TGF-β-Mediated Epithelial-Mesenchymal Transition and Cancer Metastasis


Abstract
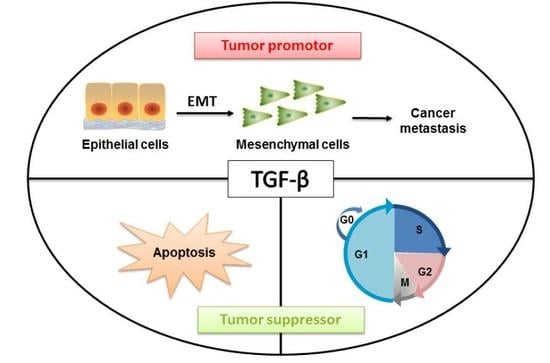
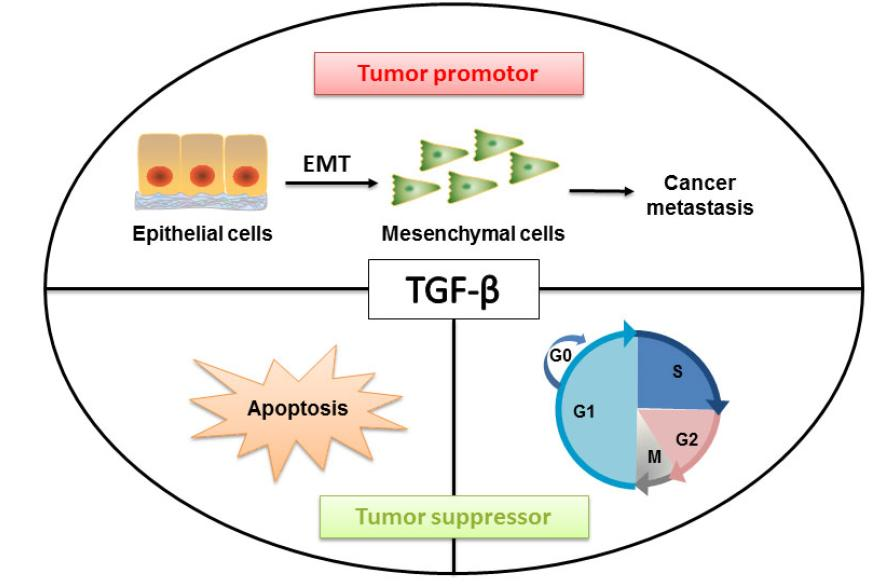
:
1. Introduction
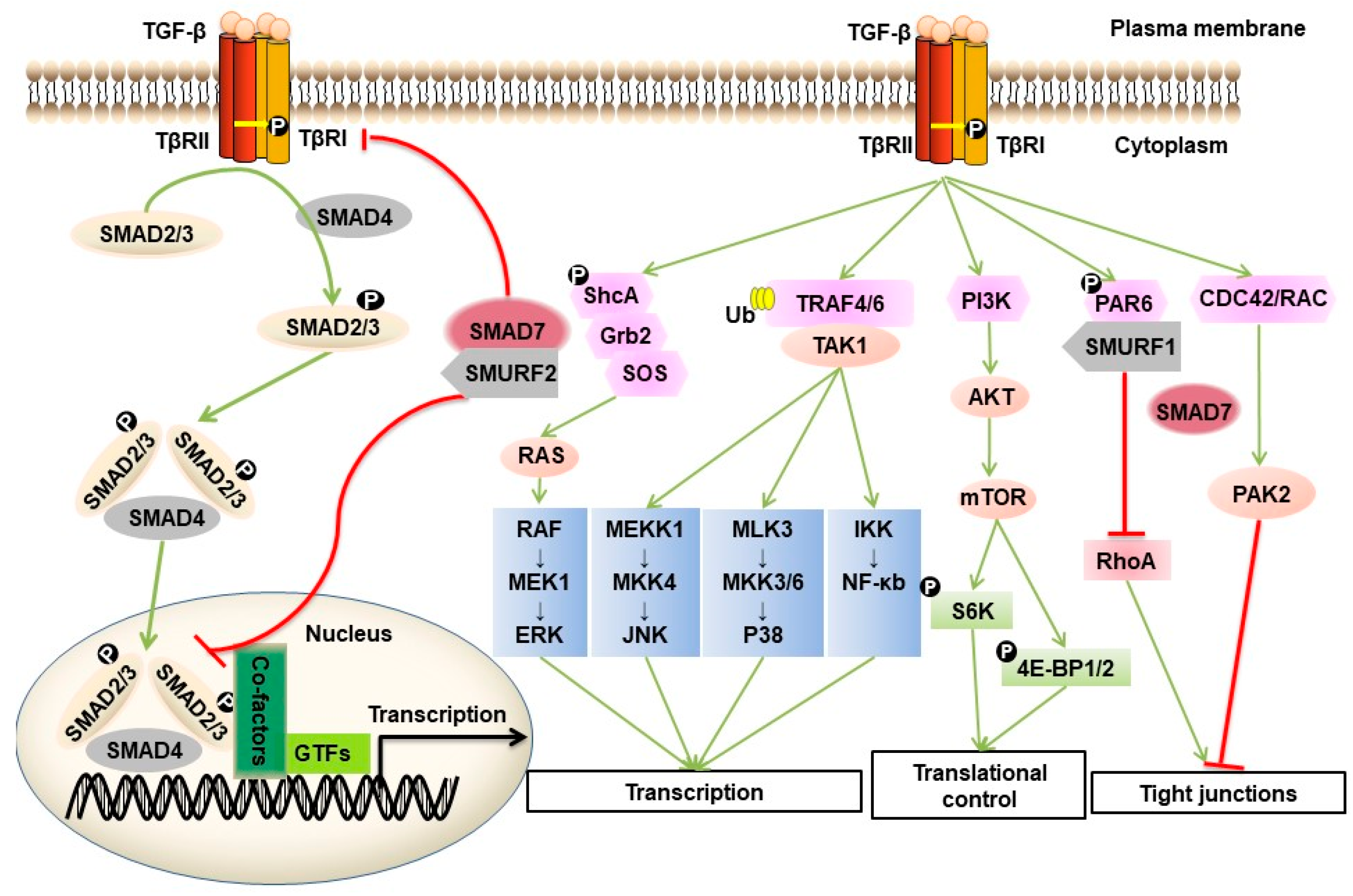
2. TGF-β and Signaling Transduction across the Plasma Membrane
3. Intracellular SMAD and Non-SMAD Signaling Pathways
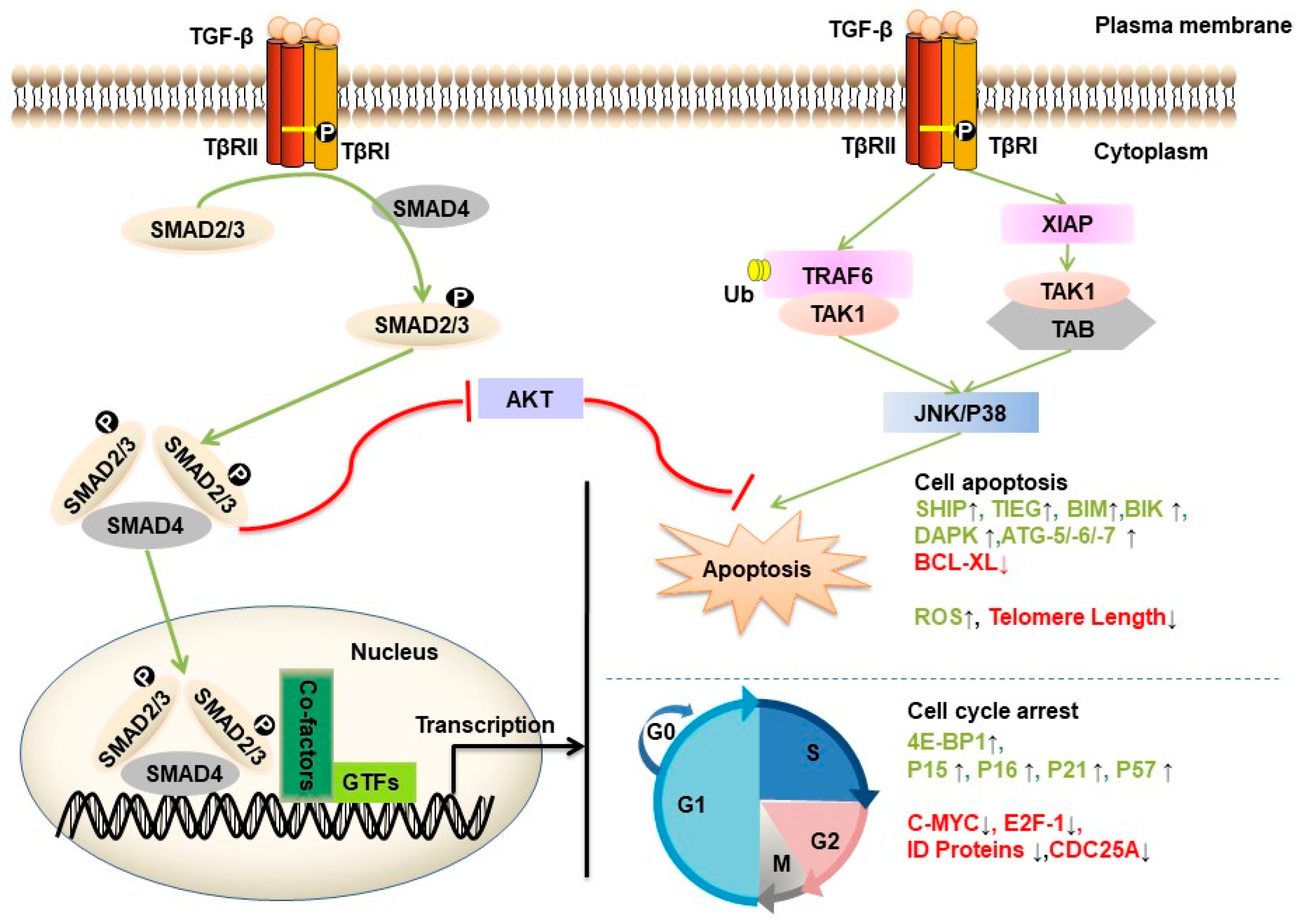
4. Tumor Suppressive Effects of TGF-β
5. TGF-β-induced Tumor Promoting Effects
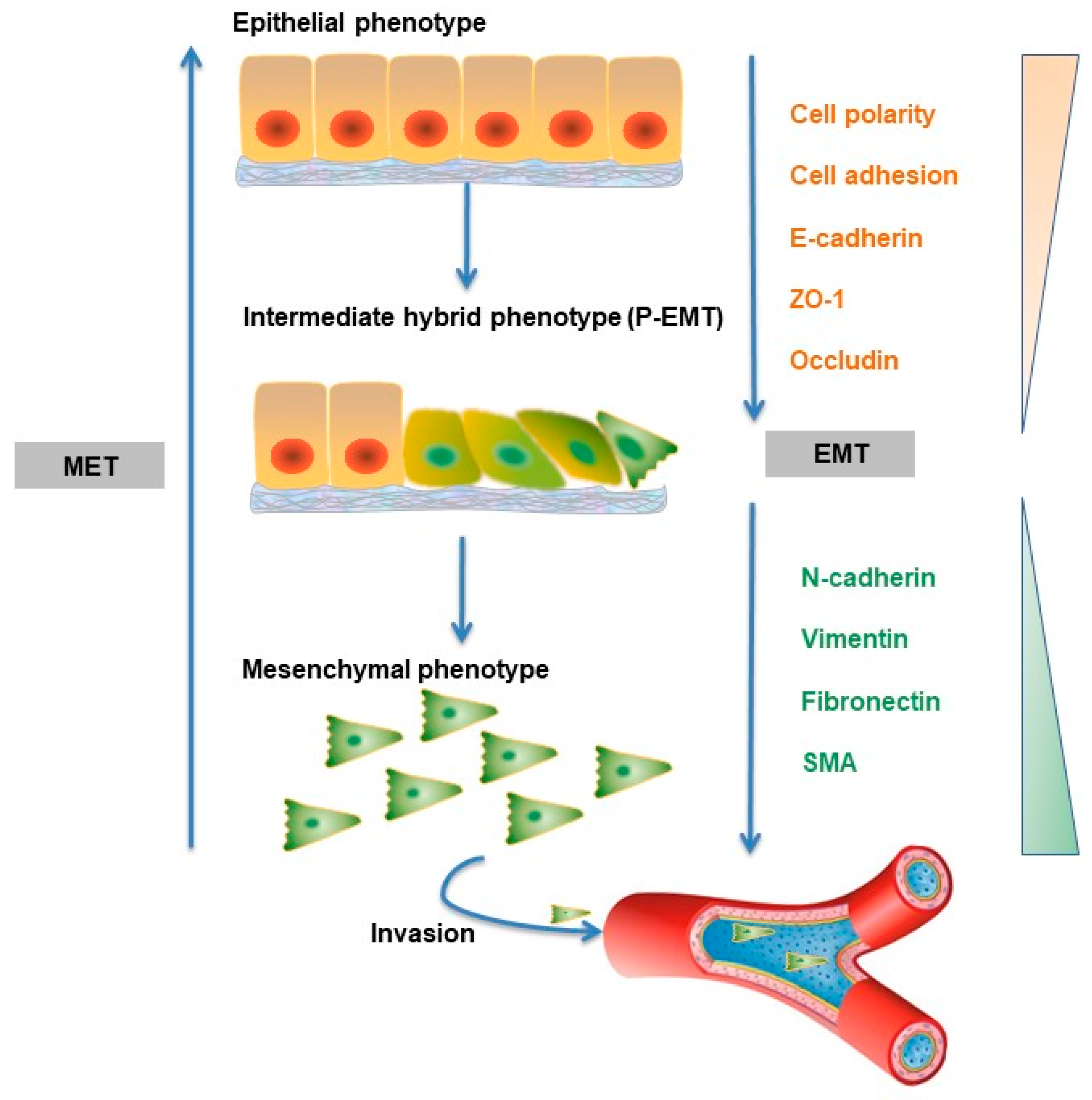
5.1. Cell Biology of TGF-β-induced EMT
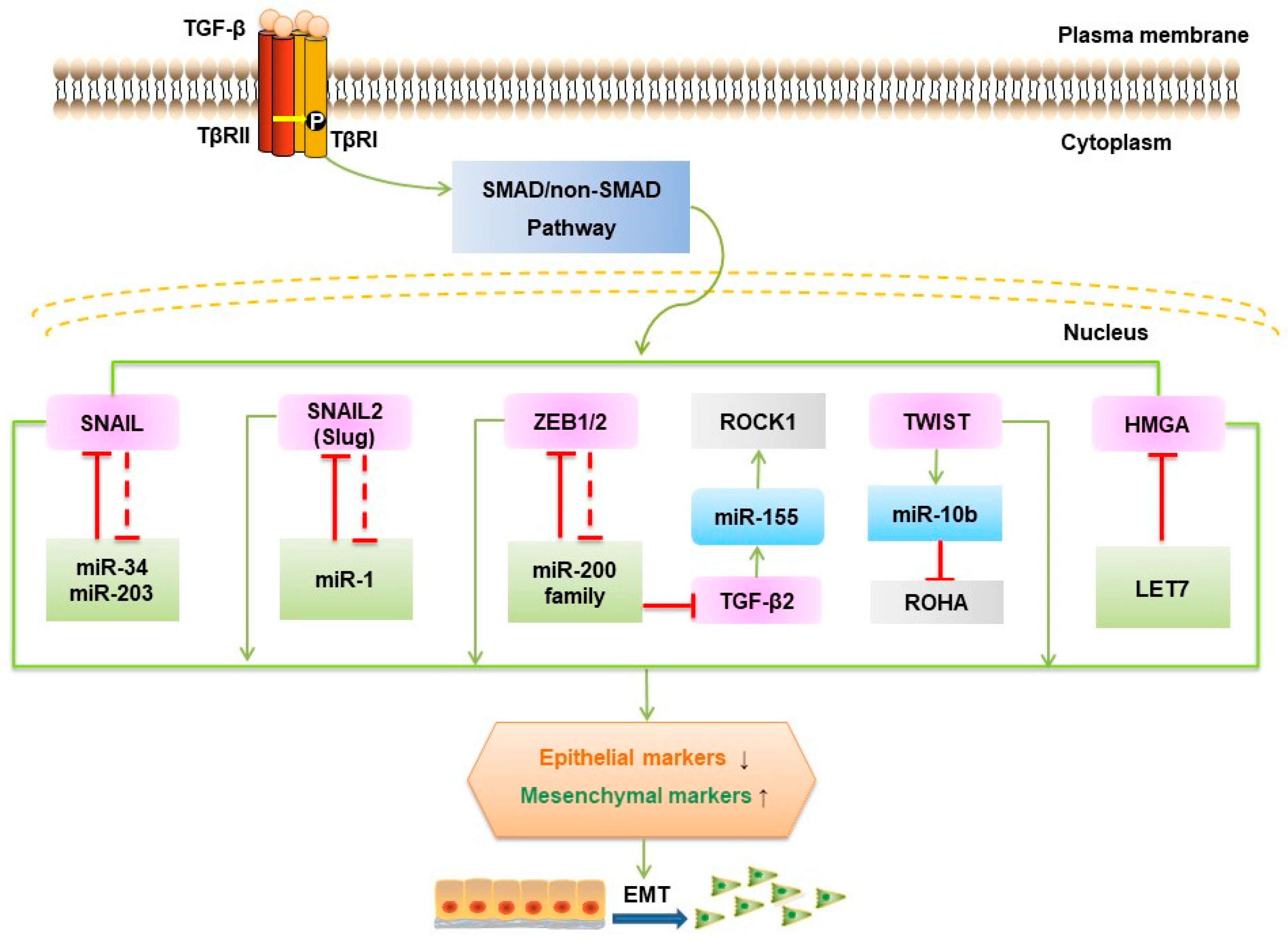
5.2. Molecular Mechanisms in TGF-β-induced EMT
5.3. MicroRNAs Involved in TGF-β-induced EMT
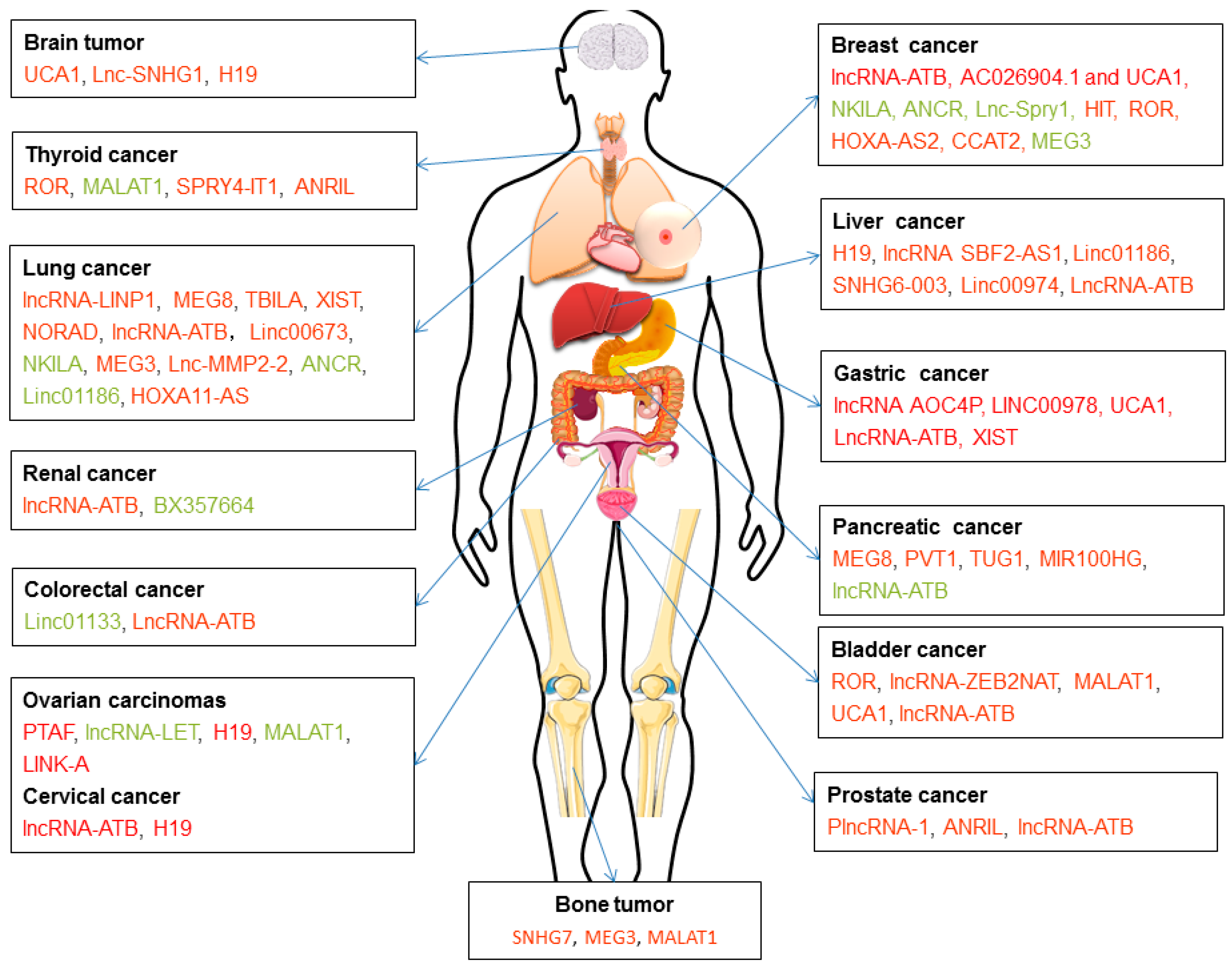
5.4. LncRNAs Involved in TGF-β-induced EMT
6. TGF-β and Metastasis
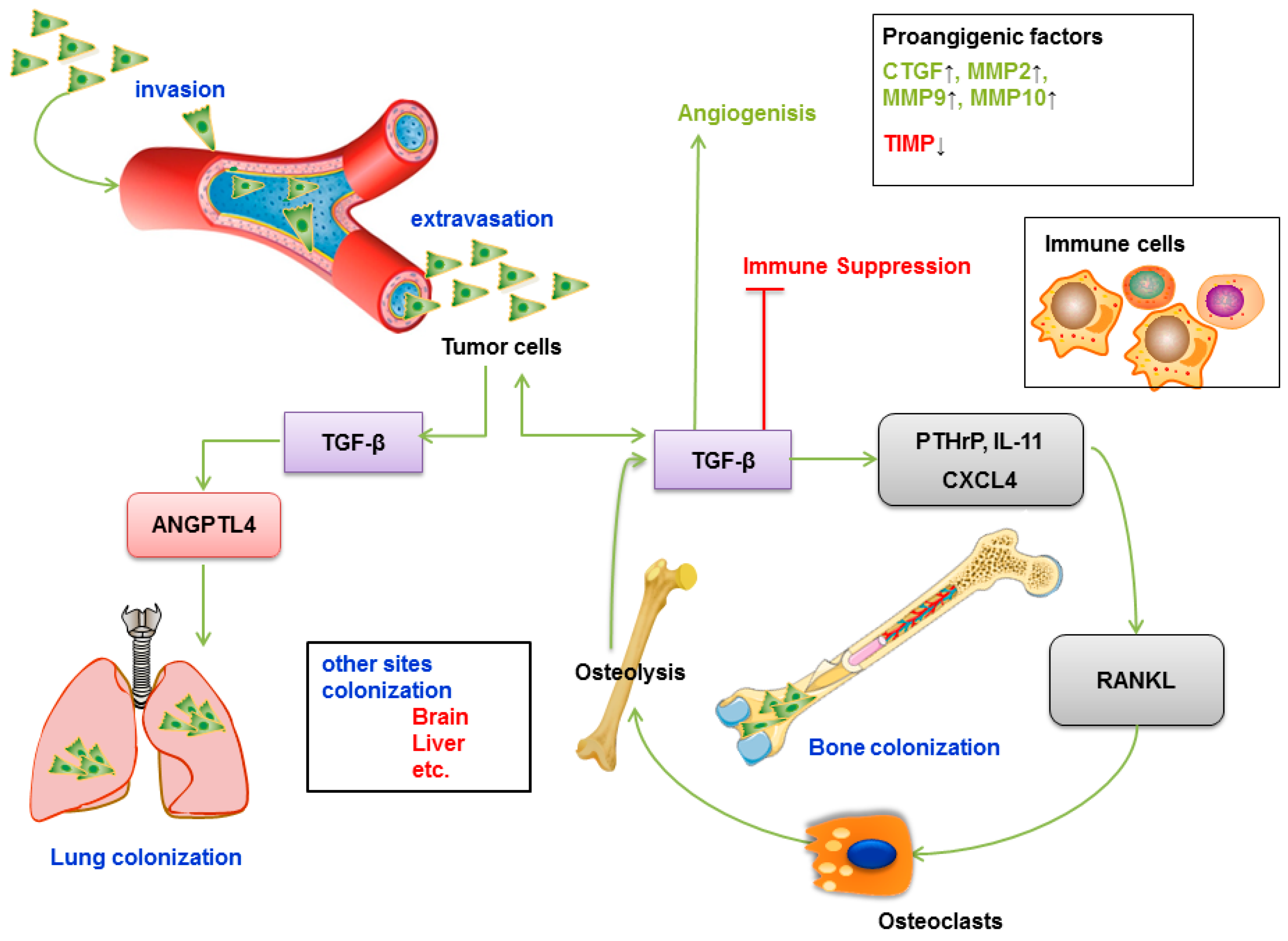
6.1. TGF-β-induced Metastasis in Tissues
6.2. TGF-β Promotes Angiogenesis
6.3. TGF-β Promotes Immune Evasion
7. Targeting the TGF-β Signaling Pathway in Cancer
8. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Faguet, G.B. A brief history of cancer: Age-old milestones underlying our current knowledge database. Int. J. Cancer 2015, 136, 2022–2036. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Fu, L. Targeting cancer stem cells: A new therapy to cure cancer patients. Am. J. Cancer Res. 2012, 2, 340–356. [Google Scholar] [PubMed]
- Farkona, S.; Diamandis, E.P.; Blasutig, I.M. Cancer immunotherapy: The beginning of the end of cancer? BMC Med. 2016, 14, 73. [Google Scholar] [CrossRef] [PubMed]
- David, C.J.; Massague, J. Contextual determinants of TGF-β action in development, immunity and cancer. Nat. Rev. Mol. Cell Biol. 2018, 19, 419–435. [Google Scholar] [CrossRef] [PubMed]
- Exposito-Villen, A.; Aranega, E.A.; Franco, D. Functional role of non-coding RNAs during epithelial-To-mesenchymal transition. Noncoding RNA 2018, 4, 14. [Google Scholar] [CrossRef]
- Derynck, R.; Budi, E.H. Specificity, versatility, and control of TGF-β family signaling. Sci. Signal 2019, 12, eaav5183. [Google Scholar] [CrossRef] [PubMed]
- De Larco, J.E.; Todaro, G.J. Growth factors from murine sarcoma virus-transformed cells. Proc. Natl. Acad. Sci. USA 1978, 75, 4001–4005. [Google Scholar] [CrossRef]
- Derynck, R.; Jarrett, J.A.; Chen, E.Y.; Eaton, D.H.; Bell, J.R.; Assoian, R.K.; Roberts, A.B.; Sporn, M.B.; Goeddel, V.D. Human transforming growth factor-β complementary DNA sequence and expression in normal and transformed cells. Nature 1985, 316, 701–705. [Google Scholar] [CrossRef]
- Sha, X.; Yang, L.; Gentry, E.L. Identification and analysis of discrete functional domains in the pro region of pre-pro-transforming growth factor β 1. J. Cell Biol. 1991, 114, 827–839. [Google Scholar] [CrossRef]
- Shi, M.; Zhu, J.; Wang, R.; Chen, X.; Mi, L.; Walz, T.; Springer, T.A. Latent TGF-β structure and activation. Nature 2011, 474, 343–349. [Google Scholar] [CrossRef]
- Cheifetz, S.; Hernandez, H.; Laiho, M.; ten Dijke, P.; Iwata, K.K.; Massague, J. Distinct transforming growth factor-β (TGF-β) receptor subsets as determinants of cellular responsiveness to three TGF-β isoforms. J. Biol. Chem. 1990, 265, 20533–20538. [Google Scholar] [PubMed]
- Dong, X.; Zhao, B.; Iacob, R.E.; Zhu, J.; Koksal, A.C.; Lu, C.; Engen, J.R.; Springer, T.A. Force interacts with macromolecular structure in activation of TGF-β. Nature 2017, 542, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Massague, J. Receptors for the TGF-β family. Cell 1992, 69, 1067–1070. [Google Scholar] [CrossRef]
- Heldin, C.H.; Moustakas, A. Signaling receptors for TGF-β family members. Cold Spring Harb. Perspect. Biol. 2016, 8, a022053. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, A.; Moss-Taylor, L.; Kim, M.J.; Ghosh, A.C.; O’Connor, M.B. TGF-β family signaling in drosophila. Cold Spring Harb. Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Savage-Dunn, C.; Padgett, R.W. The TGF-β family in caenorhabditis elegans. Cold Spring Harb. Perspect. Biol. 2017, 9, a022178. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Gelbart, W.M.; Harland, R.M.; Heldin, C.H.; Kern, S.E.; Massagué, J.; Melton, D.A.; Mlodzik, M.; Padgett, R.W.; Roberts, A.B.; et al. Nomenclature: Vertebrate mediators of TGF-β family signals. Cell 1996, 87, 173. [Google Scholar] [CrossRef]
- Wrana, J.L. Signaling by the TGF-β superfamily. Cold Spring Harb. Perspect. Biol. 2013, 5, a011197. [Google Scholar] [CrossRef] [PubMed]
- Hill, C.S. Transcriptional control by the SMADs. Cold Spring Harb. Perspect. Biol. 2016, 8, a022079. [Google Scholar] [CrossRef]
- Miyazawa, K.; Miyazono, K. Regulation of TGF-β family signaling by inhibitory SMADs. Cold Spring Harb. Perspect. Biol. 2017, 9, a022095. [Google Scholar] [CrossRef]
- Zhang, Y.E. Non-SMAD signaling pathways of the TGF-β family. Cold Spring Harb. Perspect. Biol. 2017, 9, a022129. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.E. Non-SMAD pathways in TGF-β signaling. Cell Res. 2009, 19, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Ravichandran, K.S. Signaling via Shc family adapter proteins. Oncogene 2001, 20, 6322–6330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamashita, M.; Fatyol, K.; Jin, C.; Wang, X.; Liu, Z.; Zhang, Y.E. TRAF6 mediates SMAD-independent activation of JNK and p38 by TGF-β. Mol. Cell. 2008, 31, 918–924. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, A.; Thakur, N.; Grimsby, S.; Marcusson, A.; von Bulow, V.; Schuster, N.; Zhang, S.; Heldin, C.H.; Landstrom, M. The type I TGF-β receptor engages TRAF6 to activate TAK1 in a receptor kinase-independent manner. Nat. Cell Biol. 2008, 10, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Ozdamar, B.; Bose, R.; Barrios-Rodiles, M.; Wang, H.R.; Zhang, Y.; Wrana, J.L. Regulation of the polarity protein Par6 by TGF-β receptors controls epithelial cell plasticity. Science 2005, 307, 1603–1609. [Google Scholar] [CrossRef] [PubMed]
- Wilkes, M.C.; Murphy, S.J.; Garamszegi, N.; Leof, E.B. Cell-type-specific activation of PAK2 by transforming growth factor β independent of SMAD2 and SMAD3. Mol. Cell Biol. 2003, 23, 8878–8889. [Google Scholar] [CrossRef]
- Bakin, A.V.; Tomlinson, A.K.; Bhowmick, N.A.; Moses, H.L.; Arteaga, C.L. Phosphatidylinositol 3-kinase function is required for transforming growth factor β-mediated epithelial to mesenchymal transition and cell migration. J. Biol. Chem. 2000, 275, 36803–36810. [Google Scholar] [CrossRef]
- Zhang, Y.; Alexander, P.B.; Wang, X.F. TGF-β family signaling in the control of cell proliferation and survival. Cold Spring Harb. Perspect. Biol. 2017, 9, a022145. [Google Scholar] [CrossRef]
- Azar, R.; Alard, A.; Susini, C.; Bousquet, C.; Pyronnet, S. 4E-BP1 is a target of SMAD4 essential for TGF-β-mediated inhibition of cell proliferation. EMBO J. 2009, 28, 3514–3522. [Google Scholar] [CrossRef]
- Baghdassarian, N.; Ffrench, M. Cyclin-dependent kinase inhibitors (CKIs) and hematological malignancies. Hematol. Cell Ther. 1996, 38, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Acharya, S.; Sahin, O.; Zhang, Q.; Saito, Y.; Yao, J.; Wang, H.; Li, P.; Zhang, L.; Lowery, F.J.; et al. 14-3-3zeta turns TGF-β’s function from tumor suppressor to metastasis promoter in breast cancer by contextual changes of SMAD partners from p53 to Gli2. Cancer Cell. 2015, 27, 177–192. [Google Scholar] [CrossRef] [PubMed]
- Iavarone, A.; Massague, J. Repression of the CDK activator Cdc25A and cell-cycle arrest by cytokine TGF-β in cells lacking the CDK inhibitor p15. Nature 1997, 387, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Ling, M.T.; Wang, X.; Tsao, S.W.; Wong, Y.C. Down-regulation of Id-1 expression is associated with TGF-β1-induced growth arrest in prostate epithelial cells. Biochim. Biophys. Acta 2002, 1570, 145–152. [Google Scholar] [CrossRef]
- Chen, C.R.; Kang, Y.; Siegel, P.M.; Massague, J. E2F4/5 and p107 as SMAD cofactors linking the TGF-β receptor to c-myc repression. Cell 2002, 110, 19–32. [Google Scholar] [CrossRef]
- Ozaki, I.; Hamajima, H.; Matsuhashi, S.; Mizuta, T. Regulation of TGF-β1-induced pro-apoptotic signaling by growth factor receptors and extracellular matrix receptor integrins in the liver. Front. Physiol. 2011, 2, 78. [Google Scholar] [CrossRef] [PubMed]
- Wiener, Z.; Band, A.M.; Kallio, P.; Hogstrom, J.; Hyvonen, V.; Kaijalainen, S.; Ritvos, O.; Haglund, C.; Kruuna, O.; Robine, S.; et al. Oncogenic mutations in intestinal adenomas regulate Bim-mediated apoptosis induced by TGF-β. Proc. Natl. Acad. Sci. USA 2014, 111, E2229–E2236. [Google Scholar] [CrossRef]
- Zhao, X.; Liu, Y.; Du, L.; He, L.; Ni, B.; Hu, J.; Zhu, D.; Chen, Q. Threonine 32 (Thr32) of FoxO3 is critical for TGF-β-induced apoptosis via Bim in hepatocarcinoma cells. Protein Cell 2015, 6, 127–138. [Google Scholar] [CrossRef]
- Spender, L.C.; O’Brien, D.I.; Simpson, D.; Dutt, D.; Gregory, C.D.; Allday, M.J.; Clark, L.J.; Inman, G.J. TGF-β induces apoptosis in human B cells by transcriptional regulation of BIK and BCL-XL. Cell Death Differ. 2009, 16, 593–602. [Google Scholar] [CrossRef]
- Ohgushi, M.; Kuroki, S.; Fukamachi, H.; O’Reilly, L.A.; Kuida, K.; Strasser, A.; Yonehara, S. Transforming growth factor β-dependent sequential activation of SMAD, Bim, and caspase-9 mediates physiological apoptosis in gastric epithelial cells. Mol. Cell. Biol. 2005, 25, 10017–10028. [Google Scholar] [CrossRef]
- Shima, Y.; Nakao, K.; Nakashima, T.; Kawakami, A.; Nakata, K.; Hamasaki, K.; Kato, Y.; Eguchi, K.; Ishii, N. Activation of caspase-8 in transforming growth factor-β-induced apoptosis of human hepatoma cells. Hepatology 1999, 30, 1215–1222. [Google Scholar] [CrossRef] [PubMed]
- Takekawa, M.; Tatebayashi, K.; Itoh, F.; Adachi, M.; Imai, K.; Saito, H. SMAD-dependent GADD45β expression mediates delayed activation of p38 MAP kinase by TGF-β. EMBO J. 2002, 21, 6473–6482. [Google Scholar] [CrossRef] [PubMed]
- Yan, F.; Wang, Y.; Wu, X.; Peshavariya, H.M.; Dusting, G.J.; Zhang, M.; Jiang, F. Nox4 and redox signaling mediate TGF-β-induced endothelial cell apoptosis and phenotypic switch. Cell Death Dis. 2014, 5, e1010. [Google Scholar] [CrossRef] [PubMed]
- Cardenas, H.; Vieth, E.; Lee, J.; Segar, M.; Liu, Y.; Nephew, K.P.; Matei, D. TGF-β induces global changes in DNA methylation during the epithelial-to-mesenchymal transition in ovarian cancer cells. Epigenetics 2014, 9, 1461–1472. [Google Scholar] [CrossRef] [PubMed]
