Pathophysiology and Diagnosis of ALS: Insights from Advances in Neurophysiological Techniques



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
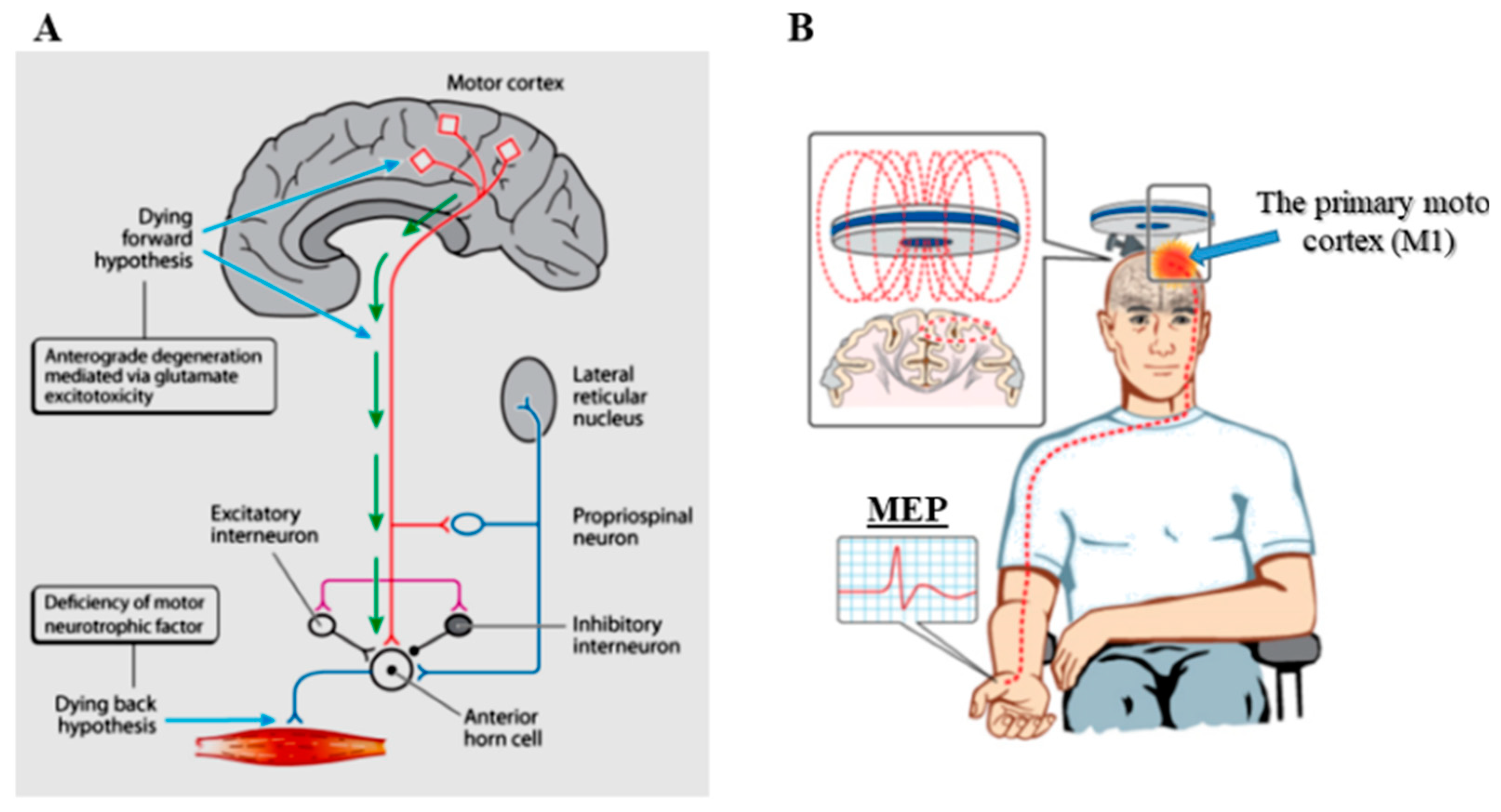
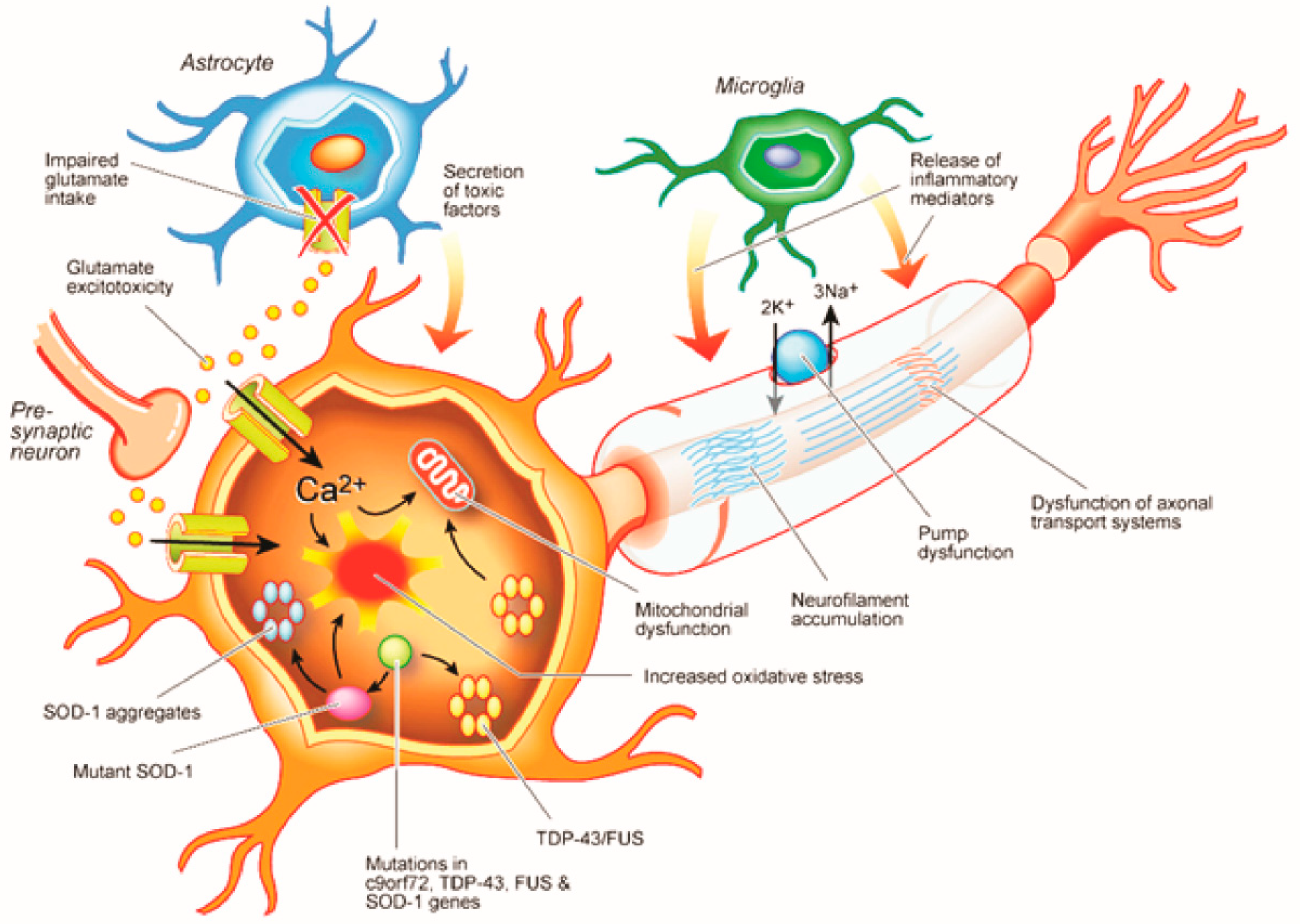
2. Cortical Physiological Dysfunction and ALS Pathogenesis
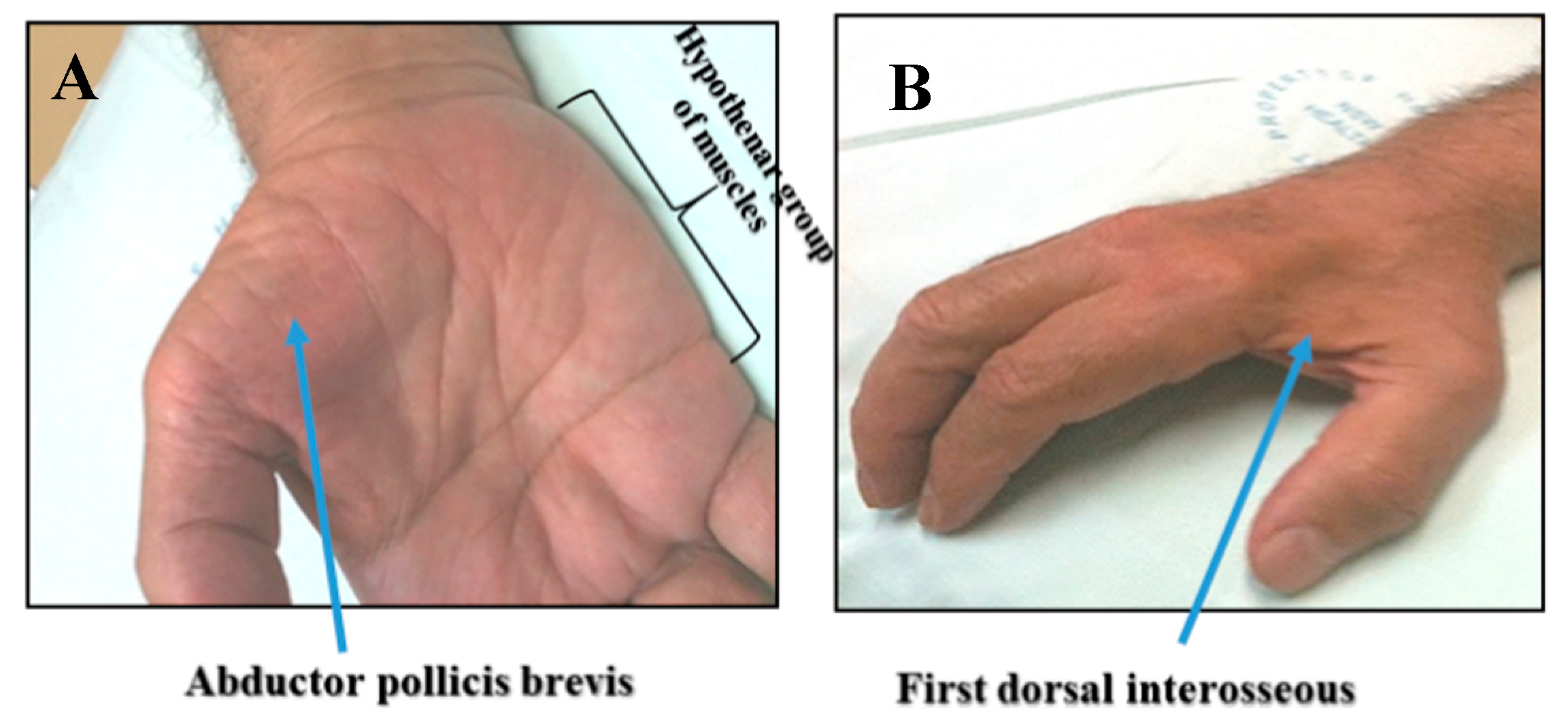
3. Clinical Insights
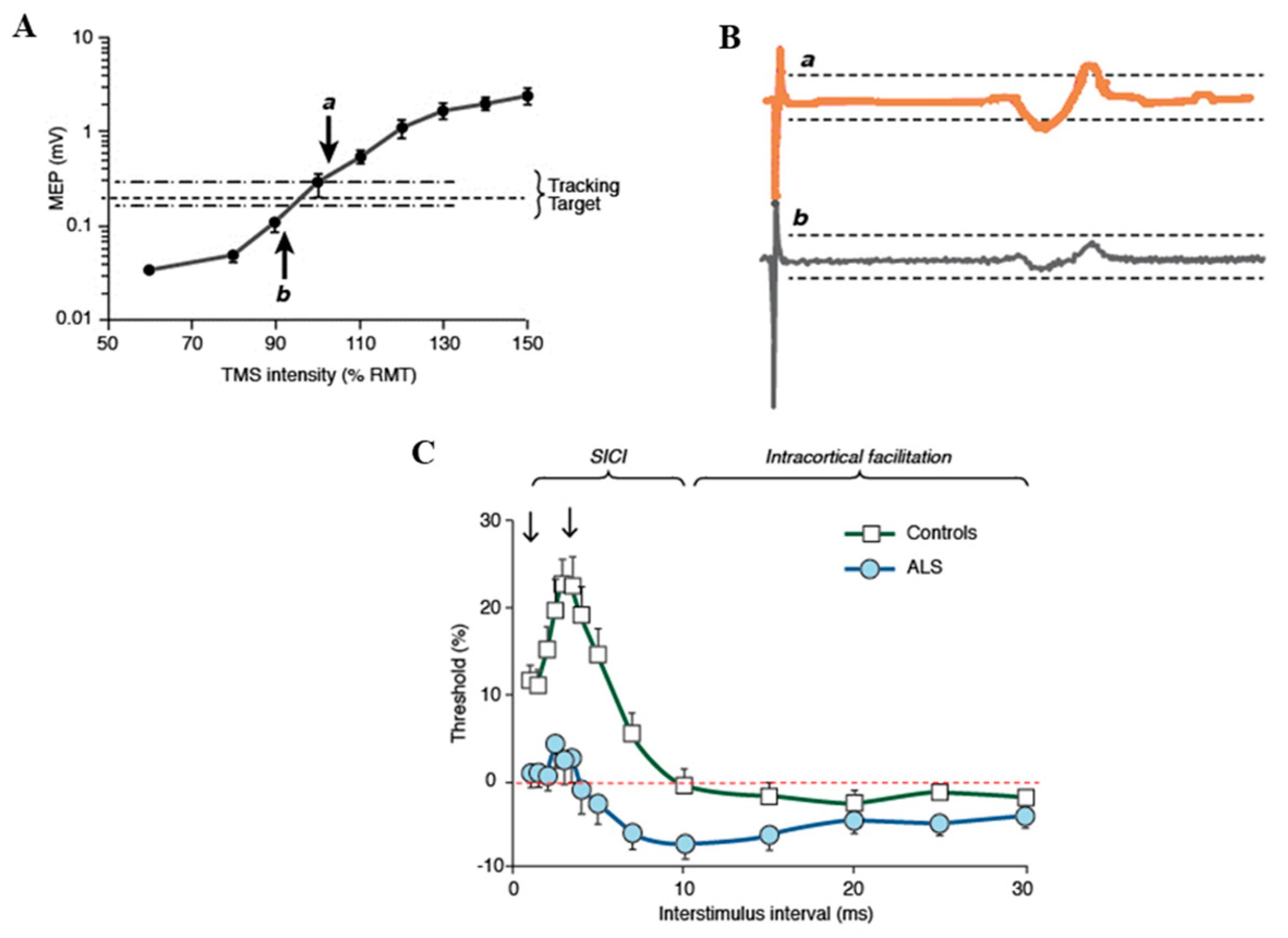
4. Pathogenic Insights from Advances in Neurophysiological (TMS) Techniques in ALS
5. Contrasting Views on ALS Pathogenesis and Site of Disease Onset
6. Novel Neurophysiological Diagnostic Biomarkers for ALS
Funding
Conflicts of Interest
References
- Brooks, B.; Miller, R.; Swash, M.; Munsat, T. El Escorial revisited: Revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Mot. Neuron Disord. 2000, 1, 293–299. [Google Scholar] [CrossRef]
- Charcot, J.; Joffroy, A. Deux cas d’atrophie musculaire progressive avec lesion de la substance grise et des faisceaux antero-lateraux de la moelle epiniere. Arch. Physiol. Neurol. Pathol. 1869, 2, 744–754. [Google Scholar]
- Geevasinga, N.; Menon, P.; Ozdinler, P.H.; Kiernan, M.C.; Vucic, S. Pathophysiological and diagnostic implications of cortical dysfunction in ALS. Nat. Rev. Neurol. 2016, 12, 651–661. [Google Scholar] [CrossRef] [PubMed]
- Vucic, S.; Rothstein, J.D.; Kiernan, M.C. Advances in treating amyotrophic lateral sclerosis: Insights from pathophysiological studies. Trends Neurosci. 2014, 37, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Dharmadasa, T.; Henderson, R.D.; Talman, P.S.; Macdonell, R.A.; Mathers, S.; Schultz, D.W.; Needham, M.; Zoing, M.; Vucic, S.; Kiernan, M.C. Motor neurone disease: Progress and challenges. Med. J. Aust. 2017, 206, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Eisen, A.; Braak, H.; Del Tredici, K.; Lemon, R.; Ludolph, A.C.; Kiernan, M.C. Cortical influences drive amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2017, 88, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Eisen, A.; Kim, S.; Pant, B. Amyotrophic lateral sclerosis (ALS): A phylogenetic disease of the corticomotoneuron? Muscle Nerve 1992, 15, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Williamson, T.L.; Cleveland, D.W. Slowing of axonal transport is a very early event in the toxicity of ALS-linked SOD1 mutants to motor neurons. Nat. Neurosci. 1999, 2, 50–56. [Google Scholar] [CrossRef]
- Fischer, L.R.; Culver, D.G.; Tennant, P.; Davis, A.A.; Wang, M.; Castellano-Sanchez, A.; Khan, J.; Polak, M.A.; Glass, J.D. Amyotrophic lateral sclerosis is a distal axonopathy: Evidence in mice and man. Exp. Neurol. 2004, 185, 232–240. [Google Scholar] [CrossRef]
- Ravits, J.; Paul, P.; Jorg, C. Focality of upper and lower motor neuron degeneration at the clinical onset of ALS. Neurology 2007, 68, 1571–1575. [Google Scholar] [CrossRef]
- Ravits, J.M.; La Spada, A.R. ALS motor phenotype heterogeneity, focality, and spread: Deconstructing motor neuron degeneration. Neurology 2009, 73, 805–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vucic, S.; Howells, J.; Trevillion, L.; Kiernan, M.C. Assessment of cortical excitability using threshold tracking techniques. Muscle Nerve 2006, 33, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Menon, P.; Geevasinga, N.; Yiannikas, C.; Howells, J.; Kiernan, M.; Vucic, S. The sensitivity and specificity of threshold-tracking transcranial magnetic stimulation for the diagnosis of amyotrophic lateral sclerosis: A prospective study. Lancet Neurol. 2015, 14, 478–484. [Google Scholar] [CrossRef]
- Al-Chalabi, A.; Calvo, A.; Chio, A.; Colville, S.; Ellis, C.M.; Hardiman, O.; Heverin, M.; Howard, R.S.; Huisman, M.H.B.; Keren, N.; et al. Analysis of amyotrophic lateral sclerosis as a multistep process: A population-based modelling study. Lancet Neurol. 2014, 13, 1108–1113. [Google Scholar] [CrossRef]
- Eisen, A.; Kiernan, M.; Mitsumoto, H.; Swash, M. Amyotrophic lateral sclerosis: A long preclinical period? J. Neurol. Neurosurg. Psychiatry 2014, 85, 1232–1238. [Google Scholar] [CrossRef] [PubMed]
- Eisen, A.A.; Shtybel, W. AAEM minimonograph #35: Clinical experience with transcranial magnetic stimulation. Muscle Nerve 1990, 13, 995–1011. [Google Scholar] [PubMed]
