Comparative Proteomics Analysis of the Seedling Root Response of Drought-sensitive and Drought-tolerant Maize Varieties to Drought Stress

 ,
,
Abstract
:
1. Introduction
2. Results
2.1. Phenotypic Differences between Chang 7-2 and TS141 Under PEG 6000 Stress
2.2. Physiological Response of Chang7-2 and TS141 to PEG 6000 Stress
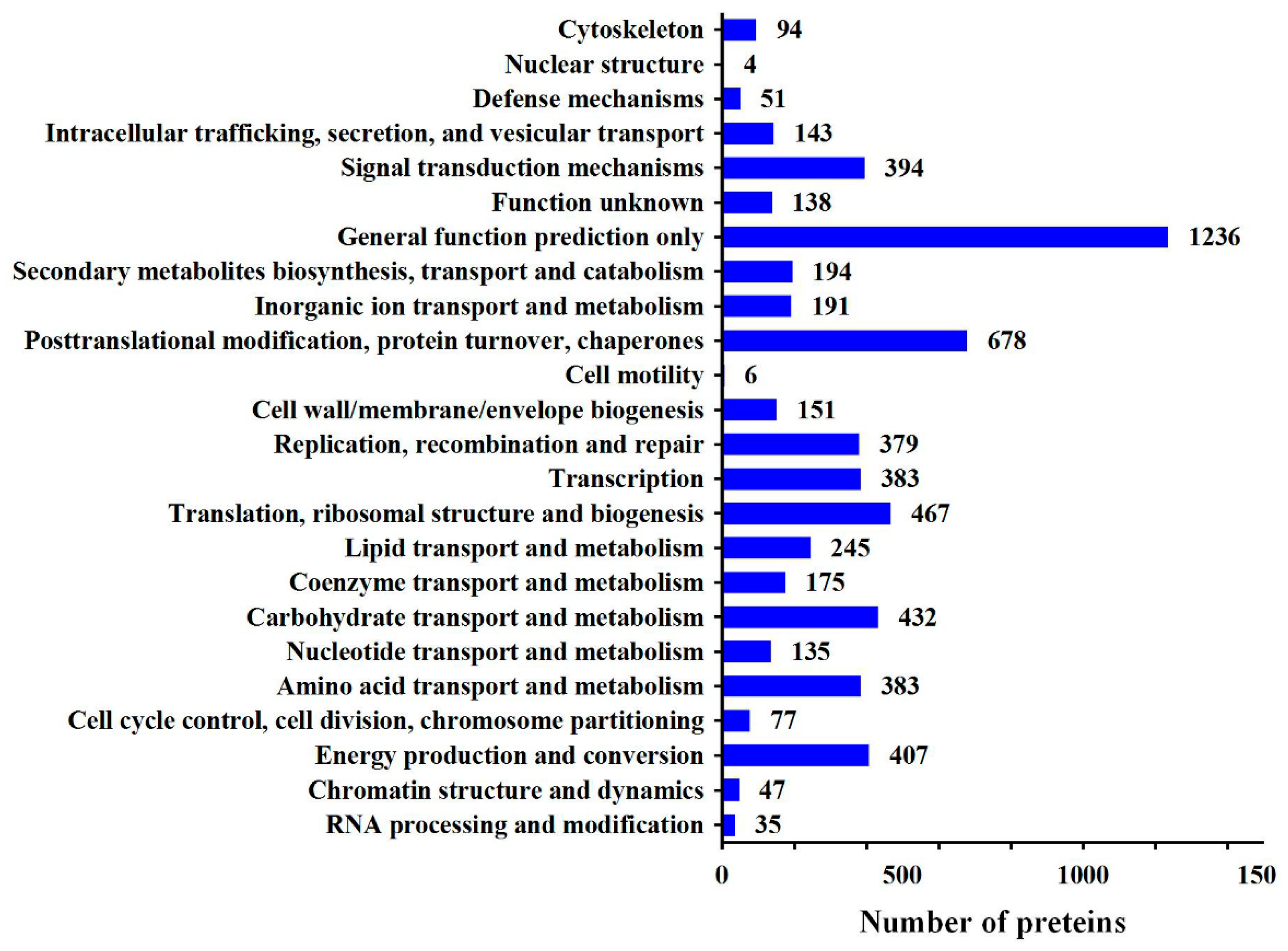
2.3. Quantitative Proteomic Analysis
2.4. iTRAQ Results
2.5. Pathway Enrichment Analysis of DEPs
2.6. qRT-PCR Analysis of DEPs
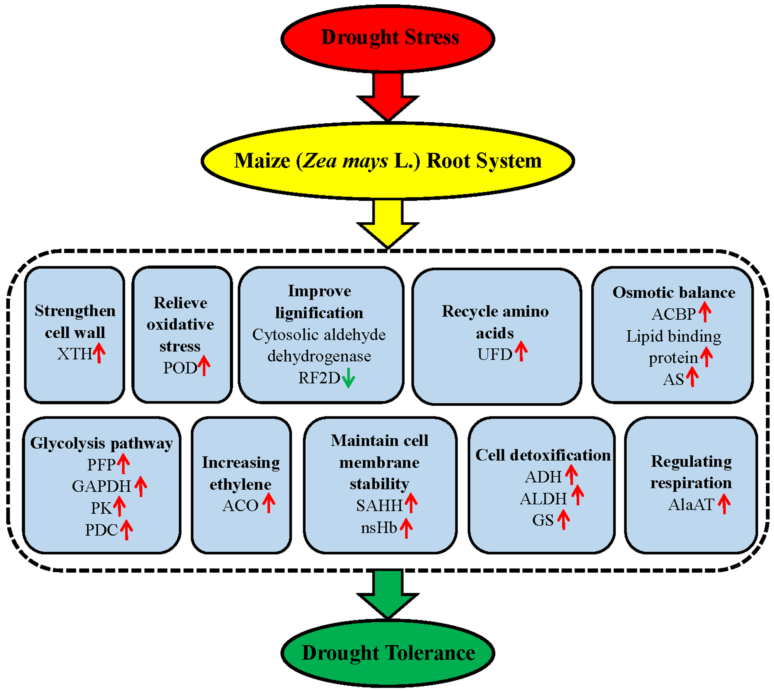
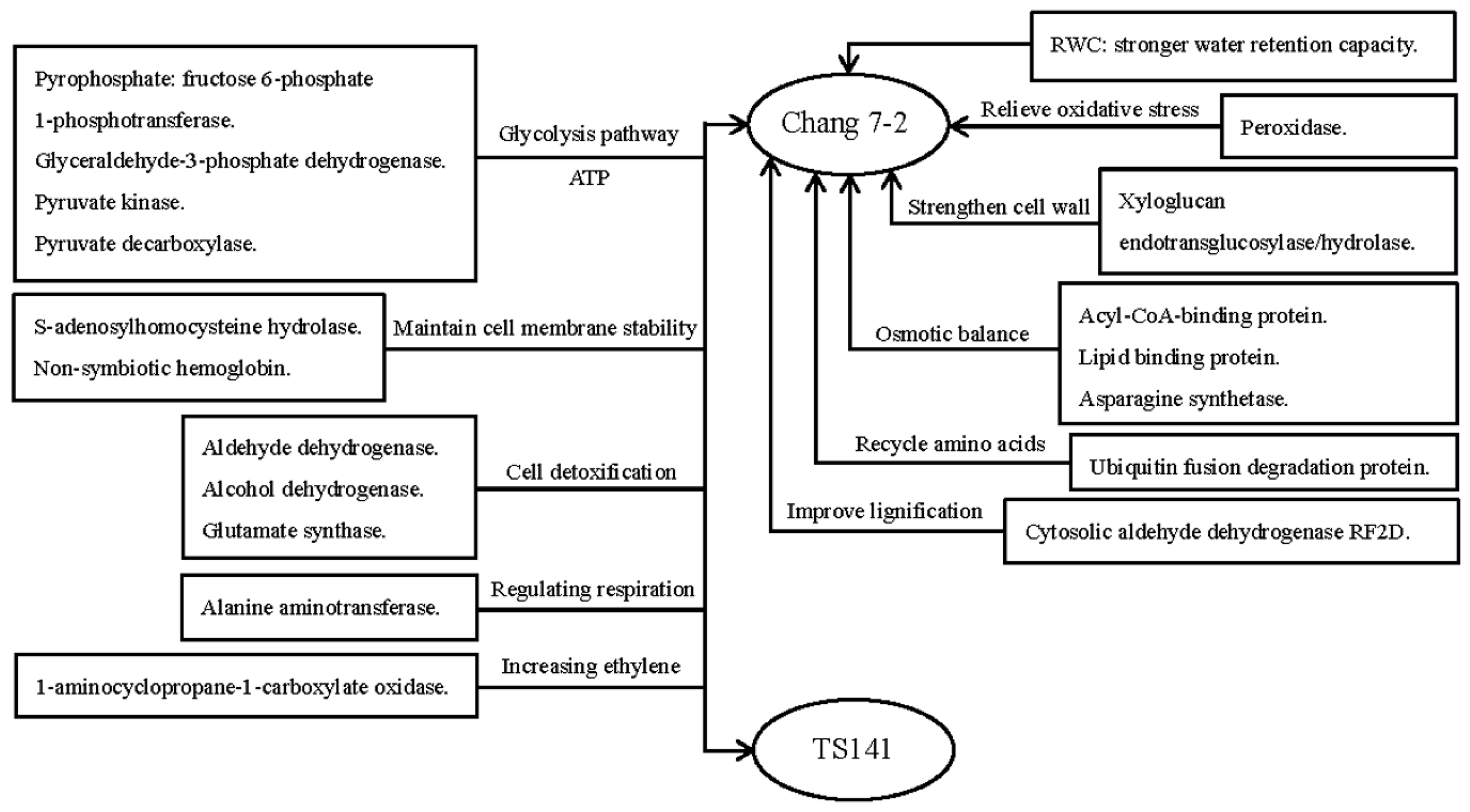
3. Discussion
3.1. Changes in Growth Parameters and Physiological Indicators
3.2. The Drought Response of Drought-Tolerance Inbred Line Chang 7-2
3.2.1. Antioxidant-related proteins are the major Drought Tolerance Signature in Chang 7-2
3.2.2. Increasing Lignin Content is Effective to Resist Drought Stress in Chang 7-2 Root System
3.2.3. Up-Regulation of Plasma Membrane Proteins could Effectively Relieve the Leaking of Cell Contents in Chang 7-2
3.2.4. Chang 7-2 could Tolerates Drought Stress by Strengthening the Cell Wall and Elongating the Roots
3.2.5. Chang 7-2 is More Efficient in Recycling Amino Acids from Proteins that have been Inactivated by Drought Stress.
3.3. The Commonality Metabolic Changes of Two Varieties
3.3.1. Proteins Involved in Cell Detoxification under Drought Stress
3.3.2. The Glycolysis/Gluconeogenesis is the Major Pathway for Maize Roots to Cope with Drought Stress
3.3.3. Maintain Cell Membrane Stability is A Common Strategy for Maize Roots under Drought Stress
3.3.4. Enhancement of Ethylene May Contribute to Enhance Drought Tolerance of Maize Roots
3.3.5. Up-Regulation of Alanine Aminotransferase (AlaAT) could Maintain the respiration and Carbon-Nitrogen Balance in Maize Roots
3.4. Proposed Molecular Model of the Response of Chang7-2 to Drought Stress
4. Materials and Methods
4.1. Plant Materials and Drought Treatment
4.2. Determination of Growth Parameters and Physiological Indicators
4.3. Protein Extraction
4.4. Protein Digestion and iTRAQ Quantification
4.5. Peptide Fractionation and LC-ESI-MS/MS Analysis
4.6. qRT-PCR
4.7. Bioinformatics Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| iTRAQ | Isobaric Tags for Relative and Absolute Quantitation |
| PEG6000 | Polyethylene glycol 6000 |
| DEPs | Differentially Expressed Proteins |
| qRT-PCR | Quantitative real-time PCR |
| 2-DE | Two-Dimensional Electrophoresis |
| CK | Control |
| D | Drought Stress |
| CCK | Chang 7-2 Control |
| CD | Chang 7-2 Drought Stress |
| TCK | TS141 Control |
| TD | TS141 Drought Stress. |
| SL | Seeding Length |
| RL | Root Length |
| SFW | Seedling Fresh Weight |
| RFW | Root Fresh Weight |
| RWC | Relative Water Content |
| REL | Relative Electrolyte Leakage |
| MDA | Malondialdehyde |
| Pro | Proline |
| SOD | Superoxide Dismutase |
| POD | Peroxidase |
| CAT | Catalase |
| COG | Cluster of Orthologous Groups of Proteins |
| GO | Gene Ontology |
