Biology and Role of Extracellular Vesicles (EVs) in the Pathogenesis of Thrombosis



Abstract
:1. Introduction
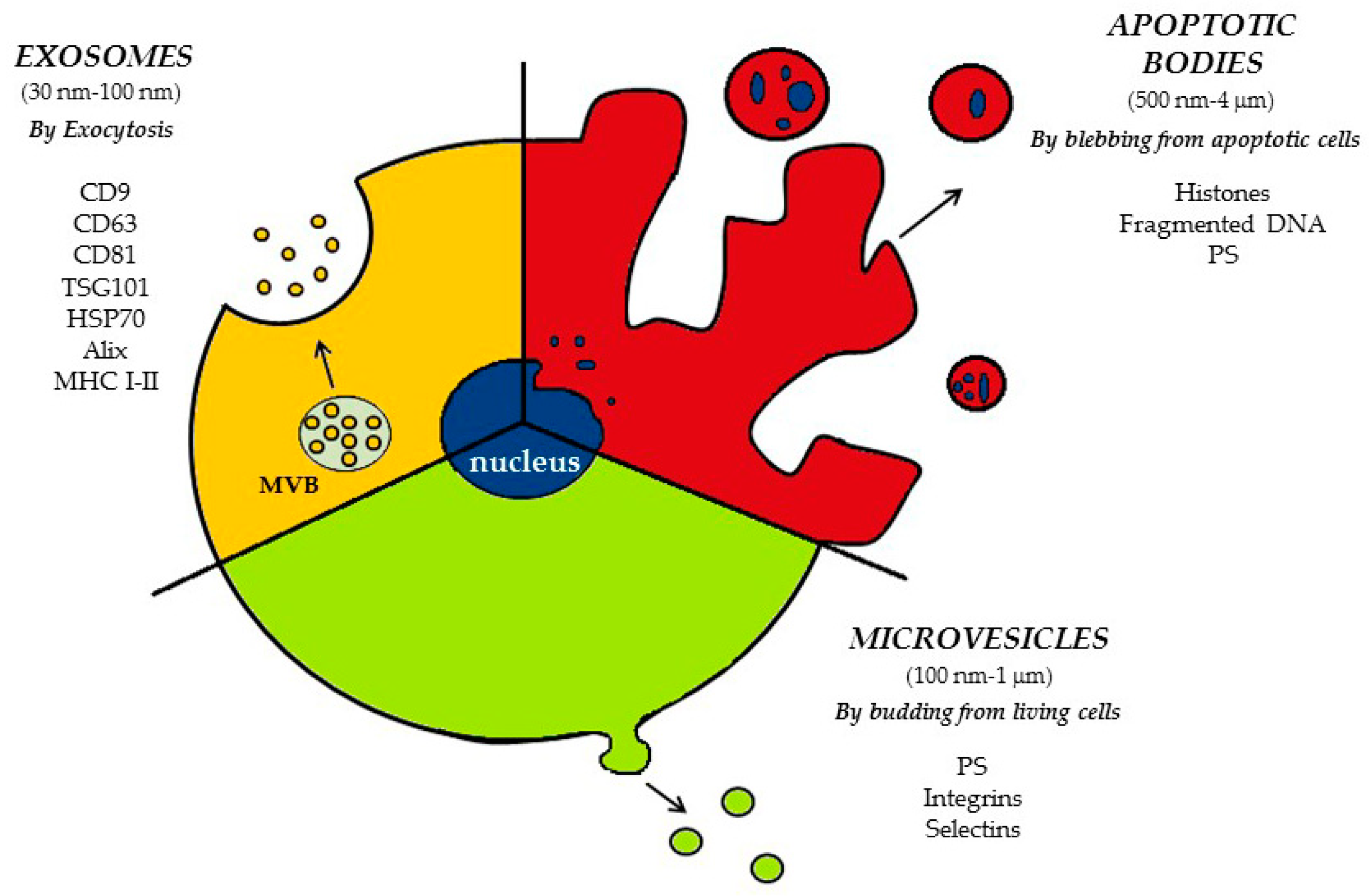
2. Classification and Biogenesis of EVs
2.1. Apoptotic Bodies (ApoBDs)
2.2. Microvesicles (MVs)
2.3. Exosomes
3. EVs in Intercellular Communication
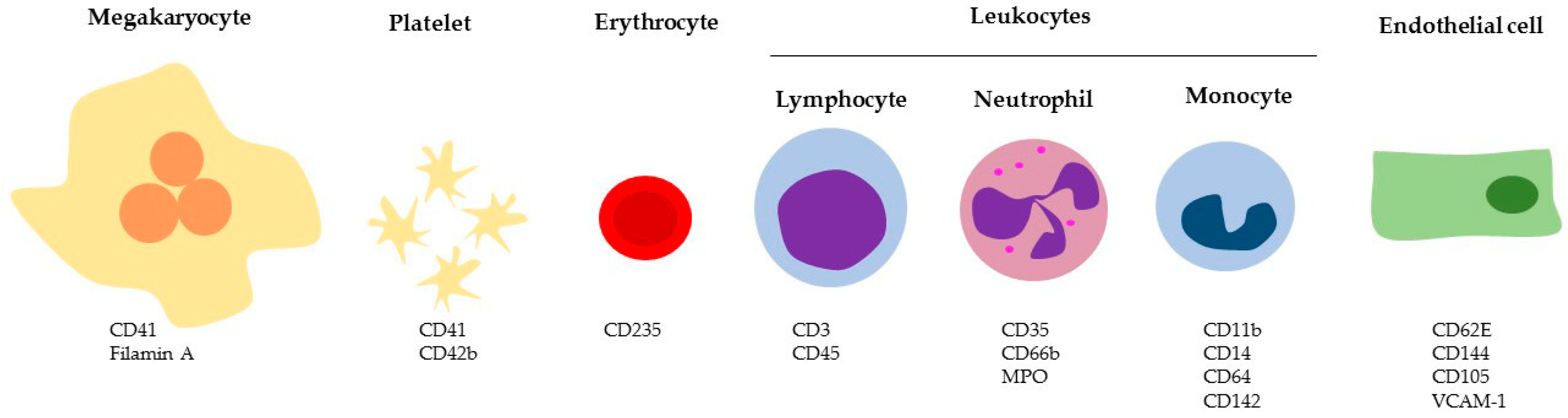
4. Circulating EVs
4.1. Megakaryocyte and Platelet-Derived Vesicles
4.2. Erythrocyte-Derived Vesicles
4.3. Leukocyte-Derived Vesicles
4.4. Endothelial-Derived Vesicles
5. Involvement of EVs in Arterial and Venous Thrombosis
5.1. Effect of EVs on Thrombosis
5.2. Pathological Function of EVs in Animal Models of Arterial and Venous Thrombosis
Pathological Function of EVs in Cancer-Associated Thrombosis
6. Clinical Applications
6.1. EVs as Biomarkers in Arterial Thrombosis
6.2. EVs as Biomarkers in Venous Thrombosis
EVs as Biomarkers in Cancer-Associated Thrombosis
6.3. Potential Therapeutic Application of EVs in Cardiovascular Diseases
7. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACS | Acute coronary syndrome |
| AFM | Atomic force microscope |
| AMI | Acute myocardial infarction |
| ApoBD | Apoptotic body |
| ARF6 | Adenosine diphosphate-ribosylation factor 6 |
| ALIX | ALG-2 interacting protein X |
| ARRCD1 | Arrestin domain-containing protein-1 |
| ATP | Adenosine triphosphate |
| C3 | Complement receptor 3 |
| CABG | Coronary-artery-by-pass-graft |
| CAD | Coronary artery disease |
| CD62L | L-selectin |
| CXCL | Chemokine (C-X-C motif) ligand |
| DLS | Dynamic light scattering |
| DVT | Deep vein thrombosis |
| EC-MV | Endothelial cell microvesicle |
| ELISA | Enzyme-linked immunosorbent assay |
| EM | Electron microscopy |
| EPC | Endothelial progenitor cell |
| ERK | Extracellular signaling-regulated kinase |
| ESC | European Society of Cardiology |
| ESCRT | Endosomal sorting complex required for transport |
| EV | Extracellular Vesicle |
| EXO | Exosome |
| FACS | Fluorescence activated cell sorting |
| FC | Flow cytometry |
| FeCl3 | Ferric chloride |
| GPIb | Glycoprotein Ib |
| GPIIb/IIIa | Glycoprotein IIb/IIIa |
| GPV | Glycoprotein V |
| GPVI | Glycoprotein VI |
| GTP | Guanosine-5′-triphosphate |
| HMGB1 | High mobility group box 1 |
| HSP70 | Heat shock proteins 70 Kd |
| ICAM-1 | Intracellular adhesion molecule 1 |
| IF | Immunofluorescence microscopy |
| ILV | Intraluminal vesicle |
| INFα | Interferon α |
| ISEV | International Society for Extracellular Vesicles |
| ISTH | International Society on Thrombosis and Haemostasis |
| IVC | Inferior vena cava |
| LDL | Low-density lipoprotein |
| LPS | Lipopolysaccharide |
| MI | Myocardial infarction |
| MLC | Myosin light-chain |
| MLCK | Myosin light-chain kinase |
| MMP | Metalloproteinase |
| MSC | Mesenchymal stem cell |
| MV | Microvesicle |
| MVB | Multivesicular body |
| NET | Neutrophil extracellular trap |
| NTA | Nanoparticle tracking analysis |
| PDI | Protein disulfide-isomerase |
| PE | Pulmonary embolism |
| PFA-100 | Platelet function analyzer |
| PKC | Protein kinase C |
| PLD | Phospholipase D |
| PMP | Platelet-derived microparticle |
| PS | Phosphatidylserine |
| PSGL-1 | P-selectin glycoprotein ligand-1 |
| PTE | Pulmonary thromboembolism |
| RNA | Ribonucleic acid |
| RPS | Resistive pulse sensing |
| STEMI | ST elevation myocardial infarction |
| TEM | Transmission electron microscopy |
| TF | Tissue factor |
| TFPI | Tissue factor pathway inhibitor |
| TM | Thrombomodulin |
| TNF-α | Tumor necrosis factor α |
| TRAP | Thrombin receptor activating peptide |
| TSG101 | Tumor susceptibility gene 101 |
| TXB2 | Thromboxane B2 |
| uPAR | Urokinase-type plasminogen activator receptor |
| VCAM-1 | Vascular cell adhesion protein 1 |
| Vps-4 | Vacuolar protein sorting-associated protein 4 |
| VTE | Venous thromboembolism |
| WB | Western blotting |
References
- Wendelboe, A.M.; Raskob, G.E. Global Burden of Thrombosis: Epidemiologic Aspects. Circ. Res. 2016, 118, 1340–1347. [Google Scholar] [CrossRef] [PubMed]
- Koupenova, M.; Kehrel, B.E.; Corkrey, H.A.; Freedman, J.E. Thrombosis and platelets: An update. Eur. Heart J. 2017, 38, 785–791. [Google Scholar] [CrossRef]
- Heit, J.A. Epidemiology of venous thromboembolism. Nat. Rev. Cardiol. 2015, 12, 464–474. [Google Scholar] [CrossRef] [PubMed]
- Prandoni, P. Venous and arterial thrombosis: Two aspects of the same disease? Clin. Epidemiol. 2009, 1, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Pol, E.; Boing, A.N.; Gool, E.L.; Nieuwland, R. Recent developments in the nomenclature, presence, isolation, detection and clinical impact of extracellular vesicles. J. Thromb. Haemost. 2016, 14, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Boulanger, C.M.; Loyer, X.; Rautou, P.E.; Amabile, N. Extracellular vesicles in coronary artery disease. Nat. Rev. Cardiol. 2017, 14, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Ridger, V.C.; Boulanger, C.M.; Angelillo-Scherrer, A.; Badimon, L.; Blanc-Brude, O.; Bochaton-Piallat, M.L.; Boilard, E.; Buzas, E.I.; Caporali, A.; Dignat-George, F.; et al. Microvesicles in vascular homeostasis and diseases. Thromb. Haemost. 2017, 117, 1296–1316. [Google Scholar] [CrossRef] [PubMed]
- Coumans, F.A.W.; Brisson, A.R.; Buzas, E.I.; Dignat-George, F.; Drees, E.E.E.; El-Andaloussi, S.; Emanueli, C.; Gasecka, A.; Hendrix, A.; Hill, A.F.; et al. Methodological Guidelines to Study Extracellular Vesicles. Circ. Res. 2017, 120, 1632–1648. [Google Scholar] [CrossRef] [PubMed]
- Sluijter, J.P.G.; Davidson, S.M.; Boulanger, C.M.; Buzas, E.I.; de Kleijn, D.P.V.; Engel, F.B.; Giricz, Z.; Hausenloy, D.J.; Kishore, R.; Lecour, S.; et al. Extracellular vesicles in diagnostics and therapy of the ischaemic heart: Position Paper from the Working Group on Cellular Biology of the Heart of the European Society of Cardiology. Cardiovasc. Res. 2018, 114, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Thery, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [PubMed]