- Lemon, R.N.; Griffiths, J. Comparing the function of the corticospinal system in different species: Organizational differences for motor specialization? Muscle Nerve 2005, 32, 261–279. [Google Scholar] [CrossRef] [PubMed]
- Vucic, S. Split elbow sign: More evidence for the importance of cortical dysfunction in ALS. J. Neurol. Neurosurg. Psychiatry 2019. [Google Scholar] [CrossRef] [PubMed]
- Wilbourn, A.J. The “split hand syndrome”. Muscle Nerve 2000, 23, 138. [Google Scholar] [CrossRef]
- Kuwabara, S.; Sonoo, M.; Komori, T.; Shimizu, T.; Hirashima, F.; Inaba, A.; Misawa, S.; Hatanaka, Y. Dissociated small hand muscle atrophy in amyotrophic lateral sclerosis: Frequency, extent, and specificity. Muscle Nerve 2008, 37, 426–430. [Google Scholar] [CrossRef]
- Menon, P.; Kiernan, M.C.; Vucic, S. ALS pathophysiology: Insights form the split-hand phenomenon. Clin. Neurophysiol. 2014, 49, 836–844. [Google Scholar] [CrossRef] [PubMed]
- Menon, P.; Kiernan, M.C.; Vucic, S. Cortical dysfunction underlies the development of the split-hand in amyotrophic lateral sclerosis. PLoS ONE 2014, 9, e87124. [Google Scholar] [CrossRef] [PubMed]
- Menon, P.; Bae, J.S.; Mioshi, E.; Kiernan, M.C.; Vucic, S. Split-hand plus sign in ALS: Differential involvement of the flexor pollicis longus and intrinsic hand muscles. Amyotroph. Lateral Scler. Front. Degener. 2013, 14, 315–318. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.S.; Menon, P.; Mioshi, E.; Kiernan, M.C.; Vucic, S. Cortical hyperexcitability and the split-hand plus phenomenon: Pathophysiological insights in ALS. Amyotroph. Lateral Scler. Front. Degener. 2014, 15, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Khalaf, R.; Martin, S.; Ellis, C.; Burman, R.; Sreedharan, J.; Shaw, C.; Leigh, P.N.; Turner, M.R.; Al-Chalabi, A. Relative preservation of triceps over biceps strength in upper limb-onset ALS: The ‘split elbow’. J. Neurol. Neurosurg. Psychiatry 2019. [Google Scholar] [CrossRef] [PubMed]
- Simon, N.G.; Lee, M.; Bae, J.S.; Mioshi, E.; Lin, C.S.; Pfluger, C.M.; Henderson, R.D.; Vucic, S.; Swash, M.; Burke, D.; et al. Dissociated lower limb muscle involvement in amyotrophic lateral sclerosis. J. Neurol. 2015, 262, 1424–1432. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.R.; Wicks, P.; Brownstein, C.A.; Massagli, M.P.; Toronjo, M.; Talbot, K.; Al-Chalabi, A. Concordance between site of onset and limb dominance in amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2011, 82, 853–854. [Google Scholar] [CrossRef] [PubMed]
- Reid, C.S.; Serrien, D.J. Handedness and the excitability of cortical inhibitory circuits. Behav. Brain Res. 2012, 230, 144–148. [Google Scholar]
- Devine, M.S.; Kiernan, M.C.; Heggie, S.; McCombe, P.A.; Henderson, R.D. Study of motor asymmetry in ALS indicates an effect of limb dominance on onset and spread of weakness, and an important role for upper motor neurons. Amyotroph. Lateral Scler. Front. Degener. 2014, 15, 481–487. [Google Scholar] [CrossRef]
- Menon, P.; Geevasinga, N.; Van Den Bos, M.; Yiannikas, C.; Kiernan, M.C.; Vucic, S. Cortical hyperexcitability and disease spread in amyotrophic lateral sclerosis. Eur. J. Neurol. 2017, 24, 816–824. [Google Scholar] [CrossRef]
- Vucic, S.; Ziemann, U.; Eisen, A.; Hallett, M.; Kiernan, M.C. Transcranial magnetic stimulation and amyotrophic lateral sclerosis: Pathophysiological insights. J. Neurol. Neurosurg. Psychiatry 2013, 84, 1161–1170. [Google Scholar] [CrossRef] [PubMed]
- Vucic, S.; van den Bos, M.; Menon, P.; Howells, J.; Dharmadasa, T.; Kiernan, M.C. Utility of threshold tracking transcranial magnetic stimulation in ALS. Clin. Neurophysiol. Pract. 2018, 3, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Rudiak, D.; Marg, E. Finding the depth of magnetic brain stimulation: A re-evaluation. Electroencephalogr. Clin. Neurophysiol. 1994, 93, 358–371. [Google Scholar] [CrossRef]
- Di Lazzaro, V.; Profice, P.; Ranieri, F.; Capone, F.; Dileone, M.; Oliviero, A.; Pilato, F. I-wave origin and modulation. Brain Stim 2012, 5, 512–525. [Google Scholar] [CrossRef] [PubMed]
- Di Lazzaro, V.; Ranieri, F.; Profice, P.; Pilato, F.; Mazzone, P.; Capone, F.; Insola, A.; Oliviero, A. Transcranial Direct Current Stimulation Effects on the Excitability of Corticospinal Axons of the Human Cerebral Cortex. Brain Stim 2013, 6, 641–643. [Google Scholar] [CrossRef] [PubMed]