| UFD | Ubiquitin Fusion Degradation Protein |
| ROS | Reactive Oxygen Species |
| CA | Coniferyl Aldehyde |
| SA | Sinapaldehyde |
| PFP | Pyrophosphate: Fructose-6-Phosphate 1-Phosphotransferase |
| PFK | Phosphofructokinase |
| PPi | Pyrophosphoric acid |
| PK | Pyruvate Kinase |
| GAPDH | Glyceraldehyde 3-Phosphate Dehydrogenase |
| PDC | Pyruvate Decarboxylase |
| ACO | 1-Amino-Cyclopropane-1-Carboxylate Oxidase |
| ACC | 1-Aminocyclopropane-1-Carboxylate |
| AlaAT | Alanine Aminotransferase |
| LC-MS/MS | Liquid Chromatography-Electrospray Ionization-Tandem Mass Spectrometry |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
References
- Lobell, D.B.; Roberts, M.J.; Schlenker, W.; Braun, N.; Little, B.B.; Rejesus, R.M.; Hammer, G.L. Greater Sensitivity to Drought Accompanies Maize Yield Increase in the U.S. Midwest. Science 2014, 344, 516–519. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, A.; Emam, Y.; Pessarakli, M. Biochemical Changes in Maize Seedlings Expose to Drought Stress Conditions at Different Nitrogen Levels. J. Plant Nutr. 2010, 33, 541–556. [Google Scholar] [CrossRef]
- Chen, D.; Wang, S.; Cao, B.; Cao, D.; Leng, G.; Li, H.; Yin, L.; Shan, L.; Deng, X. Genotypic Variation in Growth and Physiological Response to Drought Stress and Re-Watering Reveals the Critical Role of Recovery in Drought Adaptation in Maize Seedlings. Front. Plant Sci. 2016, 6, 1241. [Google Scholar] [CrossRef] [PubMed]
- Bunce, J. Leaf Transpiration Efficiency of Some Drought-Resistant Maize Lines. Crop Sci. 2010, 50, 1409–1413. [Google Scholar] [CrossRef]
- Mao, H.; Wang, H.; Liu, S.; Li, Z.; Yang, X.; Yan, J.; Li, J.; Tran, L.-S.P.; Qin, F. A transposable element in a NAC gene is associated with drought tolerance in maize seedlings. Nat. Commun. 2015, 6, 8326. [Google Scholar] [CrossRef]
- Zenda, T.; Liu, S.; Wang, X.; Jin, H.; Liu, G.; Duan, H. Comparative Proteomic and Physiological Analyses of Two Divergent Maize Inbred Lines Provide More Insights into Drought-Stress Tolerance Mechanisms. Int. J. Mol. Sci. 2018, 19, 3225. [Google Scholar] [CrossRef]
- Yang, L.T.; Qi, Y.P.; Lu, Y.B.; Guo, P.; Sang, W.; Feng, H.; Zhang, H.X.; Chen, L.S. iTRAQ protein profile analysis of Citrus sinensis roots in response to long-term boron-deficiency. J. Proteom. 2013, 93, 179–206. [Google Scholar] [CrossRef]
- Nogueira, F.C.S.; Palmisano, G.; Schwämmle, V.; Campos, F.A.P.; Larsen, M.R.; Domont, G.B.; Roepstorff, P. Performance of Isobaric and Isotopic Labeling in Quantitative Plant Proteomics. J. Proteome Res. 2012, 11, 3046–3052. [Google Scholar] [CrossRef]
- Zhang, X.H.; Rao, X.L.; Shi, H.T.; Li, R.-J.; Lu, Y.T. Overexpression of a cytosolic glyceraldehyde-3-phosphate dehydrogenase gene OsGAPC3 confers salt tolerance in rice. Plant Cell Tissue Organ Cult. (PCTOC) 2011, 107, 1. [Google Scholar] [CrossRef]
- Zhang, C.; Shi, S. Physiological and Proteomic Responses of Contrasting Alfalfa (Medicago sativa L.) Varieties to PEG-Induced Osmotic Stress. Front. Plant Sci. 2018, 9, 242. [Google Scholar] [CrossRef]