- Van der Pol, E.; Sturk, A.; van Leeuwen, T.; Nieuwland, R.; Coumans, F. Standardization of extracellular vesicle measurements by flow cytometry through vesicle diameter approximation. J. Thromb. Haemost. 2018, 16, 1236–1245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arraud, N.; Linares, R.; Tan, S.; Gounou, C.; Pasquet, J.M.; Mornet, S.; Brisson, A.R. Extracellular vesicles from blood plasma: Determination of their morphology, size, phenotype and concentration. J. Thromb. Haemost. 2014, 12, 614–627. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Ouyang, Y.; Wang, Z.; Zhang, R.; Huang, P.H.; Chen, C.; Li, H.; Li, P.; Quinn, D.; Dao, M.; et al. Isolation of exosomes from whole blood by integrating acoustics and microfluidics. Proc. Natl. Acad. Sci. USA 2017, 114, 10584–10589. [Google Scholar] [CrossRef] [Green Version]
- Chiriaco, M.S.; Bianco, M.; Nigro, A.; Primiceri, E.; Ferrara, F.; Romano, A.; Quattrini, A.; Furlan, R.; Arima, V.; Maruccio, G. Lab-on-Chip for Exosomes and Microvesicles Detection and Characterization. Sensors (Basel) 2018, 18, 3175. [Google Scholar] [CrossRef]
- Chandler, W.L. Measurement of microvesicle levels in human blood using flow cytometry. Cytom. B Clin. Cytom. 2016, 90, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Hoog, J.L.; Lotvall, J. Diversity of extracellular vesicles in human ejaculates revealed by cryo-electron microscopy. J. Extracell. Vesicles 2015, 4, 28680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lasser, C.; Jang, S.C.; Lotvall, J. Subpopulations of extracellular vesicles and their therapeutic potential. Mol. Asp. Med. 2018, 60, 1–14. [Google Scholar] [CrossRef]
- Zabeo, D.; Cvjetkovic, A.; Lasser, C.; Schorb, M.; Lotvall, J.; Hoog, J.L. Exosomes purified from a single cell type have diverse morphology. J. Extracell. Vesicles 2017, 6, 1329476. [Google Scholar] [CrossRef]
- Szatanek, R.; Baj-Krzyworzeka, M.; Zimoch, J.; Lekka, M.; Siedlar, M.; Baran, J. The Methods of Choice for Extracellular Vesicles (EVs) Characterization. Int. J. Mol. Sci. 2017, 18, 1153. [Google Scholar] [CrossRef]
- Dragovic, R.A.; Gardiner, C.; Brooks, A.S.; Tannetta, D.S.; Ferguson, D.J.; Hole, P.; Carr, B.; Redman, C.W.; Harris, A.L.; Dobson, P.J.; et al. Sizing and phenotyping of cellular vesicles using Nanoparticle Tracking Analysis. Nanomedicine 2011, 7, 780–788. [Google Scholar] [CrossRef] [Green Version]
- Paone, S.; Baxter, A.A.; Hulett, M.D.; Poon, I.K.H. Endothelial cell apoptosis and the role of endothelial cell-derived extracellular vesicles in the progression of atherosclerosis. Cell. Mol. Life Sci. 2019, 76, 1093–1106. [Google Scholar] [CrossRef] [PubMed]
- Arandjelovic, S.; Ravichandran, K.S. Phagocytosis of apoptotic cells in homeostasis. Nat. Immunol. 2015, 16, 907–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berda-Haddad, Y.; Robert, S.; Salers, P.; Zekraoui, L.; Farnarier, C.; Dinarello, C.A.; Dignat-George, F.; Kaplanski, G. Sterile inflammation of endothelial cell-derived apoptotic bodies is mediated by interleukin-1alpha. Proc. Natl. Acad. Sci. USA 2011, 108, 20684–20689. [Google Scholar] [CrossRef] [PubMed]
- Zernecke, A.; Bidzhekov, K.; Noels, H.; Shagdarsuren, E.; Gan, L.; Denecke, B.; Hristov, M.; Koppel, T.; Jahantigh, M.N.; Lutgens, E.; et al. Delivery of microRNA-126 by apoptotic bodies induces CXCL12-dependent vascular protection. Sci. Signal. 2009, 2, ra81. [Google Scholar] [CrossRef] [PubMed]
- Caruso, S.; Poon, I.K.H. Apoptotic Cell-Derived Extracellular Vesicles: More Than Just Debris. Front. Immunol. 2018, 9, 1486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, P. The nature and significance of platelet products in human plasma. Br. J. Haematol. 1967, 13, 269–288. [Google Scholar] [CrossRef] [PubMed]
- Stachowiak, J.C.; Brodsky, F.M.; Miller, E.A. A cost-benefit analysis of the physical mechanisms of membrane curvature. Nat. Cell Biol. 2013, 15, 1019–1027. [Google Scholar] [CrossRef]
- Cocucci, E.; Meldolesi, J. Ectosomes and exosomes: Shedding the confusion between extracellular vesicles. Trends Cell Biol. 2015, 25, 364–372. [Google Scholar] [CrossRef]
- Nabhan, J.F.; Hu, R.; Oh, R.S.; Cohen, S.N.; Lu, Q. Formation and release of arrestin domain-containing protein 1-mediated microvesicles (ARMMs) at plasma membrane by recruitment of TSG101 protein. Proc. Natl. Acad. Sci. USA 2012, 109, 4146–4151. [Google Scholar] [CrossRef] [Green Version]
- Tricarico, C.; Clancy, J.; D’Souza-Schorey, C. Biology and biogenesis of shed microvesicles. Small GTPases 2017, 8, 220–232. [Google Scholar] [CrossRef]
- Muralidharan-Chari, V.; Clancy, J.; Plou, C.; Romao, M.; Chavrier, P.; Raposo, G.; D’Souza-Schorey, C. ARF6-regulated shedding of tumor cell-derived plasma membrane microvesicles. Curr. Biol. 2009, 19, 1875–1885. [Google Scholar] [CrossRef] [PubMed]
- Hafiane, A.; Daskalopoulou, S.S. Extracellular vesicles characteristics and emerging roles in atherosclerotic cardiovascular disease. Metabolism 2018, 85, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Bissig, C.; Gruenberg, J. ALIX and the multivesicular endosome: ALIX in Wonderland. Trends Cell Biol. 2014, 24, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Trajkovic, K.; Hsu, C.; Chiantia, S.; Rajendran, L.; Wenzel, D.; Wieland, F.; Schwille, P.; Brugger, B.; Simons, M. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 2008, 319, 1244–1247. [Google Scholar] [CrossRef] [PubMed]
- Hessvik, N.P.; Llorente, A. Current knowledge on exosome biogenesis and release. Cell. Mol. Life Sci. 2018, 75, 193–208. [Google Scholar] [CrossRef]
- Juan, T.; Furthauer, M. Biogenesis and function of ESCRT-dependent extracellular vesicles. Semin. Cell Dev. Biol. 2018, 74, 66–77. [Google Scholar] [CrossRef]
- Kowal, J.; Tkach, M.; Thery, C. Biogenesis and secretion of exosomes. Curr. Opin. Cell Biol. 2014, 29, 116–125. [Google Scholar] [CrossRef] [Green Version]
- Tetta, C.; Ghigo, E.; Silengo, L.; Deregibus, M.C.; Camussi, G. Extracellular vesicles as an emerging mechanism of cell-to-cell communication. Endocrine 2013, 44, 11–19. [Google Scholar] [CrossRef]