- Ziemann, U.; Reis, J.; Schwenkreis, P.; Rosanova, M.; Strafella, A.; Badawy, R.; Muller-Dahlhaus, F. TMS and drugs revisited 2014. Clin. Neurophysiol. 2015, 126, 1847–1868. [Google Scholar] [CrossRef] [PubMed]
- Vucic, S.; Kiernan, M.C. Transcranial Magnetic Stimulation for the Assessment of Neurodegenerative Disease. Neurotherapeutics 2017, 14, 91–106. [Google Scholar] [CrossRef]
- Rossini, P.M.; Burke, D.; Chen, R.; Cohen, L.G.; Daskalakis, Z.; Di Iorio, R.; Di Lazzaro, V.; Ferreri, F.; Fitzgerald, P.B.; George, M.S.; et al. Non-invasive electrical and magnetic stimulation of the brain, spinal cord, roots and peripheral nerves: Basic principles and procedures for routine clinical and research application. An updated report from an I.F.C.N. Committee. Clin. Neurophysiol. 2015, 126, 1071–1107. [Google Scholar] [CrossRef] [PubMed]
- Kujirai, T.; Caramia, M.D.; Rothwell, J.C.; Day, B.L.; Thompson, P.D.; Ferbert, A.; Wroe, S.; Asselman, P.; Marsden, C.D. Corticocortical inhibition in human motor cortex. J. Physiol. 1993, 471, 501–519. [Google Scholar] [CrossRef]
- Nakamura, H.; Kitagawa, H.; Kawaguchi, Y.; Tsuji, H. Intracortical facilitation and inhibition after transcranial magnetic stimulation in conscious humans. J. Physiol. 1997, 498, 817–823. [Google Scholar] [CrossRef]
- Ziemann, U.; Rothwell, J.C.; Ridding, M.C. Interaction between intracortical inhibition and facilitation in human motor cortex. J. Physiol. 1996, 496, 873–881. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Cros, D.; Curra, A.; Di Lazzaro, V.; Lefaucheur, J.P.; Magistris, M.R.; Mills, K.; Rosler, K.M.; Triggs, W.J.; Ugawa, Y.; et al. The clinical diagnostic utility of transcranial magnetic stimulation: Report of an IFCN committee. Clin. Neurophysiol. 2008, 119, 504–532. [Google Scholar] [CrossRef] [PubMed]
- Hanajima, R.; Ugawa, Y.; Terao, Y.; Sakai, K.; Furubayashi, T.; Machii, K.; Kanazawa, I. Paired-pulse magnetic stimulation of the human motor cortex: Differences among I waves. J. Physiol. 1998, 509, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Kiers, L.; Cros, D.; Chiappa, K.H.; Fang, J. Variability of motor potentials evoked by transcranial magnetic stimulation. Electroencephalogr. Clin. Neurophysiol. 1993, 89, 415–423. [Google Scholar] [CrossRef]
- Fisher, R.J.; Nakamura, Y.; Bestmann, S.; Rothwell, J.C.; Bostock, H. Two phases of intracortical inhibition revealed by transcranial magnetic threshold tracking. Exp. Brain Res. 2002, 143, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Samusyte, G.; Bostock, H.; Rothwell, J.; Koltzenburg, M. Short-interval intracortical inhibition: Comparison between conventional and threshold-tracking techniques. Brain Stimul. 2018, 11, 806–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziemann, U. TMS and drugs. Clin. Neurophysiol. 2004, 115, 1717–1729. [Google Scholar] [CrossRef]
- Stefan, K.; Kunesch, E.; Benecke, R.; Classen, J. Effects of riluzole on cortical excitability in patients with amyotrophic lateral sclerosis. Ann. Neurol. 2001, 49, 536–539. [Google Scholar] [CrossRef]
- Zanette, G.; Tamburin, S.; Manganotti, P.; Refatti, N.; Forgione, A.; Rizzuto, N. Different mechanisms contribute to motor cortex hyperexcitability in amyotrophic lateral sclerosis. Clin. Neurophysiol. 2002, 113, 1688–1697. [Google Scholar] [CrossRef]
- Vucic, S.; Kiernan, M.C. Novel threshold tracking techniques suggest that cortical hyperexcitability is an early feature of motor neuron disease. Brain 2006, 129, 2436–2446. [Google Scholar] [CrossRef]
- Vucic, S.; Kiernan, M.C. Cortical excitability testing distinguishes Kennedy’s disease from amyotrophic lateral sclerosis. Clin. Neurophysiol. 2008, 119, 1088–1096. [Google Scholar] [CrossRef] [PubMed]
- Vucic, S.; Nicholson, G.A.; Kiernan, M.C. Cortical hyperexcitability may precede the onset of familial amyotrophic lateral sclerosis. Brain 2008, 131, 1540–1550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menon, P.; Kiernan, M.C.; Vucic, S. Cortical hyperexcitability precedes lower motor neuron dysfunction in ALS. Clin. Neurophysiol. 2015, 126, 803–809. [Google Scholar] [CrossRef] [PubMed]