- Pan, L.; Yang, Z.; Wang, J.; Wang, P.; Ma, X.; Zhou, M.; Li, J.; Gang, N.; Feng, G.; Zhao, J.; et al. Comparative proteomic analyses reveal the proteome response to short-term drought in Italian ryegrass (Lolium multiflorum). PLoS ONE 2017, 12, e0184289. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Wang, Y.; He, Q.; Li, H.; Zhang, X.; Zhang, F. Comparative proteomics illustrates the complexity of drought resistance mechanisms in two wheat (Triticum aestivum L.) cultivars under dehydration and rehydration. BMC Plant Biol. 2016, 16, 188. [Google Scholar] [CrossRef]
- Sengupta, D.; Kannan, M.; Reddy, A.R. A root proteomics-based insight reveals dynamic regulation of root proteins under progressive drought stress and recovery in Vigna radiata (L.) Wilczek. Planta 2011, 233, 1111–1127. [Google Scholar] [CrossRef]
- Avramova, V.; Nagel, K.A.; AbdElgawad, H.; Bustos, D.; DuPlessis, M.; Fiorani, F.; Beemster, G.T.S. Screening for drought tolerance of maize hybrids by multi-scale analysis of root and shoot traits at the seedling stage. J. Exp. Bot. 2016, 67, 2453–2466. [Google Scholar] [CrossRef]
- Regier, N.; Streb, S.; Cocozza, C.; Schaub, M.; Cherubini, P.; Zeeman, S.C.; Frey, B. Drought tolerance of two black poplar (Populus nigra L.) clones: Contribution of carbohydrates and oxidative stress defence. Plant Cell Environ. 2009, 32, 1724–1736. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Sharp, R.E. Complexity and coordination of root growth at low water potentials: Recent advances from transcriptomic and proteomic analyses. Plant Cell Environ. 2010, 33, 590–603. [Google Scholar] [CrossRef]
- Tuberosa, R.; Sanguineti, M.C.; Landi, P.; Michela Giuliani, M.; Salvi, S.; Conti, S. Identification of QTLs for root characteristics in maize grown in hydroponics and analysis of their overlap with QTLs for grain yield in the field at two water regimes. Plant Mol. Biol. 2002, 48, 697–712. [Google Scholar] [CrossRef] [PubMed]
- Moussa, H.R.; Abdel-Aziz, S.M.; Mans, B.J.; Andersen, J.F.; Francischetti, I.M.; Valenzuela, J.G.; Schwan, T.G.; Pham, V.M.; Garfield, M.K.; Hammer, C.H.; et al. Comparative response of drought tolerant and drought sensitive maize genotypes to water stress. Aust. J. Crop Sci. 2008, 1, 31–36. [Google Scholar]
- Sairam, R.K.; Rao, K.V.; Srivastava, G.C. Differential response of wheat genotypes to long term salinity stress in relation to oxidative stress, antioxidant activity and osmolyte concentration. Plant Sci. 2002, 163, 1037–1046. [Google Scholar] [CrossRef]
- Kumar, S.G.; Reddy, A.M.; Sudhakar, C. NaCl effects on proline metabolism in two high yielding genotypes of mulberry (Morus alba L.) with contrasting salt tolerance. Plant Sci. 2003, 165, 1245–1251. [Google Scholar] [CrossRef]
- Miller, G.A.D.; Suzuki, N.; Ciftci-Yilmaz, S.; Mittler, R.O.N. Reactive oxygen species homeostasis and signalling during drought and salinity stresses. Plant Cell Environ. 2010, 33, 453–467. [Google Scholar] [CrossRef] [PubMed]