- Valadi, H.; Ekstrom, K.; Bossios, A.; Sjostrand, M.; Lee, J.J.; Lotvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, A.; Takahashi, Y.; Nishikawa, M.; Sano, K.; Morishita, M.; Charoenviriyakul, C.; Saji, H.; Takakura, Y. Accelerated growth of B16BL6 tumor in mice through efficient uptake of their own exosomes by B16BL6 cells. Cancer Sci. 2017, 108, 1803–1810. [Google Scholar] [CrossRef]
- Del Conde, I.; Shrimpton, C.N.; Thiagarajan, P.; Lopez, J.A. Tissue-factor-bearing microvesicles arise from lipid rafts and fuse with activated platelets to initiate coagulation. Blood 2005, 106, 1604–1611. [Google Scholar] [CrossRef] [PubMed]
- Costa Verdera, H.; Gitz-Francois, J.J.; Schiffelers, R.M.; Vader, P. Cellular uptake of extracellular vesicles is mediated by clathrin-independent endocytosis and macropinocytosis. J. Control. Release 2017, 266, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Svensson, K.J.; Christianson, H.C.; Wittrup, A.; Bourseau-Guilmain, E.; Lindqvist, E.; Svensson, L.M.; Morgelin, M.; Belting, M. Exosome uptake depends on ERK1/2-heat shock protein 27 signaling and lipid Raft-mediated endocytosis negatively regulated by caveolin-1. J. Biol. Chem. 2013, 288, 17713–17724. [Google Scholar] [CrossRef] [PubMed]
- Nanbo, A.; Kawanishi, E.; Yoshida, R.; Yoshiyama, H. Exosomes derived from Epstein-Barr virus-infected cells are internalized via caveola-dependent endocytosis and promote phenotypic modulation in target cells. J. Virol. 2013, 87, 10334–10347. [Google Scholar] [CrossRef] [PubMed]
- Van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Forlow, S.B.; McEver, R.P.; Nollert, M.U. Leukocyte-leukocyte interactions mediated by platelet microparticles under flow. Blood 2000, 95, 1317–1323. [Google Scholar] [PubMed]
- Baj-Krzyworzeka, M.; Majka, M.; Pratico, D.; Ratajczak, J.; Vilaire, G.; Kijowski, J.; Reca, R.; Janowska-Wieczorek, A.; Ratajczak, M.Z. Platelet-derived microparticles stimulate proliferation, survival, adhesion, and chemotaxis of hematopoietic cells. Exp. Hematol. 2002, 30, 450–459. [Google Scholar] [CrossRef]
- Boilard, E. Extracellular vesicles and their content in bioactive lipid mediators: More than a sack of microRNA. J. Lipid Res. 2018, 59, 2037–2046. [Google Scholar] [CrossRef] [PubMed]
- Deregibus, M.C.; Cantaluppi, V.; Calogero, R.; Lo Iacono, M.; Tetta, C.; Biancone, L.; Bruno, S.; Bussolati, B.; Camussi, G. Endothelial progenitor cell derived microvesicles activate an angiogenic program in endothelial cells by a horizontal transfer of mRNA. Blood 2007, 110, 2440–2448. [Google Scholar] [CrossRef]
- Jiang, J.; Kao, C.Y.; Papoutsakis, E.T. How do megakaryocytic microparticles target and deliver cargo to alter the fate of hematopoietic stem cells? J. Control. Release 2017, 247, 1–18. [Google Scholar] [CrossRef]
- Alexander, M.; Hu, R.; Runtsch, M.C.; Kagele, D.A.; Mosbruger, T.L.; Tolmachova, T.; Seabra, M.C.; Round, J.L.; Ward, D.M.; O’Connell, R.M. Exosome-delivered microRNAs modulate the inflammatory response to endotoxin. Nat. Commun. 2015, 6, 7321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, S.A.; Xie, Y.; Fu, Z.; Wang, Y.; Wang, J.A.; Xiang, M. Emerging role of exosome-mediated intercellular communication in vascular remodeling. Oncotarget 2017, 8, 25700–25712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laffont, B.; Corduan, A.; Rousseau, M.; Duchez, A.C.; Lee, C.H.; Boilard, E.; Provost, P. Platelet microparticles reprogram macrophage gene expression and function. Thromb. Haemost. 2016, 115, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Silverman-Gavrila, R.; Silverman-Gavrila, L.; Bendeck, M.P. Cell division fidelity is altered during the vascular response to injury: Its novel role in atherosclerosis progression. Am. J. Pathol. 2013, 182, 628–639. [Google Scholar] [CrossRef] [PubMed]
- Arraud, N.; Gounou, C.; Turpin, D.; Brisson, A.R. Fluorescence triggering: A general strategy for enumerating and phenotyping extracellular vesicles by flow cytometry. Cytom. A 2016, 89, 184–195. [Google Scholar] [CrossRef] [PubMed]
- Boilard, E.; Duchez, A.C.; Brisson, A. The diversity of platelet microparticles. Curr. Opin. Hematol. 2015, 22, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Holnthoner, W.; Bonstingl, C.; Hromada, C.; Muehleder, S.; Zipperle, J.; Stojkovic, S.; Redl, H.; Wojta, J.; Schochl, H.; Grillari, J.; et al. Endothelial Cell-derived Extracellular Vesicles Size-dependently Exert Procoagulant Activity Detected by Thromboelastometry. Sci. Rep. 2017, 7, 3707. [Google Scholar] [CrossRef] [PubMed]
- Pugholm, L.H.; Baek, R.; Sondergaard, E.K.; Revenfeld, A.L.; Jorgensen, M.M.; Varming, K. Phenotyping of Leukocytes and Leukocyte-Derived Extracellular Vesicles. J. Immunol. Res. 2016, 2016, 6391264. [Google Scholar] [CrossRef] [PubMed]
- McArthur, K.; Chappaz, S.; Kile, B.T. Apoptosis in megakaryocytes and platelets: The life and death of a lineage. Blood 2018, 131, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Ponomareva, A.A.; Nevzorova, T.A.; Mordakhanova, E.R.; Andrianova, I.A.; Rauova, L.; Litvinov, R.I.; Weisel, J.W. Intracellular origin and ultrastructure of platelet-derived microparticles. J. Thromb. Haemost. 2017, 15, 1655–1667. [Google Scholar] [CrossRef] [Green Version]
- Shai, E.; Rosa, I.; Parguina, A.F.; Motahedeh, S.; Varon, D.; Garcia, A. Comparative analysis of platelet-derived microparticles reveals differences in their amount and proteome depending on the platelet stimulus. J. Proteom. 2012, 76. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.T.; McIntyre, T.M. Lipopolysaccharide signaling without a nucleus: Kinase cascades stimulate platelet shedding of proinflammatory IL-1beta-rich microparticles. J. Immunol. 2011, 186, 5489–5496. [Google Scholar] [CrossRef] [PubMed]