- Vucic, S.; Kiernan, M.C. Abnormalities in cortical and peripheral excitability in flail arm variant amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2007, 78, 849–852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibuya, K.; Park, S.B.; Geevasinga, N.; Menon, P.; Howells, J.; Simon, N.G.; Huynh, W.; Noto, Y.; Gotz, J.; Kril, J.J.; et al. Motor cortical function determines prognosis in sporadic ALS. Neurology 2016, 87, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Vucic, S.; Lin, C.S.-Y.; Cheah, B.C.; Murray, J.; Menon, P.; Krishnan, A.V.; Kiernan, M.C. Riluzole exerts central and peripheral modulating effects in amyotrophic lateral sclerosis. Brain 2013, 136, 1361–1370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacomblez, L.; Bensimon, G.; Leigh, P.N.; Guillet, P.; Meininger, V. Dose-ranging study of riluzole in amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis/Riluzole Study Group II. Lancet 1996, 347, 1425–1431. [Google Scholar] [CrossRef]
- Bensimon, G.; Lacomblez, L.; Meininger, V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N. Engl. J. Med. 1994, 330, 585–591. [Google Scholar] [CrossRef]
- Geevasinga, N.; Menon, P.; Ng, K.; Van Den Bos, M.; Byth, K.; Kiernan, M.C.; Vucic, S. Riluzole exerts transient modulating effects on cortical and axonal hyperexcitability in ALS. Amyotroph. Lateral Scler. Front. Degener. 2016, 17, 1–9. [Google Scholar] [CrossRef]
- Kuo, M.T. Redox regulation of multidrug resistance in cancer chemotherapy: Molecular mechanisms and therapeutic opportunities. Antioxid. Redox Signal. 2009, 11, 99–133. [Google Scholar] [CrossRef]
- Williams, K.L.; Fifita, J.A.; Vucic, S.; Durnall, J.C.; Kiernan, M.C.; Blair, I.P.; Nicholson, G.A. Pathophysiological insights into ALS with C9ORF72 expansions. J. Neurol. Neurosurg. Psychiatry 2013, 84, 931–935. [Google Scholar] [CrossRef] [PubMed]
- Geevasinga, N.; Menon, P.; Nicholson, G.A.; Ng, K.; Howells, J.; Kril, J.J.; Yiannikas, C.; Kiernan, M.C.; Vucic, S. Cortical Function in Asymptomatic Carriers and Patients With C9orf72 Amyotrophic Lateral Sclerosis. Jama Neurol. 2015, 72, 1268–1274. [Google Scholar] [CrossRef] [PubMed]
- Vucic, S.; Kiernan, M.C. Upregulation of persistent sodium conductances in familial ALS. J. Neurol. Neurosurg. Psychiatry 2010, 81, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhang, L.; Liang, B.; Schroeder, D.; Zhang, Z.; Cox, G.A.; Li, Y.; Lin, D.T. Hyperactive Somatostatin Interneurons Contribute to Excitotoxicity in Neurodegenerative Disorders. Nat. Neurosci. 2016, 19, 557–559. [Google Scholar] [CrossRef] [PubMed]
- Tokimura, H.; Ridding, M.C.; Tokimura, Y.; Amassian, V.E.; Rothwell, J.C. Short latency facilitation between pairs of threshold magnetic stimuli applied to human motor cortex. Electroencephalogr. Clin. Neurophysiol. 1996, 101, 263–272. [Google Scholar] [CrossRef]
- Van den Bos, M.A.J.; Menon, P.; Howells, J.; Geevasinga, N.; Kiernan, M.C.; Vucic, S. Physiological Processes Underlying Short Interval Intracortical Facilitation in the Human Motor Cortex. Front. Neurosci. 2018, 12, 240. [Google Scholar] [CrossRef] [PubMed]
- Ziemann, U.; Tergau, F.; Wischer, S.; Hildebrandt, J.; Paulus, W. Pharmacological control of facilitatory I-wave interaction in the human motor cortex. A paired transcranial magnetic stimulation study. Electroencephalogr. Clin. Neurophysiol. 1998, 109, 321–330. [Google Scholar] [CrossRef]
- Di Lazzaro, V.; Rothwell, J.C.; Oliviero, A.; Profice, P.; Insola, A.; Mazzone, P.; Tonali, P. Intracortical origin of the short latency facilitation produced by pairs of threshold magnetic stimuli applied to human motor cortex. Exp. Brain Res. 1999, 129, 494–499. [Google Scholar] [CrossRef]
- van den Bos, M.A.J.; Menon, P.; Geevasinga, N.; Kiernan, M.C.; Vucic, S. 6. ALS disability is predicted by a shift in the balance between short latency facilitatory and inhibitory circuits. Clin. Neurophysiol. 2018, 129, e3. [Google Scholar] [CrossRef]
- Van den Bos, M.A.J.; Higashihara, M.; Geevasinga, N.; Menon, P.; Kiernan, M.C.; Vucic, S. Imbalance of cortical facilitatory and inhibitory circuits underlies hyperexcitability in ALS. Neurology 2018, 91, e1669–e1676. [Google Scholar] [CrossRef]
- Desiato, M.T.; Caramia, M.D. Towards a neurophysiological marker of amyotrophic lateral sclerosis as revealed by changes in cortical excitability. Electroencephalogr. Clin. Neurophysiol. 1997, 105, 1–7. [Google Scholar] [CrossRef]