- Kang, G.Z.; Li, G.Z.; Liu, G.Q.; Xu, W.; Peng, X.Q.; Wang, C.Y.; Zhu, Y.J.; Guo, T.C. Exogenous salicylic acid enhances wheat drought tolerance by influence on the expression of genes related to ascorbate-glutathione cycle. Biol. Plant. 2013, 57, 718–724. [Google Scholar] [CrossRef]
- Koh, J.; Chen, G.; Yoo, M.-J.; Zhu, N.; Dufresne, D.; Erickson, J.E.; Shao, H.; Chen, S. Comparative Proteomic Analysis of Brassica napus in Response to Drought Stress. J. Proteome Res. 2015, 14, 3068–3081. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Xing, X.J.; Tian, Y.S.; Peng, R.H.; Xue, Y.; Zhao, W.; Yao, Q.H. Transgenic Arabidopsis Plants Expressing Tomato Glutathione S-Transferase Showed Enhanced Resistance to Salt and Drought Stress. PLoS ONE 2015, 10, e0136960. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Bae, J.M.; Huh, G.H. Transcriptional regulation of the cinnamyl alcohol dehydrogenase gene from sweetpotato in response to plant developmental stage and environmental stress. Plant Cell Rep. 2010, 29, 779–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Zhang, X.; Lu, C.; Zeng, X.; Li, Y.; Fu, D.; Wu, G. Non-specific lipid transfer proteins in plants: Presenting new advances and an integrated functional analysis. J. Exp. Bot. 2015, 66, 5663–5681. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; Chye, M.-L. New roles for acyl-CoA-binding proteins (ACBPs) in plant development, stress responses and lipid metabolism. Prog. Lipid Res. 2011, 50, 141–151. [Google Scholar] [CrossRef]
- Lea, P.J.; Sodek, L.; Parry, M.A.J.; Shewry, P.R.; Halford, N.G. Asparagine in plants. Ann. Appl. Biol. 2007, 150, 1–26. [Google Scholar] [CrossRef]
- Wang, H.; Liu, D.; Sun, J.; Zhang, A. Asparagine synthetase gene TaASN1 from wheat is up-regulated by salt stress, osmotic stress and ABA. J. Plant Physiol. 2005, 162, 81–89. [Google Scholar] [CrossRef]
- Cosgrove, D.J. Growth of the plant cell wall. Nat. Rev. Mol. Cell Biol. 2005, 6, 850–861. [Google Scholar] [CrossRef]
- Zhu, X.F.; Shi, Y.Z.; Lei, G.J.; Fry, S.C.; Zhang, B.C.; Zhou, Y.H.; Braam, J.; Jiang, T.; Xu, X.Y.; Mao, C.Z.; et al. XTH31, encoding an in vitro XEH/XET-active enzyme, regulates aluminum sensitivity by modulating in vivo XET action, cell wall xyloglucan content, and aluminum binding capacity in Arabidopsis. Plant Cell 2012, 24, 4731–4747. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.K.; Kim, J.E.; Park, J.A.; Eom, T.J.; Kim, W.T. Constitutive expression of abiotic stress-inducible hot pepper CaXTH3, which encodes a xyloglucan endotransglucosylase/hydrolase homolog, improves drought and salt tolerance in transgenic Arabidopsis plants. FEBS Lett. 2006, 580, 3136–3144. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.A.; Chang, R.Z.; Qiu, L.J. Overexpression of soybean ubiquitin-conjugating enzyme gene GmUBC2 confers enhanced drought and salt tolerance through modulating abiotic stress-responsive gene expression in Arabidopsis. Plant Mol. Biol. 2010, 72, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Gechev, T.S.; Dinakar, C.; Benina, M.; Toneva, V.; Bartels, D. Molecular mechanisms of desiccation tolerance in resurrection plants. Cell. Mol. Life Sci. 2012, 69, 3175–3186. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, S.M.; Andrade, M.O.; Gomes, A.P.S.; DaMatta, F.M.; Baracat-Pereira, M.C.; Fontes, E.P.B. Arabidopsis and tobacco plants ectopically expressing the soybean antiquitin-like ALDH7 gene display enhanced tolerance to drought, salinity, and oxidative stress. J. Exp. Bot. 2006, 57, 1909–1918. [Google Scholar] [CrossRef] [PubMed]
- Sunkar, R.; Bartels, D.; Kirch, H.H. Overexpression of a stress-inducible aldehyde dehydrogenase gene from Arabidopsis thaliana in transgenic plants improves stress tolerance. Plant J. 2003, 35, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Taira, M.; Valtersson, U.; Burkhardt, B.; Ludwig, R.A. Arabidopsis thaliana GLN2-encoded glutamine synthetase is dual targeted to leaf mitochondria and chloroplasts. Plant Cell 2004, 16, 2048–2058. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, Y.; Xu, Y.; Zhang, X.; Peng, Y.; Ma, X.; Huang, L.; Yan, Y. Physiological and iTRAQ-Based Proteomic Analyses Reveal the Function of Spermidine on Improving Drought Tolerance in White Clover. J. Proteome Res. 2016, 15, 1563–1579. [Google Scholar] [CrossRef] [PubMed]
- Gong, X.; Qu, C.; Liu, C.; Hong, M.; Wang, L.; Hong, F. Effects of Manganese Deficiency and Added Cerium on Nitrogen Metabolism of Maize. Biol. Trace Elem. Res. 2011, 144, 1240–1250. [Google Scholar] [CrossRef] [PubMed]
- Prinsi, B.; Espen, L. Mineral nitrogen sources differently affect root glutamine synthetase isoforms and amino acid balance among organs in maize. BMC Plant Biol. 2015, 15, 96. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Greenway, H.; Colmer, T.D.; Millar, A.H. Protein synthesis by rice coleoptiles during prolonged anoxia: Implications for glycolysis, growth and energy utilization. Ann. Bot. 2005, 96, 703–715. [Google Scholar] [CrossRef] [PubMed]
- Rius, S.P.; Casati, P.; Iglesias, A.A.; Gomez-Casati, D.F. Characterization of an Arabidopsis thaliana mutant lacking a cytosolic non-phosphorylating glyceraldehyde-3-phosphate dehydrogenase. Plant Mol. Biol. 2006, 61, 945–957. [Google Scholar] [CrossRef] [PubMed]
- Rochat, T.; Boudebbouze, S.; Gratadoux, J.J.; Blugeon, S.; Gaudu, P.; Langella, P.; Maguin, E. Proteomic analysis of spontaneous mutants of Lactococcus lactis: Involvement of GAPDH and arginine deiminase pathway in H2O2 resistance. Proteomics 2012, 12, 1792–1805. [Google Scholar] [CrossRef] [PubMed]
- Chugh, V.; Kaur, N.; Gupta, A.K. Role of antioxidant and anaerobic metabolism enzymes in providing tolerance to maize (Zea mays L.) seedlings against waterlogging. Indian J. Biochem. Biophys. 2011, 48, 346–352. [Google Scholar] [PubMed]