- Boilard, E.; Pare, G.; Rousseau, M.; Cloutier, N.; Dubuc, I.; Levesque, T.; Borgeat, P.; Flamand, L. Influenza virus H1N1 activates platelets through FcgammaRIIA signaling and thrombin generation. Blood 2014, 123, 2854–2863. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.; Reade, M.C.; Hyland, R.A.; Tan, S.; Marks, D.C. In vitro comparison of cryopreserved and liquid platelets: Potential clinical implications. Transfusion 2015, 55, 838–847. [Google Scholar] [CrossRef] [PubMed]
- Reininger, A.J.; Heijnen, H.F.; Schumann, H.; Specht, H.M.; Schramm, W.; Ruggeri, Z.M. Mechanism of platelet adhesion to von Willebrand factor and microparticle formation under high shear stress. Blood 2006, 107, 3537–3545. [Google Scholar] [CrossRef] [PubMed]
- Zarà, M.; Guidetti, G.F.; Boselli, D.; Villa, C.; Canobbio, I.; Seppi, C.; Visconte, C.; Canino, J.; Torti, M. Release of Prometastatic Platelet-Derived Microparticles Induced by Breast Cancer Cells: A Novel Positive Feedback Mechanism for Metastasis. TH Open 2017, 1, 155–163. [Google Scholar] [CrossRef]
- Melki, I.; Tessandier, N.; Zufferey, A.; Boilard, E. Platelet microvesicles in health and disease. Platelets 2017, 28, 214–221. [Google Scholar] [CrossRef]
- Cramer, E.M.; Norol, F.; Guichard, J.; Breton-Gorius, J.; Vainchenker, W.; Masse, J.M.; Debili, N. Ultrastructure of platelet formation by human megakaryocytes cultured with the Mpl ligand. Blood 1997, 89, 2336–2346. [Google Scholar]
- Flaumenhaft, R.; Dilks, J.R.; Richardson, J.; Alden, E.; Patel-Hett, S.R.; Battinelli, E.; Klement, G.L.; Sola-Visner, M.; Italiano, J.E., Jr. Megakaryocyte-derived microparticles: Direct visualization and distinction from platelet-derived microparticles. Blood 2009, 113, 1112–1121. [Google Scholar] [CrossRef]
- Italiano, J.E., Jr.; Mairuhu, A.T.; Flaumenhaft, R. Clinical relevance of microparticles from platelets and megakaryocytes. Curr. Opin. Hematol. 2010, 17, 578–584. [Google Scholar] [CrossRef] [Green Version]
- Flaumenhaft, R.; Mairuhu, A.T.; Italiano, J.E. Platelet- and megakaryocyte-derived microparticles. Semin. Thromb. Hemost. 2010, 36, 881–887. [Google Scholar] [CrossRef] [PubMed]
- Brambilla, M.; Facchinetti, L.; Canzano, P.; Rossetti, L.; Ferri, N.; Balduini, A.; Abbonante, V.; Boselli, D.; De Marco, L.; Di Minno, M.N.; et al. Human megakaryocytes confer tissue factor to a subset of shed platelets to stimulate thrombin generation. Thromb. Haemost. 2015, 114, 579–592. [Google Scholar] [CrossRef] [PubMed]
- Chiva-Blanch, G.; Laake, K.; Myhre, P.; Bratseth, V.; Arnesen, H.; Solheim, S.; Badimon, L.; Seljeflot, I. Platelet-, monocyte-derived and tissue factor-carrying circulating microparticles are related to acute myocardial infarction severity. PLoS ONE 2017, 12, e0172558. [Google Scholar] [CrossRef] [PubMed]
- Muller, I.; Klocke, A.; Alex, M.; Kotzsch, M.; Luther, T.; Morgenstern, E.; Zieseniss, S.; Zahler, S.; Preissner, K.; Engelmann, B. Intravascular tissue factor initiates coagulation via circulating microvesicles and platelets. FASEB J. 2003, 17, 476–478. [Google Scholar] [CrossRef]
- Nomura, S.; Shimizu, M. Clinical significance of procoagulant microparticles. J. Intensive Care 2015, 3, 2. [Google Scholar] [CrossRef]
- Boilard, E.; Blanco, P.; Nigrovic, P.A. Platelets: Active players in the pathogenesis of arthritis and SLE. Nat. Rev. Rheumatol. 2012, 8, 534–542. [Google Scholar] [CrossRef]
- Scherlinger, M.; Sisirak, V.; Richez, C.; Lazaro, E.; Duffau, P.; Blanco, P. New Insights on Platelets and Platelet-Derived Microparticles in Systemic Lupus Erythematosus. Curr. Rheumatol. Rep. 2017, 19, 48. [Google Scholar] [CrossRef]
- Tao, S.C.; Guo, S.C.; Zhang, C.Q. Platelet-derived Extracellular Vesicles: An Emerging Therapeutic Approach. Int. J. Biol. Sci. 2017, 13, 828–834. [Google Scholar] [CrossRef] [Green Version]
- Varon, D.; Shai, E. Platelets and their microparticles as key players in pathophysiological responses. J. Thromb. Haemost. 2015, 13, 40–46. [Google Scholar] [CrossRef]
- Goubran, H.A.; Burnouf, T.; Stakiw, J.; Seghatchian, J. Platelet microparticle: A sensitive physiological “fine tuning” balancing factor in health and disease. Transfus. Apher. Sci. 2015, 52, 12–18. [Google Scholar] [CrossRef]
- Heijnen, H.F.; Schiel, A.E.; Fijnheer, R.; Geuze, H.J.; Sixma, J.J. Activated platelets release two types of membrane vesicles: Microvesicles by surface shedding and exosomes derived from exocytosis of multivesicular bodies and alpha-granules. Blood 1999, 94, 3791–3799. [Google Scholar] [PubMed]
- Guo, S.C.; Tao, S.C.; Yin, W.J.; Qi, X.; Yuan, T.; Zhang, C.Q. Exosomes derived from platelet-rich plasma promote the re-epithelization of chronic cutaneous wounds via activation of YAP in a diabetic rat model. Theranostics 2017, 7, 81–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Tan, M.; Xiang, Q.; Zhou, Z.; Yan, H. Thrombin-activated platelet-derived exosomes regulate endothelial cell expression of ICAM-1 via microRNA-223 during the thrombosis-inflammation response. Thromb. Res. 2017, 154, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.; Yan, H.B.; Li, J.N.; Li, W.K.; Fu, Y.Y.; Chen, W.; Zhou, Z. Thrombin Stimulated Platelet-Derived Exosomes Inhibit Platelet-Derived Growth Factor Receptor-Beta Expression in Vascular Smooth Muscle Cells. Cell. Physiol. Biochem. 2016, 38, 2348–2365. [Google Scholar] [CrossRef] [PubMed]
- Dempsey, E.; Dervin, F.; Maguire, P.B. Platelet Derived Exosomes Are Enriched for Specific microRNAs Which Regulate WNT Signalling in Endothelial Cells. Blood 2014, 124, 2760. [Google Scholar]
- Dervin, F.; Wynne, K.; Maguire, P.B. Human Platelet Exosome Proteomics Leads to the Identification of WNT Positive Exosomes Which Impact Canonical WNT Signalling in Target Cells. Blood 2014, 124, 2758. [Google Scholar]
- Chen, Y.; Li, G.; Liu, Y.; Werth, V.P.; Williams, K.J.; Liu, M.L. Translocation of Endogenous Danger Signal HMGB1 From Nucleus to Membrane Microvesicles in Macrophages. J. Cell. Physiol. 2016, 231, 2319–2326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, W.P.; Tigges, J.C.; Toxavidis, V.; Ghiran, I. Red Blood Cells: A Source of Extracellular Vesicles. Methods Mol. Biol. 2017, 1660, 15–22. [Google Scholar] [CrossRef]