- Prout, A.J.; Eisen, A. The cortical silent period and ALS. Muscle Nerve 1994, 17, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Siciliano, G.; Manca, M.L.; Sagliocco, L.; Pastorini, E.; Pellegrinetti, A.; Sartucci, F.; Sabatini, A.; Murri, L. Cortical silent period in patients with amyotrophic lateral sclerosis. J. Neurol. Sci. 1999, 169, 93–97. [Google Scholar] [CrossRef]
- Vucic, S.; Nicholson, G.A.; Kiernan, M.C. Cortical excitability in hereditary motor neuronopathy with pyramidal signs: Comparison with ALS. J. Neurol. Neurosurg. Psychiatry 2010, 81, 97–100. [Google Scholar] [CrossRef] [PubMed]
- Wittstock, M.; Wolters, A.; Benecke, R. Transcallosal inhibition in amyotrophic lateral sclerosis. Clin. Neurophysiol. 2007, 118, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Geevasinga, N.; Korgaonkar, M.S.; Menon, P.; Van den Bos, M.; Gomes, L.; Foster, S.; Kiernan, M.C.; Vucic, S. Brain functional connectome abnormalities in amyotrophic lateral sclerosis are associated with disability and cortical hyperexcitability. Eur. J. Neurol. 2017, 24, 1507–1517. [Google Scholar] [CrossRef]
- Geevasinga, N.; Menon, P.; Sue, C.M.; Kumar, K.R.; Ng, K.; Yiannikas, C.; Kiernan, M.C.; Vucic, S. Cortical excitability changes distinguish the motor neuron disease phenotypes from hereditary spastic paraplegia. Eur. J. Neurol. 2015, 22, 826–831. [Google Scholar] [CrossRef] [PubMed]
- Grieve, S.M.; Menon, P.; Korgaonkar, M.S.; Gomes, L.; Foster, S.; Kiernan, M.C.; Vucic, S. Potential structural and functional biomarkers of upper motor neuron dysfunction in ALS. Amyotroph. Lateral Scler. Front. Degener. 2015, 17, 1–8. [Google Scholar]
- Mills, K.R. The natural history of central motor abnormalities in amyotrophic lateral sclerosis. Brain 2003, 126, 2558–2566. [Google Scholar] [CrossRef] [Green Version]
- Menon, P.; Geevasinga, N.; Yiannikas, C.; Kiernan, M.C.; Vucic, S. Cortical contributions to the flail leg syndrome: Pathophysiological insights. Amyotroph. Lateral Scler. Front. Degener. 2016, 17, 389–396. [Google Scholar] [CrossRef]
- Vucic, S.; Cheah, B.C.; Yiannikas, C.; Kiernan, M.C. Cortical excitability distinguishes ALS from mimic disorders. Clin. Neurophysiol. 2011, 122, 1860–1866. [Google Scholar] [CrossRef] [PubMed]
- Brouwer, B.; Ashby, P. Corticospinal projections to upper and lower limb spinal motoneurons in man. Electroencephalogr. Clin. Neurophysiol. 1990, 76, 509–519. [Google Scholar] [CrossRef]
- Chen, R.; Tam, A.; Butefisch, C.; Corwell, B.; Ziemann, U.; Rothwell, J.C.; Cohen, L.G. Intracortical inhibition and facilitation in different representations of the human motor cortex. J. Neurophysiol. 1998, 80, 2870–2881. [Google Scholar] [CrossRef] [PubMed]
- Macdonell, R.A.; Shapiro, B.E.; Chiappa, K.H.; Helmers, S.L.; Cros, D.; Day, B.J.; Shahani, B.T. Hemispheric threshold differences for motor evoked potentials produced by magnetic coil stimulation. Neurology 1991, 41, 1441–1444. [Google Scholar] [CrossRef] [PubMed]
- Boroojerdi, B.; Battaglia, F.; Muellbacher, W.; Cohen, L.G. Mechanisms influencing stimulus-response properties of the human corticospinal system. Clin. Neurophysiol. 2001, 112, 931–937. [Google Scholar] [CrossRef]
- Lang, N.; Rothkegel, H.; Peckolt, H.; Deuschl, G. Effects of lacosamide and carbamazepine on human motor cortex excitability: A double-blind, placebo-controlled transcranial magnetic stimulation study. Seizure 2013, 22, 726–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sommer, M.; Gileles, E.; Knappmeyer, K.; Rothkegel, H.; Polania, R.; Paulus, W. Carbamazepine reduces short-interval interhemispheric inhibition in healthy humans. Clin. Neurophysiol. 2012, 123, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Mavroudakis, N.; Caroyer, J.M.; Brunko, E.; Zegers de Beyl, D. Effects of diphenylhydantoin on motor potentials evoked with magnetic stimulation. Electroencephalogr. Clin. Neurophysiol. 1994, 93, 428–433. [Google Scholar] [CrossRef]
- Di Lazzaro, V.; Oliviero, A.; Profice, P.; Pennisi, M.A.; Pilato, F.; Zito, G.; Dileone, M.; Nicoletti, R.; Pasqualetti, P.; Tonali, P.A. Ketamine increases human motor cortex excitability to transcranial magnetic stimulation. J. Physiol. 2003, 547, 485–496. [Google Scholar] [CrossRef]