- Ouyang, B.; Fei, Z.; Joung, J.G.; Kolenovsky, A.; Koh, C.; Nowak, J.; Caplan, A.; Keller, W.A.; Cui, Y.; Cutler, A.J.; et al. Transcriptome profiling and methyl homeostasis of an Arabidopsis mutant deficient in S-adenosylhomocysteine hydrolase1 (SAHH1). Plant Mol. Biol. 2012, 79, 315–331. [Google Scholar] [CrossRef] [PubMed]
- Weretilnyk, E.A.; Alexander, K.J.; Drebenstedt, M.; Snider, J.D.; Summers, P.S.; Moffatt, B.A. Maintaining methylation activities during salt stress. The involvement of adenosine kinase. Plant Physiol. 2001, 125, 856–865. [Google Scholar] [CrossRef] [PubMed]
- Li, S.S.; Zhu, C.; Teng, K.; Liu, C.L.; Zeng, H.M. Functional Identification of SAHH Gene from Kalanchoe daigremontiana. J. Agric. Biotechnol. 2016, 24, 538–547. [Google Scholar]
- Hebelstrup, K.H.; Igamberdiev, A.U.; Hill, R.D. Metabolic effects of hemoglobin gene expression in plants. Gene 2007, 398, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Seregélyes, C.; Barna, B.; Hennig, J.; Konopka, D.; Pasternak, T.P.; Lukács, N.; Fehér, A.; Horváth, G.V.; Dudits, D. Phytoglobins can interfere with nitric oxide functions during plant growth and pathogenic responses: A transgenic approach. Plant Sci. 2003, 165, 541–550. [Google Scholar] [CrossRef]
- Ohwaki, Y.; Kawagishi-Kobayashi, M.; Wakasa, K.; Fujihara, S.; Yoneyama, T. Induction of Class-1 Non-symbiotic Hemoglobin Genes by Nitrate, Nitrite and Nitric Oxide in Cultured Rice Cells. Plant Cell Physiol. 2005, 46, 324–331. [Google Scholar] [CrossRef]
- Gupta, K.J.; Hebelstrup, K.H.; Mur, L.A.J.; Igamberdiev, A.U. Plant hemoglobins: Important players at the crossroads between oxygen and nitric oxide. FEBS Lett. 2011, 585, 3843–3849. [Google Scholar] [CrossRef] [PubMed]
- Dordas, C. Nonsymbiotic hemoglobins and stress tolerance in plants. Plant Sci. 2009, 176, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, A.; Sakurao, S.-h.; Fukunaga, K.; Matsubara, T.; Ueda-Hashimoto, M.; Tsukamoto, S.; Takahashi, M.; Morikawa, H. Three distinct Arabidopsis hemoglobins exhibit peroxidase-like activity and differentially mediate nitrite-dependent protein nitration. FEBS Lett. 2004, 572, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Bharalee, R.; Bhorali, P.; Bandyopadhyay, T.; Gohain, B.; Agarwal, N.; Ahmed, P.; Saikia, H.; Borchetia, S.; Kalita, M.C.; et al. Identification of drought tolerant progenies in tea by gene expression analysis. Funct. Integr. Genom. 2012, 12, 543–563. [Google Scholar] [CrossRef] [PubMed]
- Wan, L.; Zhang, J.; Zhang, H.; Zhang, Z.; Quan, R.; Zhou, S.; Huang, R. Transcriptional activation of OsDERF1 in OsERF3 and OsAP2-39 negatively modulates ethylene synthesis and drought tolerance in rice. PLoS ONE 2011, 6, e25216. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Ma, X.; Li, C.; Zhang, W.; Xia, G.; Wang, M. A wheat aminocyclopropane-1-carboxylate oxidase gene, TaACO1, negatively regulates salinity stress in Arabidopsis thaliana. Plant Cell Rep. 2014, 33, 1815–1827. [Google Scholar] [CrossRef] [PubMed]