- Evanno, E.; Godet, J.; Piccirilli, N.; Guilhot, J.; Milin, S.; Gombert, J.M.; Fouchaq, B.; Roche, J. Tri-methylation of H3K79 is decreased in TGF-β1-induced epithelial-to-mesenchymal transition in lung cancer. Clin. Epigenetics 2017, 9, 80. [Google Scholar] [CrossRef] [PubMed]
- Cassar, L.; Nicholls, C.; Pinto, A.R.; Chen, R.; Wang, L.; Li, H.; Liu, J.P. TGF-β receptor mediated telomerase inhibition, telomere shortening and breast cancer cell senescence. Protein Cell 2017, 8, 39–54. [Google Scholar] [CrossRef] [PubMed]
- Kiyono, K.; Suzuki, H.I.; Matsuyama, H.; Morishita, Y.; Komuro, A.; Kano, M.R.; Sugimoto, K.; Miyazono, K. Autophagy is activated by TGF-β and potentiates TGF-β-mediated growth inhibition in human hepatocellular carcinoma cells. Cancer Res. 2009, 69, 8844–8852. [Google Scholar] [CrossRef]
- Suzuki, H.I.; Kiyono, K.; Miyazono, K. Regulation of autophagy by transforming growth factor-β (TGF-β) signaling. Autophagy 2010, 6, 645–647. [Google Scholar] [CrossRef]
- Korkut, A.; Zaidi, S.; Kanchi, R.S.; Rao, S.; Gough, N.R.; Schultz, A.; Li, X.; Lorenzi, P.L.; Berger, A.C.; Robertson, G.; et al. A pan-cancer analysis reveals high-frequency genetic alterations in mediators of signaling by the TGF-β superfamily. Cell Syst. 2018, 7, 422–437.e7. [Google Scholar] [CrossRef]
- Hahn, S.A.; Schutte, M.; Hoque, A.T.; Moskaluk, C.A.; da Costa, L.T.; Rozenblum, E.; Weinstein, C.L.; Fischer, A.; Yeo, C.J.; Hruban, R.H.; et al. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science 1996, 271, 350–353. [Google Scholar] [CrossRef]
- Markowitz, S.; Wang, J.; Myeroff, L.; Parsons, R.; Sun, L.; Lutterbaugh, J.; Fan, R.S.; Zborowska, E.; Kinzler, K.W.; Vogelstein, B.; et al. Inactivation of the type II TGF-β receptor in colon cancer cells with microsatellite instability. Science 1995, 268, 1336–1338. [Google Scholar] [CrossRef] [PubMed]
- Macias-Silva, M.; Abdollah, S.; Hoodless, P.A.; Pirone, R.; Attisano, L.; Wrana, J.L. MADR2 is a substrate of the TGF-β receptor and its phosphorylation is required for nuclear accumulation and signaling. Cell 1996, 87, 1215–1224. [Google Scholar] [CrossRef]
- Yang, L.; Pang, Y.; Moses, H.L. TGF-β and immune cells: An important regulatory axis in the tumor microenvironment and progression. Trends Immunol. 2010, 31, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Pinto, M.; Oliveira, C.; Cirnes, L.; Machado, J.C.; Ramires, M.; Nogueira, A.; Carneiro, F.; Seruca, R. Promoter methylation of TGF-β receptor I and mutation of TGF-β receptor II are frequent events in MSI sporadic gastric carcinomas. J. Pathol. 2003, 200, 32–38. [Google Scholar] [CrossRef]
- Bruna, A.; Darken, R.S.; Rojo, F.; Ocana, A.; Penuelas, S.; Arias, A.; Paris, R.; Tortosa, A.; Mora, J.; Baselga, J.; et al. High TGF-β-SMAD activity confers poor prognosis in glioma patients and promotes cell proliferation depending on the methylation of the PDGF-B gene. Cancer Cell 2007, 11, 147–160. [Google Scholar] [CrossRef] [PubMed]
- Caja, L.; Dituri, F.; Mancarella, S.; Caballero-Diaz, D.; Moustakas, A.; Giannelli, G.; Fabregat, I. TGF-β and the tissue microenvironment: Relevance in fibrosis and cancer. Int. J. Mol. Sci. 2018, 19, 1294. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Quintanilla, M.; Cano, A. Transforming growth factor β-1 induces snail transcription factor in epithelial cell lines: Mechanisms for epithelial mesenchymal transitions. J. Biol. Chem. 2003, 278, 21113–21123. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. Emt: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef]
- Tsai, J.H.; Yang, J. Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes Dev. 2013, 27, 2192–2206. [Google Scholar] [CrossRef]
- van Staalduinen, J.; Baker, D.; Dijke, P.t.; van Dam, H. Epithelial-mesenchymal-transition-inducing transcription factors: New targets for tackling chemoresistance in cancer? Oncogene 2018, 37, 6195–6211. [Google Scholar] [CrossRef] [PubMed]
- Jolly, M.K.; Ware, K.E.; Gilja, S.; Somarelli, J.A.; Levine, H. EMT and MET: Necessary or permissive for metastasis? Mol. Oncol. 2017, 11, 755–769. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.; Choi, H.; el Rayes, T.; Ryu, S.; Troeger, J.; et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Aiello, N.M.; Brabletz, T.; Kang, Y.; Nieto, M.A.; Weinberg, R.A.; Stanger, B.Z. Upholding a role for EMT in pancreatic cancer metastasis. Nature 2017, 547, E7–E8. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Kang, Y. Probing the fifty shades of EMT in metastasis. Trends Cancer 2016, 2, 65–67. [Google Scholar] [CrossRef] [PubMed]
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S.; van Keymeulen, A.; Brown, D.; Moers, V.; Lemaire, S.; et al. Identification of the tumour transition states occurring during EMT. Nature 2018, 556, 463–468. [Google Scholar] [CrossRef]
- Nikitorowicz-Buniak, J.; Denton, C.P.; Abraham, D.; Stratton, R. Partially evoked epithelial-mesenchymal transition (EMT) is associated with increased TGF-β signaling within lesional scleroderma skin. PLoS ONE 2015, 10, e0134092. [Google Scholar]
- Puram, S.V.; Parikh, A.S.; Tirosh, I. Single cell RNA-seq highlights a role for a partial EMT in head and neck cancer. Mol. Cell Oncol. 2018, 5, e1448244. [Google Scholar] [CrossRef]
- Deckers, M.; van Dinther, M.; Buijs, J.; Que, I.; Lowik, C.; van der Pluijm, G.; Dijke, P.t. The tumor suppressor SMAD4 is required for transforming growth factor β-induced epithelial to mesenchymal transition and bone metastasis of breast cancer cells. Cancer Res. 2006, 66, 2202–2209. [Google Scholar] [CrossRef]
- Vincent, T.; Neve, E.P.; Johnson, J.R.; Kukalev, A.; Rojo, F.; Albanell, J.; Pietras, K.; Virtanen, I.; Philipson, L.; Leopold, P.L.; et al. A SNAIL1-SMAD3/4 transcriptional repressor complex promotes TGF-β mediated epithelial-mesenchymal transition. Nat. Cell Biol. 2009, 11, 943–950. [Google Scholar] [CrossRef] [PubMed]
- Hoot, K.E.; Lighthall, J.; Han, G.; Lu, S.L.; Li, A.; Ju, W.; Kulesz-Martin, M.; Bottinger, E.; Wang, X.J. Keratinocyte-specific SMAD2 ablation results in increased epithelial-mesenchymal transition during skin cancer formation and progression. J. Clin. Investig. 2008, 118, 2722–2732. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Lei, R.; Zhuang, X.; Li, X.; Li, G.; Lev, S.; Segura, M.F.; Zhang, X.; Hu, G. MicroRNA-182 targets SMAD7 to potentiate TGF-β-induced epithelial-mesenchymal transition and metastasis of cancer cells. Nat. Commun. 2016, 7, 13884. [Google Scholar] [CrossRef] [PubMed]
- Kuang, J.; Li, L.; Guo, L.; Su, Y.; Wang, Y.; Xu, Y.; Wang, X.; Meng, S.; Lei, L.; Xu, L.; et al. RNF8 promotes epithelial-mesenchymal transition of breast cancer cells. J. Exp. Clin. Cancer Res. 2016, 35, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zavadil, J.; Bitzer, M.; Liang, D.; Yang, Y.C.; Massimi, A.; Kneitz, S.; Piek, E.; Bottinger, E.P. Genetic programs of epithelial cell plasticity directed by transforming growth factor-β. Proc. Natl. Acad. Sci. USA 2001, 98, 6686–6691. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.K.; Pardoux, C.; Hall, M.C.; Lee, P.S.; Warburton, D.; Qing, J.; Smith, S.M.; Derynck, R. TGF-β activates Erk MAP kinase signalling through direct phosphorylation of ShcA. EMBO J. 2007, 26, 3957–3967. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.; Robinson, M.; Smith, E.; Huntley, S.; Prime, S.; Paterson, I. Induction of an epithelial to mesenchymal transition in human immortal and malignant keratinocytes by TGF-β1 involves MAPK, SMAD and AP-1 signalling pathways. J. Cell Biochem. 2005, 95, 918–931. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.; Zeng, L.; Liu, Y.; DeFea, K.; Schwartz, M.A.; Chien, S.; Shyy, J.Y. Rho-ROCK-LIMK-cofilin pathway regulates shear stress activation of sterol regulatory element binding proteins. Circ. Res. 2003, 92, 1296–1304. [Google Scholar] [CrossRef] [PubMed]
- Landstrom, M. The TAK1-TRAF6 signalling pathway. Int. J. Biochem. Cell Biol. 2010, 42, 585–589. [Google Scholar] [CrossRef]
- Song, J.; Landstrom, M. TGF-β activates PI3K-AKT signaling via TRAF6. Oncotarget 2017, 8, 99205–99206. [Google Scholar] [CrossRef]
- Xue, G.; Restuccia, D.F.; Lan, Q.; Hynx, D.; Dirnhofer, S.; Hess, D.; Ruegg, C.; Hemmings, B.A. Akt/PKB-mediated phosphorylation of TWIST1 promotes tumor metastasis via mediating cross-talk between PI3K/Akt and TGF-β signaling axes. Cancer Dis. 2012, 2, 248–259. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Teruya-Feldstein, J.; Weinberg, R.A. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature 2007, 449, 682–688. [Google Scholar] [CrossRef] [PubMed]
- Werden, S.J.; Sphyris, N.; Sarkar, T.R.; Paranjape, A.N.; LaBaff, A.M.; Taube, J.H.; Hollier, B.G.; Ramirez-Pena, E.Q.; Soundararajan, R.; den Hollander, P.; et al. Phosphorylation of serine 367 of FOXC2 by p38 regulates ZEB1 and breast cancer metastasis, without impacting primary tumor growth. Oncogene 2016, 35, 5977–5988. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Li, Y.C.; Wu, L.; Yu, G.T.; Zhang, W.F.; Huang, C.F.; Sun, Z.J. TRAF6 regulates tumour metastasis through EMT and CSC phenotypes in head and neck squamous cell carcinoma. J. Cell. Mol. Med. 2018, 22, 1337–1349. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, D.M.; Medici, D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci. Signal 2014, 7, re8. [Google Scholar] [CrossRef] [PubMed]
- Nishita, M.; Hashimoto, M.K.; Ogata, S.; Laurent, M.N.; Ueno, N.; Shibuya, H.; Cho, K.W. Interaction between Wnt and TGF-β signalling pathways during formation of Spemann’s organizer. Nature 2000, 403, 781–785. [Google Scholar] [CrossRef] [PubMed]
- Medici, D.; Hay, E.D.; Goodenough, D.A. Cooperation between snail and LEF-1 transcription factors is essential for TGF-β1-induced epithelial-mesenchymal transition. Mol. Biol. Cell. 2006, 17, 1871–1879. [Google Scholar] [CrossRef] [PubMed]
- Jamora, C.; Lee, P.; Kocieniewski, P.; Azhar, M.; Hosokawa, R.; Chai, Y.; Fuchs, E. A signaling pathway involving TGF-β2 and snail in hair follicle morphogenesis. PLoS Biol. 2005, 3, e11. [Google Scholar] [CrossRef]
- Murillo-Garzon, V.; Gorrono-Etxebarria, I.; Akerfelt, M.; Puustinen, M.C.; Sistonen, L.; Nees, M.; Carton, J.; Waxman, J.; Kypta, R.M. Frizzled-8 integrates Wnt-11 and transforming growth factor-β signaling in prostate cancer. Nat. Commun. 2018, 9, 1747. [Google Scholar] [CrossRef]