- Caramia, M.D.; Cicinelli, P.; Paradiso, C.; Mariorenzi, R.; Zarola, F.; Bernardi, G.; Rossini, P.M. Excitability changes of muscular responses to magnetic brain stimulation in patients with central motor disorders. Electroencephalogr. Clin. Neurophysiol. 1991, 81, 243–250. [Google Scholar] [CrossRef]
- Kohara, N.; Kaji, R.; Kojima, Y.; Mills, K.R.; Fujii, H.; Hamano, T.; Kimura, J.; Takamatsu, N.; Uchiyama, T. Abnormal excitability of the corticospinal pathway in patients with amyotrophic lateral sclerosis: A single motor unit study using transcranial magnetic stimulation. Electroencephalogr. Clin. Neurophysiol. 1996, 101, 32–41. [Google Scholar] [CrossRef]
- Mills, K.R.; Nithi, K.A. Corticomotor threshold is reduced in early sporadic amyotrophic lateral sclerosis. Muscle Nerve 1997, 20, 1137–1141. [Google Scholar] [CrossRef]
- Eisen, A.; Weber, M. The motor cortex and amyotrophic lateral sclerosis. Muscle Nerve 2001, 24, 564–573. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, K.; Simon, N.G.; Geevasinga, N.; Menon, P.; Howells, J.; Park, S.B.; Huynh, W.; Noto, Y.I.; Vucic, S.; Kiernan, M.C. The evolution of motor cortical dysfunction in amyotrophic lateral sclerosis. Clin. Neurophysiol. 2017, 128, 1075–1082. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Highton-Williamson, E.; Caga, J.; Matamala, J.M.; Dharmadasa, T.; Howells, J.; Zoing, M.C.; Shibuya, K.; Geevasinga, N.; Vucic, S.; et al. Primary lateral sclerosis and the amyotrophic lateral sclerosis-frontotemporal dementia spectrum. J. Neurol. 2018, 265, 1819–1828. [Google Scholar] [CrossRef] [PubMed]
- Ziemann, U. Cortical threshold and excitability measurements. In Clinical Neurophysiology of Motor Neuron Diseases. Handbook of Clinical Neurophysiology; Eisen, A., Ed.; Elsevier: Amsterdam, The Netherlands, 2004; pp. 317–335. [Google Scholar]
- Boillee, S.; Vande Velde, C.; Cleveland, D.W. ALS: A disease of motor neurons and their nonneuronal neighbors. Neuron 2006, 52, 39–59. [Google Scholar] [CrossRef] [PubMed]
- Rothstein, J.D.; Van Kammen, M.; Levey, A.I.; Martin, L.J.; Kuncl, R.W. Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann. Neurol. 1995, 38, 73–84. [Google Scholar] [CrossRef]
- Trotti, D.; Rolfs, A.; Danbolt, N.C.; Brown, R.H., Jr.; Hediger, M.A. SOD1 mutants linked to amyotrophic lateral sclerosis selectively inactivate a glial glutamate transporter. Nat. Neurosci. 1999, 2, 848. [Google Scholar] [CrossRef]
- Anderson, C.M.; Swanson, R.A. Astrocyte glutamate transport: Review of properties, regulation, and physiological functions. Glia 2000, 32, 1–14. [Google Scholar] [CrossRef]
- Boston-Howes, W.; Gibb, S.L.; Williams, E.O.; Pasinelli, P.; Brown, R.H., Jr.; Trotti, D. Caspase-3 cleaves and inactivates the glutamate transporter EAAT2. J. Biol. Chem. 2006, 281, 14076–14084. [Google Scholar] [CrossRef]
- Gibb, S.L.; Boston-Howes, W.; Lavina, Z.S.; Gustincich, S.; Brown, R.H., Jr.; Pasinelli, P.; Trotti, D. A Caspase-3-cleaved Fragment of the Glial Glutamate Transporter EAAT2 Is Sumoylated and Targeted to Promyelocytic Leukemia Nuclear Bodies in Mutant SOD1-linked Amyotrophic Lateral Sclerosis. J. Biol. Chem. 2007, 282, 32480–32490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothstein, J.D.; Patel, S.; Regan, M.R.; Haenggeli, C.; Huang, Y.H.; Bergles, D.E.; Jin, L.; Dykes Hoberg, M.; Vidensky, S.; Chung, D.S.; et al. Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature 2005, 433, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Pieri, M.; Carunchio, I.; Curcio, L.; Mercuri, N.B.; Zona, C. Increased persistent sodium current determines cortical hyperexcitability in a genetic model of amyotrophic lateral sclerosis. Exp. Neurol. 2009, 215, 368–379. [Google Scholar] [CrossRef] [PubMed]
- Saba, L.; Viscomi, M.T.; Caioli, S.; Pignataro, A.; Bisicchia, E.; Pieri, M.; Molinari, M.; Ammassari-Teule, M.; Zona, C. Altered Functionality, Morphology, and Vesicular Glutamate Transporter Expression of Cortical Motor Neurons from a Presymptomatic Mouse Model of Amyotrophic Lateral Sclerosis. Cereb Cortex 2015, 26, 1512–1528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fogarty, M.J.; Noakes, P.G.; Bellingham, M.C. Motor Cortex Layer V Pyramidal Neurons Exhibit Dendritic Regression, Spine Loss, and Increased Synaptic Excitation in the Presymptomatic hSOD1G93A Mouse Model of Amyotrophic Lateral Sclerosis. J. Neurosci. 2015, 35, 643–647. [Google Scholar] [CrossRef] [PubMed]