- Liepman, A.H.; Olsen, L.J. Alanine Aminotransferase Homologs Catalyze the Glutamate:Glyoxylate Aminotransferase Reaction in Peroxisomes of Arabidopsis. Plant Physiol. 2003, 131, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Kendziorek, M.; Paszkowski, A.; Zagdańska, B. Differential regulation of alanine aminotransferase homologues by abiotic stresses in wheat (Triticum aestivum L.) seedlings. Plant Cell Rep. 2012, 31, 1105–1117. [Google Scholar] [CrossRef] [PubMed]
- Gupta, K.J.; Zabalza, A.; van Dongen, J.T. Regulation of respiration when the oxygen availability changes. Physiol. Plant 2009, 137, 383–391. [Google Scholar] [CrossRef]
- Rocha, M.; Sodek, L.; Licausi, F.; Hameed, M.W.; Dornelas, M.C.; van Dongen, J.T. Analysis of alanine aminotransferase in various organs of soybean (Glycine max) and in dependence of different nitrogen fertilisers during hypoxic stress. Amino Acids 2010, 39, 1043–1053. [Google Scholar] [CrossRef]
- Galmés, J.; Flexas, J.; Savé, R.; Medrano, H. Water relations and stomatal characteristics of Mediterranean plants with different growth forms and leaf habits: Responses to water stress and recovery. Plant Soil 2007, 290, 139–155. [Google Scholar] [CrossRef]
- Liu, Y.; Du, H.; He, X.; Huang, B.; Wang, Z. Identification of differentially expressed salt-responsive proteins in roots of two perennial grass species contrasting in salinity tolerance. J. Plant Physiol. 2012, 169, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Madhava Rao, K.V.; Sresty, T.V.S. Antioxidative parameters in the seedlings of pigeonpea (Cajanus cajan (L.) Millspaugh) in response to Zn and Ni stresses. Plant Sci. 2000, 157, 113–128. [Google Scholar] [CrossRef]
- Bates, L.S.; Waldren, R.P.; Teare, I.D. Rapid determination of free proline for water-stress studies. Plant Soil 1973, 39, 205–207. [Google Scholar] [CrossRef]
- Beauchamp, C.; Fridovich, I. Superoxide dismutase: Improved assays and an assay applicable to acrylamide gels. Anal. Biochem. 1971, 44, 276–287. [Google Scholar] [CrossRef]
- Han, L.B.; Song, G.L.; Zhang, X. Preliminary observations on physiological responses of three turfgrass species to traffic stress. HortTechnology 2008, 18, 139–143. [Google Scholar] [CrossRef]
- Siminis, C.I.; Kanellis, A.K.; Roubelakis-Angelakis, K.A. Catalase Is Differentially Expressed in Dividing and Nondividing Protoplasts. Plant Physiol. 1994, 105, 1375–1383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, S.H.; Park, G.W.; Kim, J.Y.; Kwon, S.O.; Choi, C.-W.; Leem, S.-H.; Kwon, K.-H.; Yoo, J.S.; Lee, C.; Kim, S.; et al. Proteomic characterization of the Pseudomonas putida KT2440 global response to a monocyclic aromatic compound by iTRAQ analysis and 1DE-MudPIT. J. Proteom. 2011, 74, 620–628. [Google Scholar] [CrossRef]