- Kanei-Ishii, C.; Ninomiya-Tsuji, J.; Tanikawa, J.; Nomura, T.; Ishitani, T.; Kishida, S.; Kokura, K.; Kurahashi, T.; Ichikawa-Iwata, E.; Kim, Y.; et al. Wnt-1 signal induces phosphorylation and degradation of c-Myb protein via TAK1, HIPK2, and NLK. Genes Dev. 2004, 18, 816–829. [Google Scholar] [CrossRef] [Green Version]
- Sjolund, J.; Bostrom, A.K.; Lindgren, D.; Manna, S.; Moustakas, A.; Ljungberg, B.; Johansson, M.; Fredlund, E.; Axelson, H. The notch and TGF-β signaling pathways contribute to the aggressiveness of clear cell renal cell carcinoma. PLoS ONE 2011, 6, e23057. [Google Scholar] [CrossRef] [PubMed]
- Zavadil, J.; Cermak, L.; Soto-Nieves, N.; Bottinger, E.P. Integration of TGF-β/SMAD and Jagged1/Notch signalling in epithelial-to-mesenchymal transition. EMBO J. 2004, 23, 1155–1165. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Feng, X.H.; Derynck, R. SMAD3 and SMAD4 cooperate with c-Jun/c-Fos to mediate TGF-β-induced transcription. Nature 1998, 394, 909–913. [Google Scholar] [CrossRef] [PubMed]
- Huber, M.A.; Kraut, N.; Beug, H. Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr. Opin. Cell Biol. 2005, 17, 548–558. [Google Scholar] [CrossRef] [PubMed]
- Olsen, S.N.; Wronski, A.; Castano, Z.; Dake, B.; Malone, C.; de Raedt, T.; Enos, M.; DeRose, Y.S.; Zhou, W.; Guerra, S.; et al. Loss of RasGAP tumor suppressors underlies the aggressive nature of luminal b breast cancers. Cancer Discov. 2017, 7, 202–217. [Google Scholar] [CrossRef] [PubMed]
- Bowman, T.; Broome, M.A.; Sinibaldi, D.; Wharton, W.; Pledger, W.J.; Sedivy, J.M.; Irby, R.; Yeatman, T.; Courtneidge, S.A.; Jove, R. Stat3-mediated Myc expression is required for Src transformation and PDGF-induced mitogenesis. Proc. Natl. Acad. Sci. USA 2001, 98, 7319–7324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamouille, S.; Subramanyam, D.; Blelloch, R.; Derynck, R. Regulation of epithelial-mesenchymal and mesenchymal-epithelial transitions by microRNAs. Curr. Opin. Cell Biol. 2013, 25, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Biol. 2008, 10, 593–601. [Google Scholar] [CrossRef]
- Diepenbruck, M.; Tiede, S.; Saxena, M.; Ivanek, R.; Kalathur, R.K.R.; Luond, F.; Meyer-Schaller, N.; Christofori, G. miR-1199-5p and Zeb1 function in a double-negative feedback loop potentially coordinating EMT and tumour metastasis. Nat. Commun. 2017, 8, 1168. [Google Scholar] [CrossRef]
- Kim, N.H.; Kim, H.S.; Li, X.Y.; Lee, I.; Choi, H.S.; Kang, S.E.; Cha, S.Y.; Ryu, J.K.; Yoon, D.; Fearon, E.R.; et al. A p53/miRNA-34 axis regulates Snail1-dependent cancer cell epithelial-mesenchymal transition. J. Cell. Biol. 2011, 195, 417–433. [Google Scholar] [CrossRef]
- Hahn, S.; Jackstadt, R.; Siemens, H.; Hunten, S.; Hermeking, H. SNAIL and miR-34a feed-forward regulation of ZNF281/ZBP99 promotes epithelial-mesenchymal transition. EMBO J. 2013, 32, 3079–3095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majid, S.; Dar, A.A.; Saini, S.; Shahryari, V.; Arora, S.; Zaman, M.S.; Chang, I.; Yamamura, S.; Tanaka, Y.; Chiyomaru, T.; et al. miRNA-34b inhibits prostate cancer through demethylation, active chromatin modifications, and AKT pathways. Clin. Cancer Res. 2013, 19, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Siemens, H.; Neumann, J.; Jackstadt, R.; Mansmann, U.; Horst, D.; Kirchner, T.; Hermeking, H. Detection of miR-34a promoter methylation in combination with elevated expression of c-Met and β-catenin predicts distant metastasis of colon cancer. Clin. Cancer Res. 2013, 19, 710–720. [Google Scholar] [CrossRef] [PubMed]
- Genovese, G.; Ergun, A.; Shukla, S.A.; Campos, B.; Hanna, J.; Ghosh, P.; Quayle, S.N.; Rai, K.; Colla, S.; Ying, H.; et al. microRNA regulatory network inference identifies miR-34a as a novel regulator of TGF-β signaling in glioblastoma. Cancer Discov. 2012, 2, 736–749. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.J.; Zhang, H.; Xing, J. Coupled reversible and irreversible bistable switches underlying TGF-β-induced epithelial to mesenchymal transition. Biophys. J. 2013, 105, 1079–1089. [Google Scholar] [CrossRef] [PubMed]
- Gregory, P.A.; Bracken, C.P.; Smith, E.; Bert, A.G.; Wright, J.A.; Roslan, S.; Morris, M.; Wyatt, L.; Farshid, G.; Lim, Y.Y.; et al. An autocrine TGF-β/ZEB/miR-200 signaling network regulates establishment and maintenance of epithelial-mesenchymal transition. Mol. Biol. Cell 2011, 22, 1686–1698. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.S.; Armenteros-Monterroso, E.; East, P.; Chakravorty, P.; Matthews, N.; Winslow, M.M.; Downward, J. HMGA2 functions as a competing endogenous RNA to promote lung cancer progression. Nature 2014, 505, 212–217. [Google Scholar] [CrossRef]
- Liao, Y.C.; Wang, Y.S.; Guo, Y.C.; Lin, W.L.; Chang, M.H.; Juo, S.H. Let-7g improves multiple endothelial functions through targeting transforming growth factor-β and SIRT-1 signaling. J. Am. Coll. Cardiol. 2014, 63, 1685–1694. [Google Scholar] [CrossRef]
- Han, X.; Yan, S.; Weijie, Z.; Feng, W.; Liuxing, W.; Mengquan, L.; Qingxia, F. Critical role of miR-10b in transforming growth factor-β1-induced epithelial-mesenchymal transition in breast cancer. Cancer Gene Ther. 2014, 21, 60–67. [Google Scholar] [CrossRef]
- Ma, L.; Reinhardt, F.; Pan, E.; Soutschek, J.; Bhat, B.; Marcusson, E.G.; Teruya-Feldstein, J.; Bell, G.W.; Weinberg, R.A. Therapeutic silencing of miR-10b inhibits metastasis in a mouse mammary tumor model. Nat. Biotechnol. 2010, 28, 341–347. [Google Scholar] [CrossRef]
- Song, L.; Liu, L.; Wu, Z.; Li, Y.; Ying, Z.; Lin, C.; Wu, J.; Hu, B.; Cheng, S.Y.; Li, M.; et al. TGF-β induces miR-182 to sustain NF-kappaB activation in glioma subsets. J. Clin. Investig. 2012, 122, 3563–3578. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.A.; Sossey-Alaoui, K.; Thompson, C.L.; Danielpour, D.; Schiemann, W.P. TGF-β upregulates miR-181a expression to promote breast cancer metastasis. J. Clin. Investig. 2013, 123, 150–163. [Google Scholar] [CrossRef] [PubMed]
- Parikh, A.; Lee, C.; Joseph, P.; Marchini, S.; Baccarini, A.; Kolev, V.; Romualdi, C.; Fruscio, R.; Shah, H.; Wang, F.; et al. microRNA-181a has a critical role in ovarian cancer progression through the regulation of the epithelial-mesenchymal transition. Nat. Commun. 2014, 5, 2977. [Google Scholar] [CrossRef] [PubMed]
- Bu, P.; Wang, L.; Chen, K.Y.; Rakhilin, N.; Sun, J.; Closa, A.; Tung, K.L.; King, S.; Varanko, A.K.; Xu, Y.; et al. miR-1269 promotes metastasis and forms a positive feedback loop with TGF-β. Nat. Commun. 2015, 6, 6879. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Ooi, L.L.; Hui, K.M. MicroRNA-216a/217-induced epithelial-mesenchymal transition targets PTEN and SMAD7 to promote drug resistance and recurrence of liver cancer. Hepatology 2013, 58, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Kong, W.; Yang, H.; He, L.; Zhao, J.J.; Coppola, D.; Dalton, W.S.; Cheng, J.Q. MicroRNA-155 is regulated by the transforming growth factor β/SMAD pathway and contributes to epithelial cell plasticity by targeting RhoA. Mol. Cell. Biol. 2008, 28, 6773–6784. [Google Scholar] [CrossRef]
- Yin, K.; Yin, W.; Wang, Y.; Zhou, L.; Liu, Y.; Yang, G.; Wang, J.; Lu, J. MiR-206 suppresses epithelial mesenchymal transition by targeting TGF-β signaling in estrogen receptor positive breast cancer cells. Oncotarget 2016, 7, 24537–24548. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.I. MicroRNA Control of TGF-β Signaling. Int. J. Mol. Sci. 2018, 19, 1901. [Google Scholar] [CrossRef]
- Zaravinos, A. The regulatory role of MicroRNAs in EMT and cancer. J. Oncol. 2015, 2015, 865816. [Google Scholar] [CrossRef]
- Romano, G.; Kwong, L.N. miRNAs, melanoma and microenvironment: An intricate network. Int. J. Mol. Sci. 2017, 18, 2354. [Google Scholar] [CrossRef]
- Musavi Shenas, M.H.; Eghbal-Fard, S.; Mehrisofiani, V.; Yazdani, N.A.; Farzam, O.R.; Marofi, F.; Yousefi, M. MicroRNAs and signaling networks involved in epithelial-mesenchymal transition. J. Cell Physiol. 2019, 234, 5775–5785. [Google Scholar] [CrossRef]
- Lin, C.W.; Kao, S.H.; Yang, P.C. The miRNAs and epithelial-mesenchymal transition in cancers. Curr. Pharm. Des. 2014, 20, 5309–5318. [Google Scholar] [CrossRef]
- Shields, E.J.; Petracovici, A.F.; Bonasio, R. lncRedibly versatile: Biochemical and biological functions of long noncoding RNAs. Biochem. J. 2019, 476, 1083–1104. [Google Scholar] [CrossRef]
- Gutschner, T.; Diederichs, S. The hallmarks of cancer: A long non-coding RNA point of view. RNA Biol. 2012, 9, 703–719. [Google Scholar] [CrossRef]
- Schmitt, A.M.; Chang, H.Y. Long noncoding RNAs in cancer pathways. Cancer Cell 2016, 29, 452–463. [Google Scholar] [CrossRef]
- Lu, H.; Yang, D.; Zhang, L.; Lu, S.; Ye, J.; Li, M.; Hu, W. Linc-pint inhibits early stage pancreatic ductal adenocarcinoma growth through TGF-β pathway activation. Oncol. Lett. 2019, 17, 4633–4639. [Google Scholar] [CrossRef]
- Shin, T.J.; Lee, K.H.; Cho, H.M.; Cho, J.Y. Concise approach for screening long non-coding RNAs functionally linked to human breast cancer associated genes. Exp. Mol. Pathol. 2019, 108, 89–96. [Google Scholar] [CrossRef]
- Shi, T.; Gao, G.; Cao, Y. Long noncoding RNAs as novel biomarkers have a promising future in cancer diagnostics. Dis. Markers 2016, 2016, 9085195. [Google Scholar] [CrossRef]
- Sakai, S.; Ohhata, T.; Kitagawa, K.; Uchida, C.; Aoshima, T.; Niida, H.; Suzuki, T.; Inoue, Y.; Miyazawa, K.; Kitagawa, M. Long noncoding RNA ELIT-1 acts as a SMAD3 cofactor to facilitate TGF-β/SMAD signaling and promote epithelial-mesenchymal transition. Cancer Res. 2019. [Google Scholar]
- He, L.; Thomson, J.M.; Hemann, M.T.; Hernando-Monge, E.; Mu, D.; Goodson, S.; Powers, S.; Cordon-Cardo, C.; Lowe, S.W.; Hannon, G.J.; et al. A microRNA polycistron as a potential human oncogene. Nature 2005, 435, 828–833. [Google Scholar] [CrossRef]
- Li, W.; Kang, Y. A new Lnc in metastasis: Long noncoding RNA mediates the prometastatic functions of TGF-β. Cancer Cell 2014, 25, 557–559. [Google Scholar] [CrossRef]
- Yuan, J.H.; Yang, F.; Wang, F.; Ma, J.Z.; Guo, Y.J.; Tao, Q.F.; Liu, F.; Pan, W.; Wang, T.T.; Zhou, C.C.; et al. A long noncoding RNA activated by TGF-β promotes the invasion-metastasis cascade in hepatocellular carcinoma. Cancer Cell 2014, 25, 666–681. [Google Scholar] [CrossRef]
- Grelet, S.; Link, L.A.; Howley, B.; Obellianne, C.; Palanisamy, V.; Gangaraju, V.K.; Diehl, J.A.; Howe, P.H. A regulated PNUTS mRNA to lncRNA splice switch mediates EMT and tumour progression. Nat. Cell Biol. 2017, 19, 1105–1115. [Google Scholar] [CrossRef] [Green Version]