- Marcoux, G.; Duchez, A.C.; Cloutier, N.; Provost, P.; Nigrovic, P.A.; Boilard, E. Revealing the diversity of extracellular vesicles using high-dimensional flow cytometry analyses. Sci. Rep. 2016, 6, 35928. [Google Scholar] [CrossRef]
- Nguyen, D.B.; Ly, T.B.; Wesseling, M.C.; Hittinger, M.; Torge, A.; Devitt, A.; Perrie, Y.; Bernhardt, I. Characterization of Microvesicles Released from Human Red Blood Cells. Cell. Physiol. Biochem. 2016, 38, 1085–1099. [Google Scholar] [CrossRef]
- Leal, J.K.F.; Adjobo-Hermans, M.J.W.; Bosman, G. Red Blood Cell Homeostasis: Mechanisms and Effects of Microvesicle Generation in Health and Disease. Front. Physiol. 2018, 9, 703. [Google Scholar] [CrossRef] [PubMed]
- Bosman, G.J.; Lasonder, E.; Groenen-Dopp, Y.A.; Willekens, F.L.; Werre, J.M. The proteome of erythrocyte-derived microparticles from plasma: New clues for erythrocyte aging and vesiculation. J. Proteom. 2012, 76, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Rubin, O.; Delobel, J.; Prudent, M.; Lion, N.; Kohl, K.; Tucker, E.I.; Tissot, J.D.; Angelillo-Scherrer, A. Red blood cell-derived microparticles isolated from blood units initiate and propagate thrombin generation. Transfusion 2013, 53, 1744–1754. [Google Scholar] [CrossRef] [PubMed]
- Usman, W.M.; Pham, T.C.; Kwok, Y.Y.; Vu, L.T.; Ma, V.; Peng, B.; Chan, Y.S.; Wei, L.; Chin, S.M.; Azad, A.; et al. Efficient RNA drug delivery using red blood cell extracellular vesicles. Nat. Commun. 2018, 9, 2359. [Google Scholar] [CrossRef] [PubMed]
- Danesh, A.; Inglis, H.C.; Jackman, R.P.; Wu, S.; Deng, X.; Muench, M.O.; Heitman, J.W.; Norris, P.J. Exosomes from red blood cell units bind to monocytes and induce proinflammatory cytokines, boosting T-cell responses in vitro. Blood 2014, 123, 687–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnstone, R.M.; Bianchini, A.; Teng, K. Reticulocyte maturation and exosome release: Transferrin receptor containing exosomes shows multiple plasma membrane functions. Blood 1989, 74, 1844–1851. [Google Scholar] [PubMed]
- Liu, M.L.; Williams, K.J.; Werth, V.P. Microvesicles in Autoimmune Diseases. Adv. Clin. Chem. 2016, 77, 125–175. [Google Scholar] [CrossRef] [PubMed]
- Nolan, S.; Dixon, R.; Norman, K.; Hellewell, P.; Ridger, V. Nitric oxide regulates neutrophil migration through microparticle formation. Am. J. Pathol. 2008, 172, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Pluskota, E.; Woody, N.M.; Szpak, D.; Ballantyne, C.M.; Soloviev, D.A.; Simon, D.I.; Plow, E.F. Expression, activation, and function of integrin alphaMbeta2 (Mac-1) on neutrophil-derived microparticles. Blood 2008, 112, 2327–2335. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.G.; Williams, J.C.; Davis, B.K.; Jacobson, K.; Doerschuk, C.M.; Ting, J.P.; Mackman, N. Monocytic microparticles activate endothelial cells in an IL-1beta-dependent manner. Blood 2011, 118, 2366–2374. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, Q.; Zhang, J.K.; Li, C.; Miao, Y.R.; Lei, Q.; Li, Q.B.; Guo, A.Y. EVmiRNA: A database of miRNA profiling in extracellular vesicles. Nucleic Acids Res. 2019, 47, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Dickhout, A.; Koenen, R.R. Extracellular Vesicles as Biomarkers in Cardiovascular Disease; Chances and Risks. Front. Cardiovasc. Med. 2018, 5, 113. [Google Scholar] [CrossRef] [PubMed]
- Mayr, M.; Grainger, D.; Mayr, U.; Leroyer, A.S.; Leseche, G.; Sidibe, A.; Herbin, O.; Yin, X.; Gomes, A.; Madhu, B.; et al. Proteomics, metabolomics, and immunomics on microparticles derived from human atherosclerotic plaques. Circ. Cardiovasc. Genet. 2009, 2, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Folkesson, M.; Li, C.; Frebelius, S.; Swedenborg, J.; Wagsater, D.; Williams, K.J.; Eriksson, P.; Roy, J.; Liu, M.L. Proteolytically active ADAM10 and ADAM17 carried on membrane microvesicles in human abdominal aortic aneurysms. Thromb. Haemost. 2015, 114, 1165–1174. [Google Scholar] [CrossRef] [PubMed]
- Rossaint, J.; Kuhne, K.; Skupski, J.; Van Aken, H.; Looney, M.R.; Hidalgo, A.; Zarbock, A. Directed transport of neutrophil-derived extracellular vesicles enables platelet-mediated innate immune response. Nat. Commun. 2016, 7, 13464. [Google Scholar] [CrossRef] [PubMed]
- Dalvi, P.; Sun, B.; Tang, N.; Pulliam, L. Immune activated monocyte exosomes alter microRNAs in brain endothelial cells and initiate an inflammatory response through the TLR4/MyD88 pathway. Sci. Rep. 2017, 7, 9954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vargas, A.; Roux-Dalvai, F.; Droit, A.; Lavoie, J.P. Neutrophil-Derived Exosomes: A New Mechanism Contributing to Airway Smooth Muscle Remodeling. Am. J. Respir. Cell Mol. Biol. 2016, 55, 450–461. [Google Scholar] [CrossRef]
- Cui, C.J.; Wang, G.J.; Yang, S.; Huang, S.K.; Qiao, R.; Cui, W. Tissue Factor-bearing MPs and the risk of venous thrombosis in cancer patients: A meta-analysis. Sci. Rep. 2018, 8, 1675. [Google Scholar] [CrossRef]
- Lan, J.; Sun, L.; Xu, F.; Liu, L.; Hu, F.; Song, D.; Hou, Z.; Wu, W.; Luo, X.; Wang, J.; et al. M2 Macrophage-Derived Exosomes Promote Cell Migration and Invasion in Colon Cancer. Cancer Res. 2019, 79, 146–158. [Google Scholar] [CrossRef]
- Hristov, M.; Erl, W.; Linder, S.; Weber, P.C. Apoptotic bodies from endothelial cells enhance the number and initiate the differentiation of human endothelial progenitor cells in vitro. Blood 2004, 104, 2761–2766. [Google Scholar] [CrossRef]
- Hromada, C.; Muhleder, S.; Grillari, J.; Redl, H.; Holnthoner, W. Endothelial Extracellular Vesicles-Promises and Challenges. Front. Physiol. 2017, 8, 275. [Google Scholar] [CrossRef] [PubMed]
- Dignat-George, F.; Boulanger, C.M. The many faces of endothelial microparticles. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Njock, M.S.; Cheng, H.S.; Dang, L.T.; Nazari-Jahantigh, M.; Lau, A.C.; Boudreau, E.; Roufaiel, M.; Cybulsky, M.I.; Schober, A.; Fish, J.E. Endothelial cells suppress monocyte activation through secretion of extracellular vesicles containing antiinflammatory microRNAs. Blood 2015, 125, 3202–3212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.T.; Chu, K.; Jung, K.H.; Kim, J.M.; Moon, H.J.; Bahn, J.J.; Im, W.S.; Sunwoo, J.; Moon, J.; Kim, M.; et al. Circulating CD62E+ microparticles and cardiovascular outcomes. PLoS ONE 2012, 7, e35713. [Google Scholar] [CrossRef] [PubMed]
