, Pittsburgh, PA, USA) using the Minifold II Slot-Blot system (Schleicher & Schuell). The membrane was incubated with pre-hybridization buffer (0.3 M NaCl, 20 nM NaPO
4 (pH6.8), 2.5× Denhard’s solution (
http://cshprotocols.cshlp.org/content/2008/12/pdb.rec11538.full?text_only=true), 10% polyethylene glycol (PEG 6000), 1% sodium dodecylsulfate) for ≥1 h. Then a
32P end-labeled probe (5 × 10
6 cpm) was added and the sample was incubated at 37 °C for >6 h with rotation. The blots were then washed once with oligo wash buffer (0.05 M NaCl, 0.02 M NaPO
4 (pH 6.8), 0.5% SDS) for 10 min at room temperature. Finally, the blots were exposed to phosphor-imaging screen for >3 h and scanned on a Molecular Dynamics Storm Scanner 860. The images were optimized for clear viewing and quantified using ImageJ.
Hybridization probes
S. cerevisiae ITS1 probe (O1663) CTCTTGTCTTCTTGCCCAGTAAAA G
S. cerevisiae ITS2 probe (O1660) AGGCCAGCAATTTCAAGTTAACTCC
K. lactis ITS1 probe CCCAGTAATCTACTCATTCATAATC
K. lactis ITS2 probe ATAGACTTGACACGCAGCCCTGCTC
Sequence alignments were made with CLC Viewer 8.0
Ribosome structure models were constructed using Chimera 2 [
26] using data from The Protein Data Bank (PDB). PDB IDs: 90S 5JPQ [
10]; 80S 4V7R [
18]; early pre-60S 5Z3G [
27]; late (cytoplasmic) pre-60S 3JCT [
28].



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