- Mondal, T.; Subhash, S.; Vaid, R.; Enroth, S.; Uday, S.; Reinius, B.; Mitra, S.; Mohammed, A.; James, A.R.; Hoberg, E.; et al. MEG3 long noncoding RNA regulates the TGF-β pathway genes through formation of RNA-DNA triplex structures. Nat. Commun. 2015, 6, 7743. [Google Scholar] [CrossRef]
- Mitra, R.; Chen, X.; Greenawalt, E.J.; Maulik, U.; Jiang, W.; Zhao, Z.; Eischen, C.M. Decoding critical long non-coding RNA in ovarian cancer epithelial-to-mesenchymal transition. Nat. Commun. 2017, 8, 1604. [Google Scholar] [CrossRef]
- Yan, X.; Zhang, D.; Wu, W.; Wu, S.; Qian, J.; Hao, Y.; Yan, F.; Zhu, P.; Wu, J.; Huang, G.; et al. Mesenchymal stem cells promote hepatocarcinogenesis via lncRNA-MUF interaction with ANXA2 and miR-34a. Cancer Res. 2017, 77, 6704–6716. [Google Scholar] [CrossRef]
- Zhang, J.; Han, C.; Ungerleider, N.; Chen, W.; Song, K.; Wang, Y.; Kwon, H.; Ma, W.; Wu, T. A transforming growth factor-β and H19 signaling axis in tumor-initiating hepatocytes that regulates hepatic carcinogenesis. Hepatology 2018, 69, 1549–1563. [Google Scholar] [CrossRef]
- Matouk, I.J.; Raveh, E.; Abu-lail, R.; Mezan, S.; Gilon, M.; Gershtain, E.; Birman, T.; Gallula, J.; Schneider, T.; Barkali, M.; et al. Oncofetal H19 RNA promotes tumor metastasis. Biochim. Biophys. Acta 2014, 1843, 1414–1426. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Ye, X.L.; Xu, J.; Cao, M.G.; Fang, Z.Y.; Li, L.Y.; Guan, G.H.; Liu, Q.; Qian, Y.H.; Xie, D. The lncRNA H19 mediates breast cancer cell plasticity during EMT and MET plasticity by differentially sponging miR-200b/c and let-7b. Sci. Signal 2017, 10, eaak9557. [Google Scholar] [CrossRef] [Green Version]
- Luo, M.; Li, Z.; Wang, W.; Zeng, Y.; Liu, Z.; Qiu, J. Long non-coding RNA H19 increases bladder cancer metastasis by associating with EZH2 and inhibiting E-cadherin expression. Cancer Lett. 2013, 333, 213–221. [Google Scholar] [CrossRef]
- Rodriguez-Mateo, C.; Torres, B.; Gutierrez, G.; Pintor-Toro, J.A. Downregulation of Lnc-Spry1 mediates TGF-β-induced epithelial-mesenchymal transition by transcriptional and posttranscriptional regulatory mechanisms. Cell Death Differ. 2017, 24, 785–797. [Google Scholar] [CrossRef]
- Song, Y.X.; Sun, J.X.; Zhao, J.H.; Yang, Y.C.; Shi, J.X.; Wu, Z.H.; Chen, X.W.; Gao, P.; Miao, Z.F.; Wang, Z.N. Non-coding RNAs participate in the regulatory network of CLDN4 via ceRNA mediated miRNA evasion. Nat. Commun. 2017, 8, 289. [Google Scholar] [CrossRef]
- Battistelli, C.; Cicchini, C.; Santangelo, L.; Tramontano, A.; Grassi, L.; Gonzalez, F.J.; de Nonno, V.; Grassi, G.; Amicone, L.; Tripodi, M. The snail repressor recruits EZH2 to specific genomic sites through the enrollment of the lncRNA HOTAIR in epithelial-to-mesenchymal transition. Oncogene 2017, 36, 942–955. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, J.; Jia, S.; Wang, Y.; Kang, Y.; Zhang, W. Down-regulation of lncRNA-ATB inhibits epithelial-mesenchymal transition of breast cancer cells by increasing miR-141-3p expression. Biochem. Cell Biol. 2018, 97, 85–90. [Google Scholar] [CrossRef]
- Li, G.Y.; Wang, W.; Sun, J.Y.; Xin, B.; Zhang, X.; Wang, T.; Zhang, Q.F.; Yao, L.B.; Han, H.; Fan, D.M.; et al. Long non-coding RNAs AC026904.1 and UCA1: A “one-two punch” for TGF-β-induced SNAI2 activation and epithelial-mesenchymal transition in breast cancer. Theranostics 2018, 8, 2846–2861. [Google Scholar] [CrossRef]
- Wu, W.; Chen, F.; Cui, X.; Yang, L.; Chen, J.; Zhao, J.; Huang, D.; Liu, J.; Yang, L.; Zeng, J.; et al. LncRNA NKILA suppresses TGF-β-induced epithelial-mesenchymal transition by blocking NF-kappaB signaling in breast cancer. Int. J. Cancer 2018, 143, 2213–2224. [Google Scholar] [CrossRef]
- Li, Z.; Dong, M.; Fan, D.; Hou, P.; Li, H.; Liu, L.; Lin, C.; Liu, J.; Su, L.; Wu, L.; et al. LncRNA ANCR down-regulation promotes TGF-β-induced EMT and metastasis in breast cancer. Oncotarget 2017, 8, 67329–67343. [Google Scholar] [CrossRef]
- Richards, E.J.; Zhang, G.; Li, Z.P.; Permuth-Wey, J.; Challa, S.; Li, Y.; Kong, W.; Dan, S.; Bui, M.M.; Coppola, D.; et al. Long non-coding RNAs (LncRNA) regulated by transforming growth factor (TGF) β: LncRNA-hit-mediated TGF-β-induced epithelial to mesenchymal transition in mammary epithelia. J. Biol. Chem. 2015, 290, 6857–6867. [Google Scholar] [CrossRef]
- Pan, Y.; Li, C.; Chen, J.; Zhang, K.; Chu, X.; Wang, R.; Chen, L. The emerging roles of long noncoding RNA ROR (lincRNA-ROR) and its possible mechanisms in human cancers. Cell Physiol. Biochem. 2016, 40, 219–229. [Google Scholar] [CrossRef]
- Eades, G.; Wolfson, B.; Zhang, Y.; Li, Q.; Yao, Y.; Zhou, Q. lincRNA-RoR and miR-145 regulate invasion in triple-negative breast cancer via targeting ARF6. Mol. Cancer Res. 2015, 13, 330–338. [Google Scholar] [CrossRef]
- Fang, Y.; Wang, J.; Wu, F.; Song, Y.; Zhao, S.; Zhang, Q. Long non-coding RNA HOXA-AS2 promotes proliferation and invasion of breast cancer by acting as a miR-520c-3p sponge. Oncotarget 2017, 8, 46090–46103. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.J.; Li, Y.; Wu, Y.Z.; Wang, Y.; Nian, W.Q.; Wang, L.L.; Li, L.C.; Luo, H.L.; Wang, D.L. Long non-coding RNA CCAT2 promotes the breast cancer growth and metastasis by regulating TGF-β signaling pathway. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 706–714. [Google Scholar]
- Zhang, C.Y.; Yu, M.S.; Li, X.; Zhang, Z.; Han, C.R.; Yan, B. Overexpression of long non-coding RNA MEG3 suppresses breast cancer cell proliferation, invasion, and angiogenesis through AKT pathway. Tumour Biol. 2017, 39, 1010428317701311. [Google Scholar] [CrossRef]
- Zhang, W.; Shi, S.; Jiang, J.; Li, X.; Lu, H.; Ren, F. LncRNA MEG3 inhibits cell epithelial-mesenchymal transition by sponging miR-421 targeting E-cadherin in breast cancer. Biomed. Pharmacother. 2017, 91, 312–319. [Google Scholar] [CrossRef]
- Fu, M.; Huang, Z.; Zang, X.; Pan, L.; Liang, W.; Chen, J.; Qian, H.; Xu, W.; Jiang, P.; Zhang, X. Long noncoding RNA LINC00978 promotes cancer growth and acts as a diagnostic biomarker in gastric cancer. Cell Prolif. 2018, 51, e12425. [Google Scholar] [CrossRef]
- Zuo, Z.K.; Gong, Y.; Chen, X.H.; Ye, F.; Yin, Z.M.; Gong, Q.N.; Huang, J.S. TGF-β1-Induced LncRNA UCA1 upregulation promotes gastric cancer invasion and migration. DNA Cell Biol. 2017, 36, 159–167. [Google Scholar] [CrossRef]
- Saito, T.; Kurashige, J.; Nambara, S.; Komatsu, H.; Hirata, H.; Ueda, M.; Sakimura, S.; Uchi, R.; Takano, Y.; Shinden, Y.; et al. A long non-coding RNA activated by transforming growth factor-β is an independent prognostic marker of gastric cancer. Ann. Surg. Oncol. 2015, 22 (Suppl. 3), S915–S922. [Google Scholar] [CrossRef]
- Chen, Y.; Wei, G.; Xia, H.; Tang, Q.; Bi, F. Long noncoding RNAATB promotes cell proliferation, migration and invasion in gastric cancer. Mol. Med. Rep. 2018, 17, 1940–1946. [Google Scholar]
- Lei, K.; Liang, X.; Gao, Y.; Xu, B.; Xu, Y.; Li, Y.; Tao, Y.; Shi, W.; Liu, J. Lnc-ATB contributes to gastric cancer growth through a MiR-141-3p/TGF-β2 feedback loop. Biochem. Biophys. Res. Commun. 2017, 484, 514–521. [Google Scholar] [CrossRef]
- Zhang, Q.; Chen, B.; Liu, P.; Yang, J. XIST promotes gastric cancer (GC) progression through TGF-β1 via targeting miR-185. J. Cell Biochem. 2018, 119, 2787–2796. [Google Scholar] [CrossRef]
- Han, Q.; Zhang, W.; Meng, J.; Ma, L.; Li, A. LncRNA-LET inhibits cell viability, migration and EMT while induces apoptosis by up-regulation of TIMP2 in human granulosa-like tumor cell line KGN. Biomed. Pharmacother. 2018, 100, 250–256. [Google Scholar] [CrossRef]
- Li, J.; Huang, Y.; Deng, X.; Luo, M.; Wang, X.; Hu, H.; Liu, C.; Zhong, M. Long noncoding RNA H19 promotes transforming growth factor-β-induced epithelial-mesenchymal transition by acting as a competing endogenous RNA of miR-370-3p in ovarian cancer cells. Onco. Targets Ther. 2018, 11, 427–440. [Google Scholar] [CrossRef]
- Li, Q.; Zhang, C.; Chen, R.; Xiong, H.; Qiu, F.; Liu, S.; Zhang, M.; Wang, F.; Wang, Y.; Zhou, X.; et al. Disrupting MALAT1/miR-200c sponge decreases invasion and migration in endometrioid endometrial carcinoma. Cancer Lett. 2016, 383, 28–40. [Google Scholar] [CrossRef]
- Hu, J.; Zhang, L.; Mei, Z.; Jiang, Y.; Yi, Y.; Liu, L.; Meng, Y.; Zhou, L.; Zeng, J.; Wu, H.; et al. Interaction of E3 ubiquitin ligase MARCH7 with long noncoding RNA MALAT1 and autophagy-related protein ATG7 promotes autophagy and invasion in ovarian cancer. Cell Physiol. Biochem. 2018, 47, 654–666. [Google Scholar] [CrossRef]
- Ma, J.; Xue, M. LINK-A lncRNA promotes migration and invasion of ovarian carcinoma cells by activating TGF-β pathway. Biosci. Rep. 2018, 38, BSR20180936. [Google Scholar] [CrossRef]
- Cao, C.; Zhang, T.; Zhang, D.; Xie, L.; Zou, X.; Lei, L.; Wu, D.; Liu, L. The long non-coding RNA, SNHG6-003, functions as a competing endogenous RNA to promote the progression of hepatocellular carcinoma. Oncogene 2017, 36, 1112–1122. [Google Scholar] [CrossRef]
- Liang, H.; Zhao, X.; Wang, C.; Sun, J.; Chen, Y.; Wang, G.; Fang, L.; Yang, R.; Yu, M.; Gu, Y.; et al. Systematic analyses reveal long non-coding RNA (PTAF)-mediated promotion of EMT and invasion-metastasis in serous ovarian cancer. Mol. Cancer 2018, 17, 96. [Google Scholar] [CrossRef]
- Zhuang, J.; Lu, Q.; Shen, B.; Huang, X.; Shen, L.; Zheng, X.; Huang, R.; Yan, J.; Guo, H. TGF-β1 secreted by cancer-associated fibroblasts induces epithelial-mesenchymal transition of bladder cancer cells through lncRNA-ZEB2NAT. Sci. Rep. 2015, 5, 11924. [Google Scholar] [CrossRef]
- Fan, Y.; Shen, B.; Tan, M.; Mu, X.; Qin, Y.; Zhang, F.; Liu, Y. TGF-β-induced upregulation of malat1 promotes bladder cancer metastasis by associating with suz12. Clin. Cancer Res. 2014, 20, 1531–1541. [Google Scholar] [CrossRef]
- Gou, L.; Liu, M.; Xia, J.; Wan, Q.; Jiang, Y.; Sun, S.; Tang, M.; Zhou, L.; He, T.; Zhang, Y. BMP9 promotes the proliferation and migration of bladder cancer cells through up-regulating lncRNA UCA1. Int. J. Mol. Sci. 2018, 19, 1116. [Google Scholar] [CrossRef]
- Zhai, X.; Xu, W. Long noncoding RNA ATB promotes proliferation, migration, and invasion in bladder cancer by suppressing MicroRNA-126. Oncol. Res. 2018, 26, 1063–1072. [Google Scholar] [CrossRef]