- Banfi, C.; Brioschi, M.; Wait, R.; Begum, S.; Gianazza, E.; Pirillo, A.; Mussoni, L.; Tremoli, E. Proteome of endothelial cell-derived procoagulant microparticles. Proteomics 2005, 5, 4443–4455. [Google Scholar] [CrossRef]
- De Jong, O.G.; Verhaar, M.C.; Chen, Y.; Vader, P.; Gremmels, H.; Posthuma, G.; Schiffelers, R.M.; Gucek, M.; van Balkom, B.W. Cellular stress conditions are reflected in the protein and RNA content of endothelial cell-derived exosomes. J. Extracell. Vesicles 2012, 1. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; He, Y.; Hou, X.; Zhang, Z.; Wang, R.; Wu, Q. Endothelial Cells Can Regulate Smooth Muscle Cells in Contractile Phenotype through the miR-206/ARF6&NCX1/Exosome Axis. PLoS ONE 2016, 11, e0152959. [Google Scholar] [CrossRef]
- Halkein, J.; Tabruyn, S.P.; Ricke-Hoch, M.; Haghikia, A.; Nguyen, N.Q.; Scherr, M.; Castermans, K.; Malvaux, L.; Lambert, V.; Thiry, M.; et al. MicroRNA-146a is a therapeutic target and biomarker for peripartum cardiomyopathy. J. Clin. Investig. 2013, 123, 2143–2154. [Google Scholar] [CrossRef]
- Ong, S.G.; Lee, W.H.; Huang, M.; Dey, D.; Kodo, K.; Sanchez-Freire, V.; Gold, J.D.; Wu, J.C. Cross talk of combined gene and cell therapy in ischemic heart disease: Role of exosomal microRNA transfer. Circulation 2014, 130, 60–69. [Google Scholar] [CrossRef]
- Van Balkom, B.W.; de Jong, O.G.; Smits, M.; Brummelman, J.; den Ouden, K.; de Bree, P.M.; van Eijndhoven, M.A.; Pegtel, D.M.; Stoorvogel, W.; Wurdinger, T.; et al. Endothelial cells require miR-214 to secrete exosomes that suppress senescence and induce angiogenesis in human and mouse endothelial cells. Blood 2013, 121, 3997–4006. [Google Scholar] [CrossRef] [Green Version]
- Owens, A.P., 3rd; Mackman, N. Microparticles in hemostasis and thrombosis. Circ. Res. 2011, 108, 1284–1297. [Google Scholar] [CrossRef] [PubMed]
- Perez-Pujol, S.; Marker, P.H.; Key, N.S. Platelet microparticles are heterogeneous and highly dependent on the activation mechanism: Studies using a new digital flow cytometer. Cytom. A 2007, 71, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Mackman, N. The many faces of tissue factor. J. Thromb. Haemost. 2009, 7, 136–139. [Google Scholar] [CrossRef]
- Grover, S.P.; Mackman, N. Tissue Factor: An Essential Mediator of Hemostasis and Trigger of Thrombosis. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 709–725. [Google Scholar] [CrossRef] [PubMed]
- Camera, M.; Frigerio, M.; Toschi, V.; Brambilla, M.; Rossi, F.; Cottell, D.C.; Maderna, P.; Parolari, A.; Bonzi, R.; De Vincenti, O.; et al. Platelet activation induces cell-surface immunoreactive tissue factor expression, which is modulated differently by antiplatelet drugs. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1690–1696. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, B.A.; Krudysz-Amblo, J.; Butenas, S. Platelet tissue factor is not expressed transiently after platelet activation. Blood 2012, 119, 4338–4339. [Google Scholar] [CrossRef] [Green Version]
- Bouchard, B.A.; Mann, K.G.; Butenas, S. No evidence for tissue factor on platelets. Blood 2010, 116, 854–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butenas, S.; Bouchard, B.A.; Brummel-Ziedins, K.E.; Parhami-Seren, B.; Mann, K.G. Tissue factor activity in whole blood. Blood 2005, 105, 2764–2770. [Google Scholar] [CrossRef] [PubMed]
- Osterud, B.; Olsen, J.O. Human platelets do not express tissue factor. Thromb. Res. 2013, 132, 112–115. [Google Scholar] [CrossRef]
- Brambilla, M.; Rossetti, L.; Zara, C.; Canzano, P.; Giesen, P.L.A.; Tremoli, E.; Camera, M. Do methodological differences account for the current controversy on tissue factor expression in platelets? Platelets 2018, 29, 406–414. [Google Scholar] [CrossRef]
- Giesen, P.L.; Rauch, U.; Bohrmann, B.; Kling, D.; Roque, M.; Fallon, J.T.; Badimon, J.J.; Himber, J.; Riederer, M.A.; Nemerson, Y. Blood-borne tissue factor: Another view of thrombosis. Proc. Natl. Acad. Sci. USA 1999, 96, 2311–2315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinauridze, E.I.; Kireev, D.A.; Popenko, N.Y.; Pichugin, A.V.; Panteleev, M.A.; Krymskaya, O.V.; Ataullakhanov, F.I. Platelet microparticle membranes have 50- to 100-fold higher specific procoagulant activity than activated platelets. Thromb. Haemost. 2007, 97, 425–434. [Google Scholar] [PubMed]
- Rauch, U.; Nemerson, Y. Circulating tissue factor and thrombosis. Curr. Opin. Hematol. 2000, 7, 273–277. [Google Scholar] [CrossRef]
- Camera, M.; Toschi, V.; Brambilla, M.; Lettino, M.; Rossetti, L.; Canzano, P.; Di Minno, A.; Tremoli, E. The Role of Tissue Factor in Atherothrombosis and Coronary Artery Disease: Insights into Platelet Tissue Factor. Semin. Thromb. Hemost. 2015, 41, 737–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hathcock, J.J.; Nemerson, Y. Platelet deposition inhibits tissue factor activity: In vitro clots are impermeable to factor Xa. Blood 2004, 104, 123–127. [Google Scholar] [CrossRef]
- Rao, L.V.; Kothari, H.; Pendurthi, U.R. Tissue factor: Mechanisms of decryption. Front. Biosci. 2012, 4, 1513–1527. [Google Scholar] [CrossRef]
- Popescu, N.I.; Lupu, C.; Lupu, F. Role of PDI in regulating tissue factor: FVIIa activity. Thromb. Res. 2010, 125, 38–41. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, C.; von Bruhl, M.L.; Manukyan, D.; Grahl, L.; Lorenz, M.; Altmann, B.; Dlugai, S.; Hess, S.; Konrad, I.; Orschiedt, L.; et al. Protein disulfide isomerase acts as an injury response signal that enhances fibrin generation via tissue factor activation. J. Clin. Investig. 2008, 118, 1110–1122. [Google Scholar] [CrossRef]