- Zhu, H.G.; Cheng, W.H.; Tian, W.G.; Li, Y.J.; Liu, F.; Xue, F.; Zhu, Q.H.; Sun, Y.Q.; Sun, J. iTRAQ-based comparative proteomic analysis provides insights into somatic embryogenesis in Gossypium hirsutum L. Plant Mol. Biol. 2018, 96, 89–102. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway | Number of DEPs(1007) | p-Value | Pathway ID |
|---|---|---|---|
| Ribosome | 54 | 0.006156876 | ko03010 |
| Glycosphingolipid biosynthesis—globo and isoglobo series | 5 | 0.006714952 | ko00603 |
| Nitrogen metabolism | 11 | 0.009075108 | ko00910 |
| Amino sugar and nucleotide sugar metabolism | 31 | 0.009913536 | ko00520 |
| Pentose phosphate pathway | 17 | 0.02244357 | ko00030 |
| Mismatch repair | 8 | 0.0247802 | ko03430 |
| Fructose and mannose metabolism | 19 | 0.02541227 | ko00051 |
| Glycolysis/Gluconeogenesis | 33 | 0.02772382 | ko00010 |
| Glycosphingolipid biosynthesis—ganglio series | 4 | 0.03880259 | ko00604 |
| Phagosome | 17 | 0.0488682 | ko04145 |
| Pathway | Number of DEPs(350) | p-Value | Pathway ID |
|---|---|---|---|
| RNA polymerase | 10 | 0.000241549 | ko03020 |
| Metabolic pathways | 125 | 0.000448353 | ko01100 |
| Carbon fixation in photosynthetic organisms | 12 | 0.001034286 | ko00710 |
| Nitrogen metabolism | 6 | 0.007834169 | ko00910 |
| Glycerophospholipid metabolism | 9 | 0.008846003 | ko00564 |
| Starch and sucrose metabolism | 15 | 0.01968736 | ko00500 |
| Amino sugar and nucleotide sugar metabolism | 13 | 0.02511713 | ko00520 |
| Glycolysis/Gluconeogenesis | 14 | 0.0361396 | ko00010 |
| Galactose metabolism | 8 | 0.04039815 | ko00052 |
| Phenylpropanoid biosynthesis | 18 | 0.04056657 | ko00940 |
| Alanine, aspartate and glutamate metabolism | 7 | 0.0461047 | ko00250 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeng, W.; Peng, Y.; Zhao, X.; Wu, B.; Chen, F.; Ren, B.; Zhuang, Z.; Gao, Q.; Ding, Y. Comparative Proteomics Analysis of the Seedling Root Response of Drought-sensitive and Drought-tolerant Maize Varieties to Drought Stress. Int. J. Mol. Sci. 2019, 20, 2793. https://doi.org/10.3390/ijms20112793
Zeng W, Peng Y, Zhao X, Wu B, Chen F, Ren B, Zhuang Z, Gao Q, Ding Y. Comparative Proteomics Analysis of the Seedling Root Response of Drought-sensitive and Drought-tolerant Maize Varieties to Drought Stress. International Journal of Molecular Sciences. 2019; 20(11):2793. https://doi.org/10.3390/ijms20112793
Chicago/Turabian StyleZeng, Wenjing, Yunling Peng, Xiaoqiang Zhao, Boyang Wu, Fenqi Chen, Bin Ren, Zelong Zhuang, Qiaohong Gao, and Yongfu Ding. 2019. "Comparative Proteomics Analysis of the Seedling Root Response of Drought-sensitive and Drought-tolerant Maize Varieties to Drought Stress" International Journal of Molecular Sciences 20, no. 11: 2793. https://doi.org/10.3390/ijms20112793
APA StyleZeng, W., Peng, Y., Zhao, X., Wu, B., Chen, F., Ren, B., Zhuang, Z., Gao, Q., & Ding, Y. (2019). Comparative Proteomics Analysis of the Seedling Root Response of Drought-sensitive and Drought-tolerant Maize Varieties to Drought Stress. International Journal of Molecular Sciences, 20(11), 2793. https://doi.org/10.3390/ijms20112793