- Jin, Y.; Cui, Z.; Li, X.; Jin, X.; Peng, J. Upregulation of long non-coding RNA PlncRNA-1 promotes proliferation and induces epithelial-mesenchymal transition in prostate cancer. Oncotarget 2017, 8, 26090–26099. [Google Scholar] [CrossRef]
- Wang, S.H.; Zhang, M.D.; Wu, X.C.; Weng, M.Z.; Zhou, D.; Quan, Z.W. Overexpression of LncRNA-ROR predicts a poor outcome in gallbladder cancer patients and promotes the tumor cells proliferation, migration, and invasion. Tumour Biol. 2016, 37, 12867–12875. [Google Scholar] [CrossRef]
- Zhao, B.; Lu, Y.L.; Yang, Y.; Hu, L.B.; Bai, Y.; Li, R.Q.; Zhang, G.Y.; Li, J.; Bi, C.W.; Yang, L.B.; et al. Overexpression of lncRNA ANRIL promoted the proliferation and migration of prostate cancer cells via regulating let-7a/TGF-β1/ SMAD signaling pathway. Cancer Biomark. 2018, 21, 613–620. [Google Scholar] [CrossRef]
- Xu, S.; Yi, X.M.; Tang, C.P.; Ge, J.P.; Zhang, Z.Y.; Zhou, W.Q. Long non-coding RNA ATB promotes growth and epithelial-mesenchymal transition and predicts poor prognosis in human prostate carcinoma. Oncol. Rep. 2016, 36, 10–22. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Wang, G.; Gao, Y.; Zhao, C.; Li, X.; Zhang, F.; Jiang, C.; Wu, B. Lnc-SNHG1 activates the TGFBR2/SMAD3 and RAB11A/Wnt/β-catenin pathway by sponging MiR-302/372/373/520 in invasive pituitary tumors. Cell Physiol. Biochem. 2018, 48, 1291–1303. [Google Scholar] [CrossRef]
- Li, Z.; Liu, H.; Zhong, Q.; Wu, J.; Tang, Z. LncRNA UCA1 is necessary for TGF-β-induced epithelial-mesenchymal transition and stemness via acting as a ceRNA for SLUG in glioma cells. FEBS Open Bio. 2018, 8, 1855–1865. [Google Scholar] [CrossRef]
- Zhang, C.; Hao, Y.; Wang, Y.; Xu, J.; Teng, Y.; Yang, X. TGF-β/SMAD4-regulated LncRNA-LINP1 inhibits epithelial-mesenchymal transition in lung cancer. Int. J. Biol. Sci. 2018, 14, 1715–1723. [Google Scholar] [CrossRef]
- Lu, Z.; Li, Y.; Che, Y.; Huang, J.; Sun, S.; Mao, S.; Lei, Y.; Li, N.; Sun, N.; He, J. The TGF-β-induced lncRNA TBILA promotes non-small cell lung cancer progression in vitro and in vivo via cis-regulating HGAL and activating S100A7/JAB1 signaling. Cancer Lett. 2018, 432, 156–168. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, G.; Cheng, Z.; Dai, L.; Jia, L.; Jing, X.; Wang, H.; Zhang, R.; Liu, M.; Jiang, T.; et al. Knockdown of LncRNA-XIST suppresses proliferation and TGF-β1-induced EMT in NSCLC through the notch-1 pathway by regulation of miR-137. Genet. Test Mol. Biomark. 2018, 22, 333–342. [Google Scholar] [CrossRef]
- Li, C.; Wan, L.; Liu, Z.; Xu, G.; Wang, S.; Su, Z.; Zhang, Y.; Zhang, C.; Liu, X.; Lei, Z.; et al. Long non-coding RNA XIST promotes TGF-β-induced epithelial-mesenchymal transition by regulating miR-367/141-ZEB2 axis in non-small-cell lung cancer. Cancer Lett. 2018, 418, 185–195. [Google Scholar] [CrossRef]
- Kawasaki, N.; Miwa, T.; Hokari, S.; Sakurai, T.; Ohmori, K.; Miyauchi, K.; Miyazono, K.; Koinuma, D. Long noncoding RNA NORAD regulates transforming growth factor-β signaling and epithelial-to-mesenchymal transition-like phenotype. Cancer Sci. 2018, 109, 2211–2220. [Google Scholar] [CrossRef]
- Cao, Y.; Luo, X.; Ding, X.; Cui, S.; Guo, C. LncRNA ATB promotes proliferation and metastasis in A549 cells by down-regulation of microRNA-494. J. Cell Biochem. 2018, 119, 6935–6942. [Google Scholar] [CrossRef]
- Lu, W.; Zhang, H.; Niu, Y.; Wu, Y.; Sun, W.; Li, H.; Kong, J.; Ding, K.; Shen, H.M.; Wu, H.; et al. Long non-coding RNA linc00673 regulated non-small cell lung cancer proliferation, migration, invasion and epithelial mesenchymal transition by sponging miR-150-5p. Mol. Cancer 2017, 16, 118. [Google Scholar] [CrossRef]
- Lu, Z.; Li, Y.; Wang, J.; Che, Y.; Sun, S.; Huang, J.; Chen, Z.; He, J. Long non-coding RNA NKILA inhibits migration and invasion of non-small cell lung cancer via NF-kappaB/Snail pathway. J. Exp. Clin. Cancer Res. 2017, 36, 54. [Google Scholar] [CrossRef]
- Terashima, M.; Tange, S.; Ishimura, A.; Suzuki, T. MEG3 long noncoding RNA contributes to the epigenetic regulation of epithelial-mesenchymal transition in lung cancer cell lines. J. Biol. Chem. 2017, 292, 82–99. [Google Scholar] [CrossRef]
- Wu, D.M.; Deng, S.H.; Liu, T.; Han, R.; Zhang, T.; Xu, Y. TGF-β-mediated exosomal lnc-MMP2-2 regulates migration and invasion of lung cancer cells to the vasculature by promoting MMP2 expression. Cancer Med. 2018, 7, 5118–5129. [Google Scholar] [CrossRef]
- Wang, S.; Lan, F.; Xia, Y. lncRA ANCR inhibits non-small cell lung cancer cell migration and invasion by inactivating TGF-β pathway. Med. Sci. Monit. 2018, 24, 6002–6009. [Google Scholar] [CrossRef]
- Hao, Y.; Yang, X.; Zhang, D.; Luo, J.; Chen, R. Long noncoding RNA LINC01186, regulated by TGF-β/SMAD3, inhibits migration and invasion through epithelial-mesenchymal-transition in lung cancer. Gene 2017, 608, 1–12. [Google Scholar] [CrossRef]
- Zhang, Y.; He, R.Q.; Dang, Y.W.; Zhang, X.L.; Wang, X.; Huang, S.N.; Huang, W.T.; Jiang, M.T.; Gan, X.N.; Xie, Y.; et al. Comprehensive analysis of the long noncoding RNA HOXA11-AS gene interaction regulatory network in NSCLC cells. Cancer Cell Int. 2016, 16, 89. [Google Scholar] [CrossRef]
- Li, Y.; Liu, G.; Li, X.; Dong, H.; Xiao, W.; Lu, S. Long non-coding RNA SBF2-AS1 promotes hepatocellular carcinoma progression through regulation of miR-140-5p-TGFBR1 pathway. Biochem. Biophys. Res. Commun. 2018, 503, 2826–2832. [Google Scholar] [CrossRef]
- Tang, J.; Zhuo, H.; Zhang, X.; Jiang, R.; Ji, J.; Deng, L.; Qian, X.; Zhang, F.; Sun, B. A novel biomarker Linc00974 interacting with KRT19 promotes proliferation and metastasis in hepatocellular carcinoma. Cell Death Dis. 2014, 5, e1549. [Google Scholar] [CrossRef]
- Zhang, X.; Feng, W.; Zhang, J.; Ge, L.; Zhang, Y.; Jiang, X.; Peng, W.; Wang, D.; Gong, A.; Xu, M. Long noncoding RNA PVT1 promotes epithelialmesenchymal transition via the TGF-β/SMAD pathway in pancreatic cancer cells. Oncol. Rep. 2018, 40, 1093–1102. [Google Scholar]
- Terashima, M.; Ishimura, A.; Wanna-Udom, S.; Suzuki, T. MEG8 long noncoding RNA contributes to epigenetic progression of the epithelial-mesenchymal transition of lung and pancreatic cancer cells. J. Biol. Chem. 2018, 293, 18016–18030. [Google Scholar] [CrossRef] [Green Version]
- Qin, C.F.; Zhao, F.L. Long non-coding RNA TUG1 can promote proliferation and migration of pancreatic cancer via EMT pathway. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 2377–2384. [Google Scholar]
- Ottaviani, S.; Stebbing, J.; Frampton, A.E.; Zagorac, S.; Krell, J.; de Giorgio, A.; Trabulo, S.M.; Nguyen, V.T.M.; Magnani, L.; Feng, H.; et al. TGF-β induces miR-100 and miR-125b but blocks let-7a through LIN28B controlling PDAC progression. Nat. Commun. 2018, 9, 1845. [Google Scholar] [CrossRef]
- Qu, S.; Yang, X.; Song, W.; Sun, W.; Li, X.; Wang, J.; Zhong, Y.; Shang, R.; Ruan, B.; Zhang, Z.; et al. Downregulation of lncRNA-ATB correlates with clinical progression and unfavorable prognosis in pancreatic cancer. Tumour Biol. 2016, 37, 3933–3938. [Google Scholar] [CrossRef]
- Liu, Y.; Qian, J.; Li, X.; Chen, W.; Xu, A.; Zhao, K.; Hua, Y.; Huang, Z.; Zhang, J.; Liang, C.; et al. Long noncoding RNA BX357664 regulates cell proliferation and epithelial-to-mesenchymal transition via inhibition of TGF-β1/p38/HSP27 signaling in renal cell carcinoma. Oncotarget 2016, 7, 81410–81422. [Google Scholar]
- Xiong, J.; Liu, Y.; Jiang, L.; Zeng, Y.; Tang, W. High expression of long non-coding RNA lncRNA-ATB is correlated with metastases and promotes cell migration and invasion in renal cell carcinoma. Jpn. J. Clin. Oncol. 2016, 46, 378–384. [Google Scholar] [CrossRef] [Green Version]
- Yue, B.; Qiu, S.; Zhao, S.; Liu, C.; Zhang, D.; Yu, F.; Peng, Z.; Yan, D. LncRNA-ATB mediated E-cadherin repression promotes the progression of colon cancer and predicts poor prognosis. J. Gastroenterol. Hepatol. 2016, 31, 595–603. [Google Scholar] [CrossRef]
- Iguchi, T.; Uchi, R.; Nambara, S.; Saito, T.; Komatsu, H.; Hirata, H.; Ueda, M.; Sakimura, S.; Takano, Y.; Kurashige, J.; et al. A long noncoding RNA, lncRNA-ATB, is involved in the progression and prognosis of colorectal cancer. Anticancer Res. 2015, 35, 1385–1388. [Google Scholar]
- Kong, J.; Sun, W.; Li, C.; Wan, L.; Wang, S.; Wu, Y.; Xu, E.; Zhang, H.; Lai, M. Long non-coding RNA LINC01133 inhibits epithelial-mesenchymal transition and metastasis in colorectal cancer by interacting with SRSF6. Cancer Lett. 2016, 380, 476–484. [Google Scholar] [CrossRef]
- Zhang, S.Z.; Cai, L.; Li, B. MEG3 long non-coding RNA prevents cell growth and metastasis of osteosarcoma. Bratisl Lek Listy 2017, 118, 632–636. [Google Scholar] [CrossRef]
- Huo, Y.; Li, Q.; Wang, X.; Jiao, X.; Zheng, J.; Li, Z.; Pan, X. MALAT1 predicts poor survival in osteosarcoma patients and promotes cell metastasis through associating with EZH2. Oncotarget 2017, 8, 46993–47006. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; Zhao, F.; Zhang, Z.; Sun, F.; Wang, M. Long noncoding RNA SNHG7 promotes the tumor growth and epithelial-to-mesenchymal transition via regulation of mir-34a signals in osteosarcoma. Cancer Biother. Radiopharm. 2018, 33, 365–372. [Google Scholar] [CrossRef]
- Zhang, R.; Hardin, H.; Huang, W.; Chen, J.; Asioli, S.; Righi, A.; Maletta, F.; Sapino, A.; Lloyd, R.V. MALAT1 long non-coding RNA expression in thyroid tissues: Analysis by in situ hybridization and real-time PCR. Endocr. Pathol. 2017, 28, 7–12. [Google Scholar] [CrossRef]
- Zhang, R.; Hardin, H.; Huang, W.; Buehler, D.; Lloyd, R.V. Long non-coding RNA linc-ROR is upregulated in papillary thyroid carcinoma. Endocr. Pathol. 2018, 29, 1–8. [Google Scholar] [CrossRef]
- Zhou, H.; Sun, Z.; Li, S.; Wang, X.; Zhou, X. LncRNA SPRY4-IT was concerned with the poor prognosis and contributed to the progression of thyroid cancer. Cancer Gene Ther. 2018, 25, 39–46. [Google Scholar] [CrossRef]
- Zhao, J.J.; Hao, S.; Wang, L.L.; Hu, C.Y.; Zhang, S.; Guo, L.J.; Zhang, G.; Gao, B.; Jiang, Y.; Tian, W.G.; et al. Long non-coding RNA ANRIL promotes the invasion and metastasis of thyroid cancer cells through TGF-β/SMAD signaling pathway. Oncotarget 2016, 7, 57903–57918. [Google Scholar] [CrossRef]
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging biological principles of metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef]
- Chiang, A.C.; Massague, J. Molecular basis of metastasis. N. Engl. J. Med. 2008, 359, 2814–2823. [Google Scholar] [CrossRef]