- Falati, S.; Liu, Q.; Gross, P.; Merrill-Skoloff, G.; Chou, J.; Vandendries, E.; Celi, A.; Croce, K.; Furie, B.C.; Furie, B. Accumulation of tissue factor into developing thrombi in vivo is dependent upon microparticle P-selectin glycoprotein ligand 1 and platelet P-selectin. J. Exp. Med. 2003, 197, 1585–1598. [Google Scholar] [CrossRef]
- Thomas, G.M.; Panicot-Dubois, L.; Lacroix, R.; Dignat-George, F.; Lombardo, D.; Dubois, C. Cancer cell-derived microparticles bearing P-selectin glycoprotein ligand 1 accelerate thrombus formation in vivo. J. Exp. Med. 2009, 206, 1913–1927. [Google Scholar] [CrossRef]
- Geddings, J.E.; Hisada, Y.; Boulaftali, Y.; Getz, T.M.; Whelihan, M.; Fuentes, R.; Dee, R.; Cooley, B.C.; Key, N.S.; Wolberg, A.S.; et al. Tissue factor-positive tumor microvesicles activate platelets and enhance thrombosis in mice. J. Thromb. Haemost. 2016, 14, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Suades, R.; Padro, T.; Vilahur, G.; Badimon, L. Circulating and platelet-derived microparticles in human blood enhance thrombosis on atherosclerotic plaques. Thromb. Haemost. 2012, 108, 1208–1219. [Google Scholar] [CrossRef] [PubMed]
- Barry, O.P.; Pratico, D.; Lawson, J.A.; FitzGerald, G.A. Transcellular activation of platelets and endothelial cells by bioactive lipids in platelet microparticles. J. Clin. Investig. 1997, 99, 2118–2127. [Google Scholar] [CrossRef] [PubMed]
- Van Der Meijden, P.E.; Van Schilfgaarde, M.; Van Oerle, R.; Renne, T.; ten Cate, H.; Spronk, H.M. Platelet- and erythrocyte-derived microparticles trigger thrombin generation via factor XIIa. J. Thromb. Haemost. 2012, 10, 1355–1362. [Google Scholar] [CrossRef] [PubMed]
- Tripisciano, C.; Weiss, R.; Eichhorn, T.; Spittler, A.; Heuser, T.; Fischer, M.B.; Weber, V. Different Potential of Extracellular Vesicles to Support Thrombin Generation: Contributions of Phosphatidylserine, Tissue Factor, and Cellular Origin. Sci. Rep. 2017, 7, 6522. [Google Scholar] [CrossRef] [PubMed]
- Rubin, O.; Crettaz, D.; Tissot, J.D.; Lion, N. Microparticles in stored red blood cells: Submicron clotting bombs? Blood Transfus. 2010, 8, 31–38. [Google Scholar] [CrossRef]
- Van Beers, E.J.; Schaap, M.C.; Berckmans, R.J.; Nieuwland, R.; Sturk, A.; van Doormaal, F.F.; Meijers, J.C.; Biemond, B.J. Circulating erythrocyte-derived microparticles are associated with coagulation activation in sickle cell disease. Haematologica 2009, 94, 1513–1519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aharon, A.; Tamari, T.; Brenner, B. Monocyte-derived microparticles and exosomes induce procoagulant and apoptotic effects on endothelial cells. Thromb. Haemost. 2008, 100, 878–885. [Google Scholar] [PubMed]
- Sabatier, F.; Roux, V.; Anfosso, F.; Camoin, L.; Sampol, J.; Dignat-George, F. Interaction of endothelial microparticles with monocytic cells in vitro induces tissue factor-dependent procoagulant activity. Blood 2002, 99, 3962–3970. [Google Scholar] [CrossRef]
- Koshiar, R.L.; Somajo, S.; Norstrom, E.; Dahlback, B. Erythrocyte-derived microparticles supporting activated protein C-mediated regulation of blood coagulation. PLoS ONE 2014, 9, e104200. [Google Scholar] [CrossRef]
- Somajo, S.; Koshiar, R.L.; Norstrom, E.; Dahlback, B. Protein S and factor V in regulation of coagulation on platelet microparticles by activated protein C. Thromb. Res. 2014, 134, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Lacroix, R.; Sabatier, F.; Mialhe, A.; Basire, A.; Pannell, R.; Borghi, H.; Robert, S.; Lamy, E.; Plawinski, L.; Camoin-Jau, L.; et al. Activation of plasminogen into plasmin at the surface of endothelial microparticles: A mechanism that modulates angiogenic properties of endothelial progenitor cells in vitro. Blood 2007, 110, 2432–2439. [Google Scholar] [CrossRef] [PubMed]
- Lacroix, R.; Plawinski, L.; Robert, S.; Doeuvre, L.; Sabatier, F.; Martinez de Lizarrondo, S.; Mezzapesa, A.; Anfosso, F.; Leroyer, A.S.; Poullin, P.; et al. Leukocyte- and endothelial-derived microparticles: A circulating source for fibrinolysis. Haematologica 2012, 97, 1864–1872. [Google Scholar] [CrossRef] [PubMed]
- Steppich, B.; Mattisek, C.; Sobczyk, D.; Kastrati, A.; Schomig, A.; Ott, I. Tissue factor pathway inhibitor on circulating microparticles in acute myocardial infarction. Thromb. Haemost. 2005, 93, 35–39. [Google Scholar] [CrossRef]
- Srikanthan, S.; Li, W.; Silverstein, R.L.; McIntyre, T.M. Exosome poly-ubiquitin inhibits platelet activation, downregulates CD36 and inhibits pro-atherothombotic cellular functions. J. Thromb. Haemost. 2014, 12, 1906–1917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gidlof, O.; van der Brug, M.; Ohman, J.; Gilje, P.; Olde, B.; Wahlestedt, C.; Erlinge, D. Platelets activated during myocardial infarction release functional miRNA, which can be taken up by endothelial cells and regulate ICAM1 expression. Blood 2013, 121, 3908–3917. [Google Scholar] [CrossRef] [Green Version]
- Al-Nedawi, K.; Szemraj, J.; Cierniewski, C.S. Mast cell-derived exosomes activate endothelial cells to secrete plasminogen activator inhibitor type 1. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1744–1749. [Google Scholar] [CrossRef]
- Perlman, R.L. Mouse models of human disease: An evolutionary perspective. Evol. Med. Public Health 2016, 2016, 170–176. [Google Scholar] [CrossRef]
- Mackman, N. Mouse models of venous thrombosis are not equal. Blood 2016, 127, 2510–2511. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, A.; Li, W.; Febbraio, M.; Espinola, R.G.; McCrae, K.R.; Cockrell, E.; Silverstein, R.L. Platelet CD36 mediates interactions with endothelial cell-derived microparticles and contributes to thrombosis in mice. J. Clin. Investig. 2008, 118, 1934–1943. [Google Scholar] [CrossRef]
- Furlan-Freguia, C.; Marchese, P.; Gruber, A.; Ruggeri, Z.M.; Ruf, W. P2X7 receptor signaling contributes to tissue factor-dependent thrombosis in mice. J. Clin. Investig. 2011, 121, 2932–2944. [Google Scholar] [CrossRef] [PubMed]
- Diaz, J.A.; Obi, A.T.; Myers, D.D., Jr.; Wrobleski, S.K.; Henke, P.K.; Mackman, N.; Wakefield, T.W. Critical review of mouse models of venous thrombosis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 556–562. [Google Scholar] [CrossRef]