- Jara, J.H.; Villa, S.R.; Khan, N.A.; Bohn, M.C.; Özdinler, P.H. AAV2 mediated retrograde transduction of corticospinal motor neurons reveals initial and selective apical dendrite degeneration in ALS. Neurobiol. Dis. 2012, 47, 174–183. [Google Scholar] [CrossRef] [Green Version]
- Özdinler, P.H.; Benn, S.; Yamamoto, T.H.; Güzel, M.; Brown, R.H.; Macklis, J.D. Corticospinal Motor Neurons and Related Subcerebral Projection Neurons Undergo Early and Specific Neurodegeneration in hSOD1G93A Transgenic ALS Mice. J. Neurosci. 2011, 31, 4166–4177. [Google Scholar] [CrossRef] [Green Version]
- Kuo, J.J.; Schonewille, M.; Siddique, T.; Schults, A.N.; Fu, R.; Bar, P.R.; Anelli, R.; Heckman, C.J.; Kroese, A.B. Hyperexcitability of cultured spinal motoneurons from presymptomatic ALS mice. J. Neurophysiol. 2004, 91, 571–575. [Google Scholar] [CrossRef]
- Imbrici, P.; D’Adamo, M.C.; Kullmann, D.M.; Pessia, M. Episodic ataxia type 1 mutations in the KCNA1 gene impair the fast inactivation properties of the human potassium channels Kv1.4-1.1/Kvbeta1.1 and Kv1.4-1.1/Kvbeta1.2. Eur. J. Neurosci. 2006, 24, 3073–3083. [Google Scholar] [CrossRef]
- Jara, J.H.; Genc, B.; Klessner, J.L.; Ozdinler, P.H. Retrograde labeling, transduction, and genetic targeting allow cellular analysis of corticospinal motor neurons: Implications in health and disease. Front. Neuroanat. 2014, 8, 16. [Google Scholar] [CrossRef]
- Jara, J.H.; Genc, B.; Cox, G.A.; Bohn, M.C.; Roos, R.P.; Macklis, J.D.; Ulupinar, E.; Ozdinler, P.H. Corticospinal Motor Neurons Are Susceptible to Increased ER Stress and Display Profound Degeneration in the Absence of UCHL1 Function. Cereb. Cortex 2015, 25, 4259–4272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wainger, B.J.; Kiskinis, E.; Mellin, C.; Wiskow, O.; Han, S.S.; Sandoe, J.; Perez, N.P.; Williams, L.A.; Lee, S.; Boulting, G.; et al. Intrinsic Membrane Hyperexcitability of Amyotrophic Lateral Sclerosis Patient-Derived Motor Neurons. Cell Rep. 2014, 7, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, J.J.; Siddique, T.; Fu, R.; Heckman, C.J. Increased persistent Na(+) current and its effect on excitability in motoneurones cultured from mutant SOD1 mice. J. Physiol. 2005, 563, 843–854. [Google Scholar] [CrossRef] [PubMed]
- Kanai, K.; Kuwabara, S.; Misawa, S.; Tamura, N.; Ogawara, K.; Nakata, M.; Sawai, S.; Hattori, T.; Bostock, H. Altered axonal excitability properties in amyotrophic lateral sclerosis: Impaired potassium channel function related to disease stage. Brain 2006, 129, 953–962. [Google Scholar] [CrossRef] [PubMed]
- Geevasinga, N.; Menon, P.; Howells, J.; Nicholson, G.A.; Kiernan, M.C.; Vucic, S. Axonal ion channel dysfunction in c9orf72 familial amyotrophic lateral sclerosis. Jama Neurol. 2015, 72, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Saxena, S.; Roselli, F.; Singh, K.; Leptien, K.; Julien, J.-P.; Gros-Louis, F.; Caroni, P. Neuroprotection through Excitability and mTOR Required in ALS Motoneurons to Delay Disease and Extend Survival. Neuron 2013, 80, 80–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leroy, F.; Lamotte d’Incamps, B.; Imhoff-Manuel, R.D.; Zytnicki, D. Early intrinsic hyperexcitability does not contribute to motoneuron degeneration in amyotrophic lateral sclerosis. eLife 2014, 3, e04046. [Google Scholar] [CrossRef]
- Haidet-Phillips, A.M.; Hester, M.E.; Miranda, C.J.; Meyer, K.; Braun, L.; Frakes, A.; Song, S.; Likhite, S.; Murtha, M.J.; Foust, K.D.; et al. Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat. Biotech. 2011, 29, 824–828. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Lee, M.-H.; Henderson, L.; Tyagi, R.; Bachani, M.; Steiner, J.; Campanac, E.; Hoffman, D.A.; von Geldern, G.; Johnson, K.; et al. Human endogenous retrovirus-K contributes to motor neuron disease. Sci. Transl. Med. 2015, 7, 307ra153. [Google Scholar] [CrossRef]