- Jung, H.Y.; Fattet, L.; Yang, J. Molecular pathways: Linking tumor microenvironment to epithelial-mesenchymal transition in metastasis. Clin. Cancer Res. 2015, 21, 962–968. [Google Scholar] [CrossRef]
- Nguyen, D.X.; Bos, P.D.; Massague, J. Metastasis: From dissemination to organ-specific colonization. Nat. Rev. Cancer 2009, 9, 274–284. [Google Scholar] [CrossRef]
- Roodman, G.D. Biology of osteoclast activation in cancer. J. Clin. Oncol. 2001, 19, 3562–3571. [Google Scholar] [CrossRef]
- Yin, J.J.; Selander, K.; Chirgwin, J.M.; Dallas, M.; Grubbs, B.G.; Wieser, R.; Massague, J.; Mundy, G.R.; Guise, T.A. TGF-β signaling blockade inhibits PTHrP secretion by breast cancer cells and bone metastases development. J. Clin. Investig. 1999, 103, 197–206. [Google Scholar] [CrossRef]
- Kang, Y.; Siegel, P.M.; Shu, W.; Drobnjak, M.; Kakonen, S.M.; Cordon-Cardo, C.; Guise, T.A.; Massague, J. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 2003, 3, 537–549. [Google Scholar] [CrossRef] [Green Version]
- Buijs, J.T.; Stayrook, K.R.; Guise, T.A. The role of TGF-β in bone metastasis: Novel therapeutic perspectives. Bonekey Rep. 2012, 1, 96. [Google Scholar] [CrossRef]
- Biswas, S.; Guix, M.; Rinehart, C.; Dugger, T.C.; Chytil, A.; Moses, H.L.; Freeman, M.L.; Arteaga, C.L. Inhibition of TGF-β with neutralizing antibodies prevents radiation-induced acceleration of metastatic cancer progression. J. Clin. Investig. 2007, 117, 1305–1313. [Google Scholar] [CrossRef]
- Padua, D.; Zhang, X.H.; Wang, Q.; Nadal, C.; Gerald, W.L.; Gomis, R.R.; Massague, J. TGF-β primes breast tumors for lung metastasis seeding through angiopoietin-like 4. Cell 2008, 133, 66–77. [Google Scholar] [CrossRef]
- Gupta, G.P.; Perk, J.; Acharyya, S.; de Candia, P.; Mittal, V.; Todorova-Manova, K.; Gerald, W.L.; Brogi, E.; Benezra, R.; Massague, J. ID genes mediate tumor reinitiation during breast cancer lung metastasis. Proc. Natl. Acad. Sci. USA 2007, 104, 19506–19511. [Google Scholar] [CrossRef]
- Zhuang, X.; Zhang, H.; Li, X.; Li, X.; Cong, M.; Peng, F.; Yu, J.; Zhang, X.; Yang, Q.; Hu, G. Differential effects on lung and bone metastasis of breast cancer by Wnt signalling inhibitor DKK1. Nat. Cell. Biol. 2017, 19, 1274–1285. [Google Scholar] [CrossRef]
- Jung, B.; Staudacher, J.J.; Beauchamp, D. Transforming growth factor β superfamily signaling in development of colorectal cancer. Gastroenterology 2017, 152, 36–52. [Google Scholar] [CrossRef]
- Steinbichler, T.B.; Dudas, J.; Riechelmann, H.; Skvortsova, II. The role of exosomes in cancer metastasis. Semin. Cancer Biol. 2017, 44, 170–181. [Google Scholar] [CrossRef]
- Carmeliet, P.; Jain, R.K. Angiogenesis in cancer and other diseases. Nature 2000, 407, 249–257. [Google Scholar] [CrossRef]
- Roberts, A.B.; Thompson, N.L.; Heine, U.; Flanders, C.; Sporn, M.B. Transforming growth factor-β: Possible roles in carcinogenesis. Br. J. Cancer 1988, 57, 594–600. [Google Scholar] [CrossRef]
- Goumans, M.J.; Valdimarsdottir, G.; Itoh, S.; Rosendahl, A.; Sideras, P.; Dijke, P.t. Balancing the activation state of the endothelium via two distinct TGF-β type I receptors. EMBO J. 2002, 21, 1743–1753. [Google Scholar] [CrossRef]
- Breier, G.; Blum, S.; Peli, J.; Groot, M.; Wild, C.; Risau, W.; Reichmann, E. Transforming growth factor-β and RAS regulate the VEGF/VEGF-receptor system during tumor angiogenesis. Int. J. Cancer 2002, 97, 142–148. [Google Scholar] [CrossRef]
- Sanchez-Elsner, T.; Botella, L.M.; Velasco, B.; Corbi, A.; Attisano, L.; Bernabeu, C. Synergistic cooperation between hypoxia and transforming growth factor-β pathways on human vascular endothelial growth factor gene expression. J. Biol. Chem. 2001, 276, 38527–38535. [Google Scholar] [CrossRef]
- Liu, D.; Li, L.; Zhang, X.X.; Wan, D.Y.; Xi, B.X.; Hu, Z.; Ding, W.C.; Zhu, D.; Wang, X.L.; Wang, W.; et al. SIX1 promotes tumor lymphangiogenesis by coordinating TGF-β signals that increase expression of VEGF-C. Cancer Res. 2014, 74, 5597–5607. [Google Scholar] [CrossRef]
- Sun, H.; Miao, C.; Liu, W.; Qiao, X.; Yang, W.; Li, L.; Li, C. TGF-β1/TβRII/SMAD3 signaling pathway promotes VEGF expression in oral squamous cell carcinoma tumor-associated macrophages. Biochem. Biophys. Res. Commun. 2018, 497, 583–590. [Google Scholar] [CrossRef]
- Groppa, E.; Brkic, S.; Bovo, E.; Reginato, S.; Sacchi, V.; di Maggio, N.; Muraro, M.G.; Calabrese, D.; Heberer, M.; Gianni-Barrera, R.; et al. VEGF dose regulates vascular stabilization through Semaphorin3A and the Neuropilin-1 + monocyte/TGF-β1 paracrine axis. EMBO Mol. Med. 2015, 7, 1366–1384. [Google Scholar] [CrossRef]
- Rak, J.; Mitsuhashi, Y.; Sheehan, C.; Tamir, A.; Viloria-Petit, A.; Filmus, J.; Mansour, S.J.; Ahn, N.G.; Kerbel, R.S. Oncogenes and tumor angiogenesis: Differential modes of vascular endothelial growth factor up-regulation in ras-transformed epithelial cells and fibroblasts. Cancer Res. 2000, 60, 490–498. [Google Scholar]
- Miyazono, K.; Ehata, S.; Koinuma, D. Tumor-promoting functions of transforming growth factor-β in progression of cancer. Ups. J. Med. Sci. 2012, 117, 143–152. [Google Scholar] [CrossRef]
- Chen, W.; Ten Dijke, P. Immunoregulation by members of the TGF-β superfamily. Nat. Rev. Immunol. 2016, 16, 723–740. [Google Scholar] [CrossRef]
- Gorelik, L.; Flavell, R.A. Abrogation of TGF-β signaling in T cells leads to spontaneous T cell differentiation and autoimmune disease. Immunity 2000, 12, 171–181. [Google Scholar] [CrossRef]
- Donkor, M.K.; Sarkar, A.; Savage, P.A.; Franklin, R.A.; Johnson, L.K.; Jungbluth, A.A.; Allison, J.P.; Li, M.O. T cell surveillance of oncogene-induced prostate cancer is impeded by T cell-derived TGF-β1 cytokine. Immunity 2011, 35, 123–134. [Google Scholar] [CrossRef]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E., III; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGF-β attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Canellas, A.; Hernando-Momblona, X.; et al. TGF-β drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef]
- Gorelik, L.; Flavell, R.A. Immune-mediated eradication of tumors through the blockade of transforming growth factor-β signaling in T cells. Nat. Med. 2001, 7, 1118–1122. [Google Scholar] [CrossRef]
- Thomas, D.A.; Massague, J. TGF-β directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 2005, 8, 369–380. [Google Scholar] [CrossRef]
- Fu, S.; Zhang, N.; Yopp, A.C.; Chen, D.; Mao, M.; Chen, D.; Zhang, H.; Ding, Y.; Bromberg, J.S. TGF-β induces Foxp3 + T-regulatory cells from CD4 + CD25 - precursors. Am. J. Transplant. 2004, 4, 1614–1627. [Google Scholar] [CrossRef]
- Worthington, J.J.; Kelly, A.; Smedley, C.; Bauche, D.; Campbell, S.; Marie, J.C.; Travis, M.A. Integrin alphavβ8-mediated TGF-β activation by effector regulatory T cells is essential for suppression of T-Cell-mediated inflammation. Immunity 2015, 42, 903–915. [Google Scholar] [CrossRef]
- Metelli, A.; Wu, B.X.; Fugle, C.W.; Rachidi, S.; Sun, S.; Zhang, Y.; Wu, J.; Tomlinson, S.; Howe, P.H.; Yang, Y.; et al. Surface expression of TGF-β docking receptor GARP promotes oncogenesis and immune tolerance in breast cancer. Cancer Res. 2016, 76, 7106–7117. [Google Scholar] [CrossRef]
- Imai, K.; Minamiya, Y.; Koyota, S.; Ito, M.; Saito, H.; Sato, Y.; Motoyama, S.; Sugiyama, T.; Ogawa, J. Inhibition of dendritic cell migration by transforming growth factor-β1 increases tumor-draining lymph node metastasis. J. Exp. Clin. Cancer Res. 2012, 31, 3. [Google Scholar] [CrossRef]
- Ito, M.; Minamiya, Y.; Kawai, H.; Saito, S.; Saito, H.; Nakagawa, T.; Imai, K.; Hirokawa, M.; Ogawa, J. Tumor-derived TGF-β-1 induces dendritic cell apoptosis in the sentinel lymph node. J. Immunol. 2006, 176, 5637–5643. [Google Scholar] [CrossRef]
- Kobie, J.J.; Wu, R.S.; Kurt, R.A.; Lou, S.; Adelman, M.K.; Whitesell, L.J.; Ramanathapuram, L.V.; Arteaga, C.L.; Akporiaye, E.T. Transforming growth factor β inhibits the antigen-presenting functions and antitumor activity of dendritic cell vaccines. Cancer Res. 2003, 63, 1860–1864. [Google Scholar]
- Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. The Yin-Yang of tumor-associated macrophages in neoplastic progression and immune surveillance. Immunol. Rev. 2008, 222, 155–161. [Google Scholar] [CrossRef]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of tumor-associated neutrophil phenotype by TGF-β: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef]
- Wang, Y.; Chu, J.; Yi, P.; Dong, W.; Saultz, J.; Wang, Y.; Wang, H.; Scoville, S.; Zhang, J.; Wu, L.C.; et al. SMAD4 promotes TGF-β-independent NK cell homeostasis and maturation and antitumor immunity. J. Clin. Investig. 2018, 128, 5123–5136. [Google Scholar] [CrossRef]
- Li, M.O.; Sanjabi, S.; Flavell, R.A. Transforming growth factor-β controls development, homeostasis, and tolerance of T cells by regulatory T cell-dependent and -independent mechanisms. Immunity 2006, 25, 455–471. [Google Scholar] [CrossRef]
- Akhurst, R.J. Targeting TGF-β signaling for therapeutic gain. Cold Spring Harb. Perspect. Biol. 2017, 9, a022301. [Google Scholar] [CrossRef]
- Colak, S.; Ten Dijke, P. Targeting TGF-β signaling in cancer. Trends Cancer 2017, 3, 56–71. [Google Scholar] [CrossRef]
- Zappa, C.; Mousa, S.A. Non-small cell lung cancer: Current treatment and future advances. Transl. Lung Cancer Res. 2016, 5, 288–300. [Google Scholar] [CrossRef]
- Jaschinski, F.; Rothhammer, T.; Jachimczak, P.; Seitz, C.; Schneider, A.; Schlingensiepen, K.H. The antisense oligonucleotide trabedersen (AP 12009) for the targeted inhibition of TGF-β2. Curr. Pharm. Biotechnol. 2011, 12, 2203–2213. [Google Scholar] [CrossRef]
- Fujiwara, Y.; Nokihara, H.; Yamada, Y.; Yamamoto, N.; Sunami, K.; Utsumi, H.; Asou, H.; Takahash, I.O.; Ogasawara, K.; Gueorguieva, I.; et al. Phase 1 study of galunisertib, a TGF-β receptor I kinase inhibitor, in Japanese patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2015, 76, 1143–1152. [Google Scholar] [CrossRef]
- Garrison, K.; Hahn, T.; Lee, W.C.; Ling, L.E.; Weinberg, A.D.; Akporiaye, E.T. The small molecule TGF-β signaling inhibitor SM16 synergizes with agonistic OX40 antibody to suppress established mammary tumors and reduce spontaneous metastasis. Cancer Immunol. Immunother. 2012, 61, 511–521. [Google Scholar] [CrossRef]
- Mohammad, K.S.; Javelaud, D.; Fournier, P.G.; Niewolna, M.; McKenna, C.R.; Peng, X.H.; Duong, V.; Dunn, L.K.; Mauviel, A.; Guise, T.A. TGF-β RI kinase inhibitor SD-208 reduces the development and progression of melanoma bone metastases. Cancer Res. 2011, 71, 175–184. [Google Scholar] [CrossRef]
- Uhl, M.; Aulwurm, S.; Wischhusen, J.; Weiler, M.; Ma, J.Y.; Almirez, R.; Mangadu, R.; Liu, Y.W.; Platten, M.; Herrlinger, U.; et al. SD-208, a novel transforming growth factor β receptor I kinase inhibitor, inhibits growth and invasiveness and enhances immunogenicity of murine and human glioma cells in vitro and in vivo. Cancer Res. 2004, 64, 7954–7961. [Google Scholar] [CrossRef]
- Akbari, A.; Amanpour, S.; Muhammadnejad, S.; Ghahremani, M.H.; Ghaffari, S.H.; Dehpour, A.R.; Mobini, G.R.; Shidfar, F.; Abastabar, M.; Khoshzaban, A.; et al. Evaluation of antitumor activity of a TGF-β receptor I inhibitor (SD-208) on human colon adenocarcinoma. Daru 2014, 22, 47. [Google Scholar] [CrossRef]
- Doi, T.; Lee, K.H.; Kim, T.M.; Ohtsu, A.; Kim, T.Y.; Ikeda, M.; Yoh, K.; Stampino, C.G.; Hirohashi, T.; Suzuki, A.; et al. A phase I study of the human anti-activin receptor-like kinase 1 antibody PF-03446962 in Asian patients with advanced solid tumors. Cancer Med. 2016, 5, 1454–1463. [Google Scholar] [CrossRef]
- Morris, J.C.; Tan, A.R.; Olencki, T.E.; Shapiro, G.I.; Dezube, B.J.; Reiss, M.; Hsu, F.J.; Berzofsky, J.A.; Lawrence, D.P. Phase I study of GC1008 (fresolimumab): A human anti-transforming growth factor-β (TGF-β) monoclonal antibody in patients with advanced malignant melanoma or renal cell carcinoma. PLoS ONE 2014, 9, e90353. [Google Scholar] [CrossRef]
- Lacouture, M.E.; Morris, J.C.; Lawrence, D.P.; Tan, A.R.; Olencki, T.E.; Shapiro, G.I.; Dezube, B.J.; Berzofsky, J.A.; Hsu, F.J.; Guitart, J. Cutaneous keratoacanthomas/squamous cell carcinomas associated with neutralization of transforming growth factor β by the monoclonal antibody fresolimumab (GC1008). Cancer Immunol. Immunother. 2015, 64, 437–446. [Google Scholar] [CrossRef]
- Tolcher, A.W.; Berlin, J.D.; Cosaert, J.; Kauh, J.; Chan, E.; Piha-Paul, S.A.; Amaya, A.; Tang, S.; Driscoll, K.; Kimbung, R.; et al. A phase 1 study of anti-TGF-β receptor type-II monoclonal antibody LY3022859 in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2017, 79, 673–680. [Google Scholar] [CrossRef]
- Chang, Z.L.; Lorenzini, M.H.; Chen, X.; Tran, U.; Bangayan, N.J.; Chen, Y.Y. Rewiring T-cell responses to soluble factors with chimeric antigen receptors. Nat. Chem. Biol. 2018, 14, 317–324. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
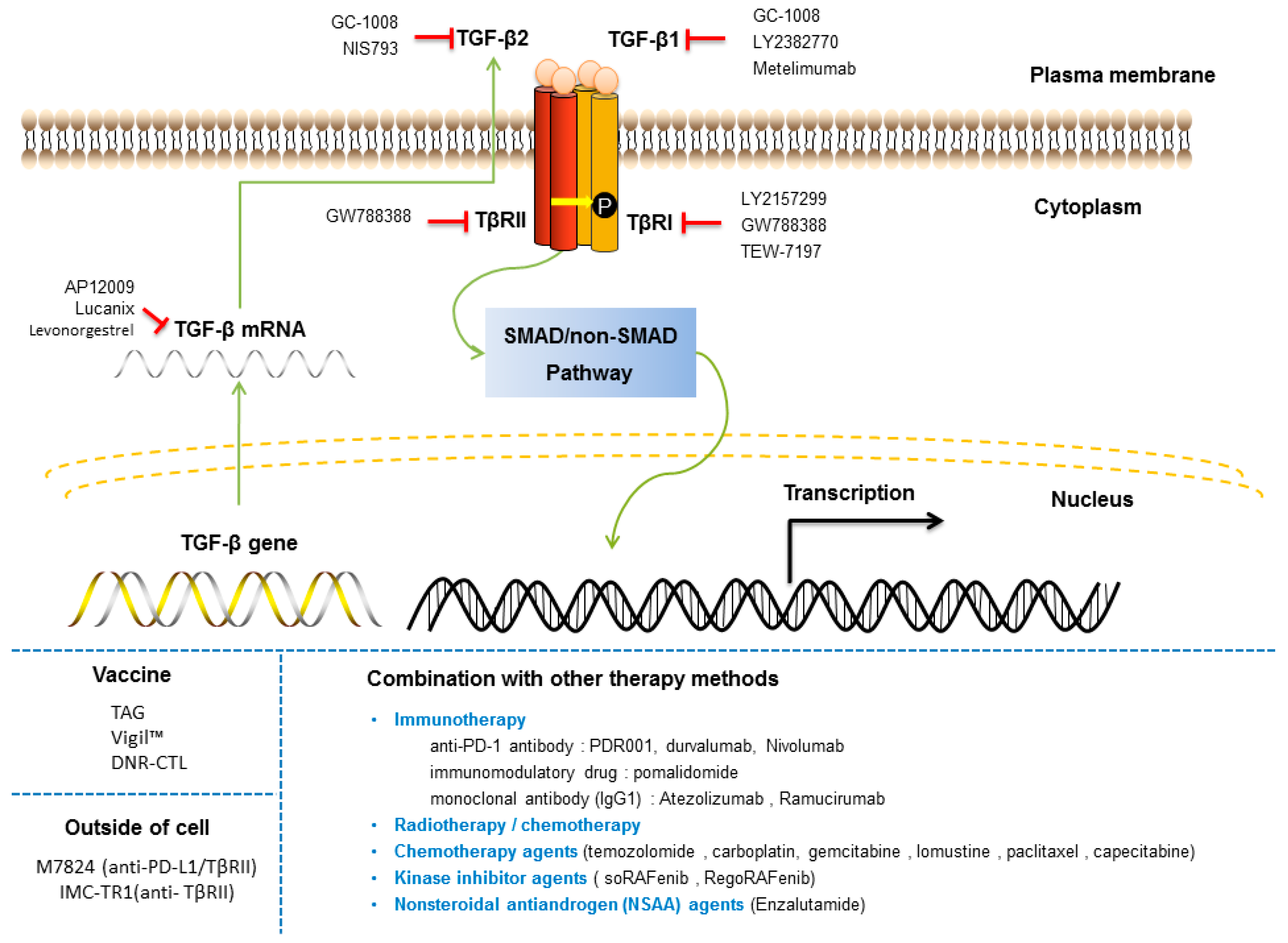{kind=link}
| Cancer Type | LncRNA | Function and Mechanism of Action | Example of Key Findings or Experiments |
|---|---|---|---|
| Breast cancer | lncRNA-ATB [144] | Functions as a sponge of miR-141-3p, increasing ZEB1 and ZEB2 expression. | Knockdown results in a morphological change of breast cancer cells from spindle-like to round shape and in a remarkable inhibition of cell migration and invasion. |
| AC026904.1 and UCA1 [145] | Functions as an enhancer RNA (eRNA) to activate Slug gene transcription in the nucleus, whereas UCA1 exerts a competitive endogenous RNA (ceRNA) for titrating miR-1 and miR-203a to promote Slug expression at the post-transcriptional level in the cytoplasm. | Knockdown of either AC026904.1 or UCA1 prolongs survival time of the nude mice bearing D3H2LN mammary tumors, these two genes exert critical roles in TGF-β-induced EMT and promote invasion in metastatic breast cancer. | |
| NKILA [146] | Suppresses TGF-β-induced EMT by blocking NF-κB signaling | Overexpression reduces TGF-β-induced tumor metastasis in vivo. | |
| ANCR [147] | Functions as a downstream effector molecule, down-regulated by TGF-β1, and is essential for TGF-β1-induced EMT by decreasing RUNX2 expression. | Ectopic expression partly attenuates the TGF-β1-induced EMT and knockdown promotes TGF-β1-induced EMT and metastasis in breast cancer. | |
| Lnc-Spry1 [141] | Functions as an immediate-early regulator of EMT that is downregulated by TGF-β, affecting the expression of TGF-β-regulated gene targets; alternative splicing by U2AF65 splicing factor; isoform switching of fibroblast growth factor receptors. | Knockdown promotes a mesenchymal-like phenotype and results in increased cell migration and invasion. | |
| lncRNA-HIT [148] | Ectopic expression disrupts tight junction by targeting E-cadherin. | Knockdown results in decrease of cell migration, invasion, tumor growth, and metastasis. | |
| linc-ROR [149,150] | Functions as a sponge of miR-145 and therefore upregulate the expression of ARF6, which regulates adhesion and invasion properties of breast tumor cells through E-cadherin. | Regulates the cancer stem cell phenotype in Triple-negative Breast Cancer, which plays a critical role in drug resistance and metastasis. | |
| HOXA-AS2 [151] | Functions as an endogenous sponge of miR-520c-3p, and controls the expression of miR-520c-3p target genes, TβR2 and RELA, in breast cancer cells. | Knockdown inhibits the progression of breast cancer cells in vitro and in vivo. | |
| CCAT2 [152] | Knockdown causes cell cycle arrested in G0/G1 phase, promotes cell apoptosis and downregulates the protein expression levels of TGF-β, Smad2 and α-SMA in breast cancer cells. | Down-regulation inhibits the proliferation, invasion and migration in breast cancer cells. | |
| MEG3 [134,153,154] | Regulates the TGF-β pathway genes through formation of RNA-DNA triplex structures, and downregulates AKT and functions as a sponge of miR-421 to regulate E-cadherin expression. | Ectopic expression inhibits in vivo tumorigenesis and angiogenesis in a nude mouse xenograft model. | |
| Gastric cancer | LINC00978 (known as MIR4435-2HG and AK001796) [155] | Knockdown inhibits the activation of TGF-β/SMAD signaling pathway and EMT in GC cells. | Knockdown inhibits the proliferation, migration and invasion and decreases the in vivo tumorigenicity of GC cells in mice. |
| UCA1 [156] | Knockdown inhibits TGF-β1-induced-EMT process and the effect could be partly restored by TGF-β1 treatment. | A potential oncogenic factor by regulating GC cells proliferation, invasion, and metastasis under TGF-β1 induction. | |
| lncRNA-ATB [157,158,159] | Induced by TGF-β and functions as a ceRNA of miR-141-3p or miR-200s. | A novel biomarker of lncRNA, correlated with increased invasion depth, more distant metastasis and advanced tumor-node-metastasis stage. | |
| XIST [160] | Functions as a competing endogenous lncRNA (ceRNA) to regulate TGF-β1 by sponging miR-185 in GC. | sh-XIST inhibited GC development in vitro. | |
| Ovarian carcinomas | LncRNA-LET [161] | Regulates EMT process and the expression of TIMP2 and activates the Wnt/β-catenin and Notch signaling pathways. | Overexpression inhibits cell viability, migration and EMT process, and increases apoptosis in KGN cells |
| H19 [162] | Functions by competing with miR-370-3p to regulate TGF-β-induced EMT in ovarian cancer. | Knockdown suppresses TGF-β-induced EMT, while H19 overexpression promotes TGF-β-induced EMT. | |
| MALAT1 [163,164] | TGF-β increases its expression by inhibiting miR-200c; MALAT1 interacts with MARCH7, which regulates TGF-β-smad2/3 pathway by interacting with TβR2, via miR-200a as a ceRNA. | Interrupts the interaction between miR-200c/MALAT1 decreases the invasive capacity of EEC cells and EMT in vitro and inhibits EEC growth and EMT-associated protein expression in vivo; This LncRNA plays an important role in TβR2-Smad2/3-MALAT1/MARCH7/ATG7 feedback loop mediated autophagy, migration and invasion in ovarian cancer. | |
| LINK-A [165] cervical cancer | TGF-β1 treatment has no effects on LINK-A expression, and there is no clear mechanism. | Overexpression increases expression of TGF-β1 in ovarian carcinoma cells and promotes cell migration and invasion and this effect can be attenuated by TGF-β1; Plasma levels are correlated with distant tumor metastasis but not tumor size. | |
| lncRNA-ATB [166] | No clear mechanism. | A promising prognostic marker that correlates with the malignant phenotypes and poor prognosis of cervical cancer | |
| PTAF [167] | Functions by competing with miR-25 and affecting SNAI2 to regulate the expression of many EMT-related protein-coding genes in OvCa. | A mediator of TGF-β signaling. Upregulation induces elevated SNAI2, which in turn promoted OvCa cell EMT and invasion; knockdown inhibits tumor progression and metastasis in an orthotopic mouse model of OvCa. | |
| Bladder cancer | lncRNA-ZEB2NAT [168] | Induced by TGF-β, and can regulate EMT process by affecting ZEB2 protein level. | Knockdown reverses CAF-CM-induced EMT and invasion of cancer cells, as well as reduces the ZEB2 protein level. |
| MALAT1 [169] | Induced by TGF-β and regulates EMT by negatively correlated E-cadherin, N-cadherin and fibronectin expression by zeste 12 (suz12) in vitro and in vivo. | Overexpression is significantly correlated with poor survival in patients with bladder cancer. Inhibition of malat1 or suz12 suppresses the migratory and invasive properties induced by TGF-β and inhibits tumor metastasis in animal models. | |
| UCA1 [170] | Induced by BMP9 through phosphorylated AKT and there are no clear mechanism. | Its BMP-9-induced expression associates with increased proliferation and migration of bladder cancer cells. The promoting effect of BMP9 is rescued after interfering with UCA1 in BMP9 overexpressed bladder cancer cells both in vitro and in vivo. | |
| lncRNA-ATB [171] | Is upregulated by TGF-β, acting as a molecular sponge of miR-126 and regulate the direct target of miR-126 (KRAS). | Its overexpression significantly promotes cell viability, migration, and invasion in T24 cells. | |
| PlncRNA-1 [172] | Regulates the cell cycle, cyclin-D1 and EMT in prostate cancer cells through the TGF-β1 pathway. | Functions as an oncogene. | |
| ROR [173] | Knockdown can reverse TGF-β1-induced-EMT phenotype in SGC-996 and Noz cells. However, there are no clear mechanism. | High expression is associated with poor prognosis in gallbladder cancer patients and knockdown inhibits cell proliferation, migration, and invasion. | |
| Prostate cancer | ANRIL [174] | Regulates let-7a/TGF-β1/Smad signaling pathway. | Overexpression promotes the proliferation and migration of prostate cancer cells. |
| lncRNA-ATB [175] | Upregulated by TGF-β, stimulates EMT associated with ZEB1 and ZNF217 expression levels via ERK and PI3K/AKT signaling pathways. | Overexpression promotes, and knockdown of lncRNA-ATB inhibits the growth of prostate cancer cells via regulations of cell cycle regulatory protein expression levels. | |