- Gould, T.W.; Buss, R.R.; Vinsant, S.; Prevette, D.; Sun, W.; Knudson, C.M.; Milligan, C.E.; Oppenheim, R.W. Complete dissociation of motor neuron death from motor dysfunction by Bax deletion in a mouse model of ALS. J. Neurosci. 2006, 26, 8774–8786. [Google Scholar] [CrossRef]
- Pun, S.; Santos, A.F.; Saxena, S.; Xu, L.; Caroni, P. Selective vulnerability and pruning of phasic motoneuron axons in motoneuron disease alleviated by CNTF. Nat. Neurosci. 2006, 9, 408–419. [Google Scholar] [CrossRef] [PubMed]
- Pagani, M.R.; Reisin, R.C.; Uchitel, O.D. Calcium signaling pathways mediating synaptic potentiation triggered by amyotrophic lateral sclerosis IgG in motor nerve terminals. J. Neurosci. 2006, 26, 2661–2672. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.R.; Kiernan, M.C.; Leigh, P.N.; Talbot, K. Biomarkers in amyotrophic lateral sclerosis. Lancet Neurol. 2009, 8, 94–109. [Google Scholar] [CrossRef]
- Gowers, W. A Manual of Diseases of the Nervous System: Spinal Cord and Nerves; Churchill: London, UK, 1888; pp. 356–381.
- Kiernan, J.; Hudson, A. Changes in sizes of cortical and lower motor neurons in amyotrophic lateral sclerosis. Brain 1991, 114, 843–853. [Google Scholar] [CrossRef] [PubMed]
- Pamphlett, R.; Kril, J.; Hng, T. Motor neuron disease: A primary disorder of corticomotoneurons? Muscle Nerve 1995, 18, 314–318. [Google Scholar] [CrossRef] [PubMed]
- Flament, D.; Goldsmith, P.; Buckley, C.J.; Lemon, R.N. Task dependence of responses in first dorsal interosseous muscle to magnetic brain stimulation in man. J. Physiol. 1993, 464, 361–378. [Google Scholar] [CrossRef] [PubMed]
- de Carvalho, M.; Dengler, R.; Eisen, A.; England, J.D.; Kaji, R.; Kimura, J.; Mills, K.; Mitsumoto, H.; Nodera, H.; Shefner, J.; et al. Electrodiagnostic criteria for diagnosis of ALS. Clin. Neurophysiol. 2008, 119, 497–503. [Google Scholar] [CrossRef]
- Geevasinga, N.; Loy, C.T.; Menon, P.; de Carvalho, M.; Swash, M.; Schrooten, M.; Van Damme, P.; Gawel, M.; Sonoo, M.; Higashihara, M.; et al. Awaji criteria improves the diagnostic sensitivity in amyotrophic lateral sclerosis: A systematic review using individual patient data. Clin. Neurophysiol. 2016, 127, 2684–2691. [Google Scholar] [CrossRef]
- Costa, J.; Swash, M.; de Carvalho, M. Awaji criteria for the diagnosis of amyotrophic lateral sclerosis: A systematic review. Arch. Neurol. 2012, 69, 1410–1416. [Google Scholar] [CrossRef]
- Geevasinga, N.; Menon, P.; Scherman, D.B.; Simon, N.; Yiannikas, C.; Henderson, R.D.; Kiernan, M.C.; Vucic, S. Diagnostic criteria in amyotrophic lateral sclerosis A multicenter prospective study. Neurology 2016, 87, 684–690. [Google Scholar] [CrossRef]
- Higashihara, M.; Sonoo, M.; Imafuku, I.; Fukutake, T.; Kamakura, K.; Inoue, K.; Hatanaka, Y.; Shimizu, T.; Tsuji, S.; Ugawa, Y. Fasciculation potentials in amyotrophic lateral sclerosis and the diagnostic yield of the Awaji algorithm. Muscle Nerve 2012, 45, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Geevasinga, N.; Howells, J.; Menon, P.; van den Bos, M.; Shibuya, K.; Matamala, J.M.; Park, S.B.; Byth, K.; Kiernan, M.C.; Vucic, S. Amyotrophic lateral sclerosis diagnostic index: Toward a personalized diagnosis of ALS. Neurology 2019, 92, e536–e547. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
van den Bos, M.A.J.; Geevasinga, N.; Higashihara, M.; Menon, P.; Vucic, S. Pathophysiology and Diagnosis of ALS: Insights from Advances in Neurophysiological Techniques. Int. J. Mol. Sci. 2019, 20, 2818. https://doi.org/10.3390/ijms20112818
van den Bos MAJ, Geevasinga N, Higashihara M, Menon P, Vucic S. Pathophysiology and Diagnosis of ALS: Insights from Advances in Neurophysiological Techniques. International Journal of Molecular Sciences. 2019; 20(11):2818. https://doi.org/10.3390/ijms20112818
Chicago/Turabian Stylevan den Bos, Mehdi A. J., Nimeshan Geevasinga, Mana Higashihara, Parvathi Menon, and Steve Vucic. 2019. "Pathophysiology and Diagnosis of ALS: Insights from Advances in Neurophysiological Techniques" International Journal of Molecular Sciences 20, no. 11: 2818. https://doi.org/10.3390/ijms20112818
APA Stylevan den Bos, M. A. J., Geevasinga, N., Higashihara, M., Menon, P., & Vucic, S. (2019). Pathophysiology and Diagnosis of ALS: Insights from Advances in Neurophysiological Techniques. International Journal of Molecular Sciences, 20(11), 2818. https://doi.org/10.3390/ijms20112818