| Brain tumor | lnc-SNHG1 [176] | Activates the TGFBR2/SMAD3 and RAB11A/Wnt/β-catenin pathways in pituitary tumor cells via sponging miR-302/372/373/520. | Promotes the progression of pituitary tumors, ectopic expression of lnc-SNHG1 promotes cell proliferation, migration, and invasion, as well as the EMT, by affecting the cell cycle and cell apoptosis in vitro and tumor growth in vivo. |
| UCA1 [177] | Functions as a ceRNA of miR-1 and miR-203a to promote Slug expression, which underlies TGF-β-induced EMT and stemness of glioma cells. | Knockdown attenuates EMT and stemness processes and their enhancement by TGF-β. | |
| Lung cancer | lncRNA-LINP1 [178] | TGF-β1 inhibits its transcription in a SMAD4-dependent manner. | Inhibits TGF-β-induced EMT and thereby controlling cancer cell migration, invasion, and stemless in lung cancer cells. |
| TBILA [179] | Induced by TGF-β and functions via cis-regulating HGAL and activating S100A7/JAB1 signaling. | Promotes non-small cell lung cancer progression in vitro and in vivo. | |
| XIST [180,181] | Functions as an endogenous sponge by directly binding to miR-137, negatively regulating its expression and regulating Notch gene expression. | Overexpression inhibits proliferation and TGF-β1-induced EMT in A549 and H1299 cells, regulating proliferation and TGF-β1-induced EMT in NSCLC, which could be involved in NSCLC progression. | |
| NORAD [182] | Affects the physical interaction of its binding partner (importin β1) with Smad3, and then inhibits the nuclear accumulation of Smad complexes in response to TGF-β. | Stimulates TGF-β signaling and regulates TGF-β-induced EMT-like phenotype in A549 cells. | |
| lncRNA-ATB [183] | Regulates EMT by down-regulating miR-494 in A549 cells, which in turn increases phosphorylated levels of AKT, JAK1, and STAT3. | Overexpression promotes proliferation, migration, and invasion of A549 cells. In contract, ATB silence shows the opposite influence. | |
| linc00673 [184] | Functions as a sponge of miR-150-5p and indirectly modulates the expression of key EMT regulator ZEB1. | Inhibition attenuates the tumorigenesis ability of A549 cells in vivo. | |
| NKILA [185] | Expression is regulated by TGF-β and regulates EMT process by inhibiting the phosphorylation of IκBα and NF-κB activation to attenuate Snail expression. | Inhibits migration, invasion and viability of NSCLC cells. Lower NKILA expression are correlated with lymph node metastasis and advanced TNM stage. | |
| MEG3 [186] | Associates with JARID2 and the regulatory regions of target genes to recruit the complex by epigenetic regulation (PRC2/JARID2/ H3K27 axis). | Knockdown inhibits TGF-β-mediated changes in cell morphology and cell motility characteristic of EMT and counteracts TGF-β-dependent changes in the expression of EMT-related genes; In contrast, overexpression enhances these effects. | |
| lnc-MMP2-2 [187] | ls highly enriched in TGF-β-mediated exosomes and might function by increasing the expression of MMP2 through its enhancer activity. | Knockdown affects lung cancer invasion and vascular permeability. | |
| ANCR [188] | Inhibits NSCLC cell migration and invasion by downregulating TGF-β1 expression, however TGF-β1 treatment shows no significant effects on ANCR expression but promotes NSCLC cell migration and invasion. | Low expression level indicates shorter postoperative survival time of NSCLC patients, whereas, ectopic expression inhibits NSCLC cell migration, invasion and downregulated TGF-β1 expression, and this effect can be attenuated by TGF- β1. | |
| LINC01186 [189] | Functions as a mediator of TGF-β signaling, is down-regulated by TGF-β1 regulating EMT by Smad3 | Knockdown promotes cell migration and invasion, whereas, overexpression prevents cell metastasis. | |
| HOXA11-AS [190] | Regulates the expression of various pathways and genes, especially DOCK8 and TGF-β pathway, however, there is no clear mechanism. | Its expression may determine the overall survival and disease-free survival of lung adenocarcinoma patients in TCGA. | |
| Liver cancer | lncRNA SBF2-AS1 [191] | Functions as a ceRNA of miR-140-5p and regulates the expression of TβR1. | Knockdown inhibits the proliferation, migration and invasion of HCC cells and attenuate the development of HCC tumor in vivo. |
| SNHG6-003 [166] | Functions as a ceRNA of miR-26a/b and thereby modulates the expression of transforming growth factor-β-activated kinase 1 (TAK1). | Ectopic expression in HCC cells promotes cell proliferation and induces drug resistance, whereas knockdown promotes apoptosis. High expression of SNHG6-003 closely correlated with tumor progression and shorter survival in HCC patients. | |
| LINC00974 [192] | Interacts with KRT19, as a sponge of miR-642, activating the Notch and TGF-β pathways. | Knockdown inhibits cell proliferation and invasion with an activation of apoptosis and cell cycle arrest both in vitro and in vitro. | |
| lncRNA-ATB [132] | Upregulated by TGF-b and can induce EMT and invasion by acting as ceRNA of miR-200 family and increasing ZEB1/2; promotes organ colonization by binding IL-11 mRNA, autocrine induction of IL-11, and triggering STAT3 signaling. | Promotes the invasion-metastasis cascade in hepatocellular carcinoma. | |
| Pancreatic cancer | PVT1 [193] | Regulates EMT process via TGF-β1/Smad signaling. | Acts as an oncogene in pancreatic cancer, knockdown of PVT1 inhibits viability, adhesion, migration and invasion. |
| MEG8 [194] | MEG8, which shares the DLK1-DIO3 locus with MEG3, can induce the recruitment of EZH2 protein to miR-34a and miR-203 genes for histone H3 methylation and transcriptional repression, inducing EMT-related cell morphological changes and increases cell motility in the absence of TGF-β by activating the gene expression program required for EMT. | Plays critical role in TGF-β-induced EMT in A549 lung cancer and Panc1 pancreatic cancer cells. | |
| TUG1 [195] | Regulates EMT process though TGF-β/Smad pathway | Overexpression increases cell proliferation and migration capacities, enhancing the proliferation and migration of pancreatic cancer cells | |
| MIR100HG [196] | Induced by TGF-β, and this gene contains miR-100, miR-125b and let-7a in its intron, through SMAD2/3. These miRNAs regulate a multitude of genes involved in the inhibition of p53 and DNA damage response pathways. | Plays prominent role in metastasis of pancreatic cancer. | |
| lncRNA-ATB [197] | No clear mechanism. | Low expression levels are correlated with lymph node metastases neural invasion, and clinical stage and worse overall survival prognoses of patients. | |
| Renal cancer | BX357664 [197,198] | Blocks the TGF-β1/p38/HSP27 pathway. | Upregulation reduces migration, invasion, and proliferation capabilities in RCC cells. |
| lncRNA-ATB [199] | No clear mechanism. | Its expression is correlated with metastases and promotes cell migration and invasion in renal cell carcinoma. Knockdown could inhibit cell proliferation, trigger apoptosis, reduce epithelial-to-mesenchymal transition program and suppress cell migration and invasion. | |
| Colorectal cancer | lncRNA-ATB [199,200,201] | Upregulated by TGF-β, and suppresses E-cadherin expression and promoting EMT process. | High expression is significantly associated with greater tumor size, depth of tumor invasion, lymphatic invasion, vascular invasion, and lymph node metastasis. |
| LINC01133 [202] | Downregulated by TGF-β and its expression is positively correlated with E-cadherin, and negatively correlated with Vimentin. Directly interacting with SRSF6, which promotes EMT and metastasis in CRC cells. | Inhibits EMT and metastasis in colorectal cancer (CRC) cells and low LIINC01133 expression in tumors with poor survival in CRC samples. | |
| Bone tumor | MEG3 [203] | Represses Notch and TGF-β signaling pathway by inhibiting Notch1, Hes1, TGF-β and N-cadherin expression, and increasing E-cadherin. | Overexpression represses cell proliferation and migration ability. |
| MALAT1 [204] | Induced by TGF-β and its overexpression decreases E-cadherin level, however, this effect was partially reversed by EZH2 knockdown. | Overexpression promotes cell metastasis, a potential diagnostic and prognostic factor in osteosarcoma. | |
| SNHG7 [205] | Inhibits tumor suppressor miR-34s signals and the targets of miR-34a, including proliferation-related Notch1, apoptosis-related BCL-2, cell cycle-related CDK6, and EMT-related SMAD4. | Knockdown delays the tumor growth in osteosarcoma tissues. | |
| Thyroid cancer | MALAT1 [206] | Induced by TGF-β and supports a role for MALAT1 in EMT in thyroid tumors. | Functions as an oncogene and as a tumor suppressor in different types of thyroid tumors. |
| ROR [207] | Functions as a competing endogenous RNA (ceRNA) of sponging miR-145. | Expression is induced by TGF-β in cells undergoing EMT. | |
| SPRY4-IT1 [208] | Knockdown increases the levels of TGF-β1 and p-Smad2/3 and this effect could be rescued by the interference of TGF-β1. | A novel prognostic factor, which correlates with poor prognosis and exhibits that silenced SPRY4-IT1 inhibited the proliferative and migratory abilities of TC cells. | |
| ANRIL [209] | Reduces p15INK4B expression through inhibiting TGF-β/Smad signaling pathway. | Promotes invasion and metastasis of TC cells, and the silencing of ANRIL inhibits the invasion and metastasis of TPC-1 cells. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hao, Y.; Baker, D.; ten Dijke, P. TGF-β-Mediated Epithelial-Mesenchymal Transition and Cancer Metastasis. Int. J. Mol. Sci. 2019, 20, 2767. https://doi.org/10.3390/ijms20112767
Hao Y, Baker D, ten Dijke P. TGF-β-Mediated Epithelial-Mesenchymal Transition and Cancer Metastasis. International Journal of Molecular Sciences. 2019; 20(11):2767. https://doi.org/10.3390/ijms20112767
Chicago/Turabian StyleHao, Yang, David Baker, and Peter ten Dijke. 2019. "TGF-β-Mediated Epithelial-Mesenchymal Transition and Cancer Metastasis" International Journal of Molecular Sciences 20, no. 11: 2767. https://doi.org/10.3390/ijms20112767
APA StyleHao, Y., Baker, D., & ten Dijke, P. (2019). TGF-β-Mediated Epithelial-Mesenchymal Transition and Cancer Metastasis. International Journal of Molecular Sciences, 20(11), 2767. https://doi.org/10.3390/ijms20112767