- Ramacciotti, E.; Hawley, A.E.; Farris, D.M.; Ballard, N.E.; Wrobleski, S.K.; Myers, D.D., Jr.; Henke, P.K.; Wakefield, T.W. Leukocyte- and platelet-derived microparticles correlate with thrombus weight and tissue factor activity in an experimental mouse model of venous thrombosis. Thromb. Haemost. 2009, 101, 748–754. [Google Scholar] [PubMed]
- Tarantino, E.; Amadio, P.; Squellerio, I.; Porro, B.; Sandrini, L.; Turnu, L.; Cavalca, V.; Tremoli, E.; Barbieri, S.S. Role of thromboxane-dependent platelet activation in venous thrombosis: Aspirin effects in mouse model. Pharmacol. Res. 2016, 107, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Biro, E.; Sturk-Maquelin, K.N.; Vogel, G.M.; Meuleman, D.G.; Smit, M.J.; Hack, C.E.; Sturk, A.; Nieuwland, R. Human cell-derived microparticles promote thrombus formation in vivo in a tissue factor-dependent manner. J. Thromb. Haemost. 2003, 1, 2561–2568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mege, D.; Mezouar, S.; Dignat-George, F.; Panicot-Dubois, L.; Dubois, C. Microparticles and cancer thrombosis in animal models. Thromb. Res. 2016, 140, 21–26. [Google Scholar] [CrossRef]
- Davila, M.; Amirkhosravi, A.; Coll, E.; Desai, H.; Robles, L.; Colon, J.; Baker, C.H.; Francis, J.L. Tissue factor-bearing microparticles derived from tumor cells: Impact on coagulation activation. J. Thromb. Haemost. 2008, 6, 1517–1524. [Google Scholar] [CrossRef]
- Lima, L.G.; Chammas, R.; Monteiro, R.Q.; Moreira, M.E.; Barcinski, M.A. Tumor-derived microvesicles modulate the establishment of metastatic melanoma in a phosphatidylserine-dependent manner. Cancer Lett. 2009, 283, 168–175. [Google Scholar] [CrossRef]
- Mezouar, S.; Darbousset, R.; Dignat-George, F.; Panicot-Dubois, L.; Dubois, C. Inhibition of platelet activation prevents the P-selectin and integrin-dependent accumulation of cancer cell microparticles and reduces tumor growth and metastasis in vivo. Int. J. Cancer 2015, 136, 462–475. [Google Scholar] [CrossRef]
- Wang, J.G.; Geddings, J.E.; Aleman, M.M.; Cardenas, J.C.; Chantrathammachart, P.; Williams, J.C.; Kirchhofer, D.; Bogdanov, V.Y.; Bach, R.R.; Rak, J.; et al. Tumor-derived tissue factor activates coagulation and enhances thrombosis in a mouse xenograft model of human pancreatic cancer. Blood 2012, 119, 5543–5552. [Google Scholar] [CrossRef] [Green Version]
- Thomas, G.M.; Brill, A.; Mezouar, S.; Crescence, L.; Gallant, M.; Dubois, C.; Wagner, D.D. Tissue factor expressed by circulating cancer cell-derived microparticles drastically increases the incidence of deep vein thrombosis in mice. J. Thromb. Haemost. 2015, 13, 1310–1319. [Google Scholar] [CrossRef] [PubMed]
- Leal, A.C.; Mizurini, D.M.; Gomes, T.; Rochael, N.C.; Saraiva, E.M.; Dias, M.S.; Werneck, C.C.; Sielski, M.S.; Vicente, C.P.; Monteiro, R.Q. Tumor-Derived Exosomes Induce the Formation of Neutrophil Extracellular Traps: Implications For The Establishment of Cancer-Associated Thrombosis. Sci. Rep. 2017, 7, 6438. [Google Scholar] [CrossRef] [PubMed]
- Giannopoulos, G.; Oudatzis, G.; Paterakis, G.; Synetos, A.; Tampaki, E.; Bouras, G.; Hahalis, G.; Alexopoulos, D.; Tousoulis, D.; Cleman, M.W.; et al. Red blood cell and platelet microparticles in myocardial infarction patients treated with primary angioplasty. Int. J. Cardiol. 2014, 176, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Amadio, P.; Baldassarre, D.; Sandrini, L.; Weksler, B.B.; Tremoli, E.; Barbieri, S.S. Effect of cigarette smoke on monocyte procoagulant activity: Focus on platelet-derived brain-derived neurotrophic factor (BDNF). Platelets 2017, 28, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Amadio, P.; Baldassarre, D.; Tarantino, E.; Zacchi, E.; Gianellini, S.; Squellerio, I.; Amato, M.; Weksler, B.B.; Tremoli, E.; Barbieri, S.S. Production of prostaglandin E2 induced by cigarette smoke modulates tissue factor expression and activity in endothelial cells. FASEB J. 2015, 29, 4001–4010. [Google Scholar] [CrossRef] [PubMed]
- Bernal-Mizrachi, L.; Jy, W.; Jimenez, J.J.; Pastor, J.; Mauro, L.M.; Horstman, L.L.; de Marchena, E.; Ahn, Y.S. High levels of circulating endothelial microparticles in patients with acute coronary syndromes. Am. Heart J. 2003, 145, 962–970. [Google Scholar] [CrossRef]
- Burnier, L.; Fontana, P.; Kwak, B.R.; Angelillo-Scherrer, A. Cell-derived microparticles in haemostasis and vascular medicine. Thromb. Haemost. 2009, 101, 439–451. [Google Scholar] [CrossRef] [PubMed]
- Chirinos, J.A.; Heresi, G.A.; Velasquez, H.; Jy, W.; Jimenez, J.J.; Ahn, E.; Horstman, L.L.; Soriano, A.O.; Zambrano, J.P.; Ahn, Y.S. Elevation of endothelial microparticles, platelets, and leukocyte activation in patients with venous thromboembolism. J. Am. Coll. Cardiol. 2005, 45, 1467–1471. [Google Scholar] [CrossRef] [PubMed]
- Chironi, G.; Simon, A.; Hugel, B.; Del Pino, M.; Gariepy, J.; Freyssinet, J.M.; Tedgui, A. Circulating leukocyte-derived microparticles predict subclinical atherosclerosis burden in asymptomatic subjects. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2775–2780. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.S.; Zhang, H.G.; Zhang, Q.J.; Xiu, R.J. Small-size circulating endothelial microparticles in coronary artery disease. PLoS ONE 2014, 9, e104528. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.H.; Chu, K.; Lee, S.T.; Park, H.K.; Bahn, J.J.; Kim, D.H.; Kim, J.H.; Kim, M.; Kun Lee, S.; Roh, J.K. Circulating endothelial microparticles as a marker of cerebrovascular disease. Ann. Neurol. 2009, 66, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Leroyer, A.S.; Isobe, H.; Leseche, G.; Castier, Y.; Wassef, M.; Mallat, Z.; Binder, B.R.; Tedgui, A.; Boulanger, C.M. Cellular origins and thrombogenic activity of microparticles isolated from human atherosclerotic plaques. J. Am. Coll. Cardiol. 2007, 49, 772–777. [Google Scholar] [CrossRef] [PubMed]
- Mallat, Z.; Benamer, H.; Hugel, B.; Benessiano, J.; Steg, P.G.; Freyssinet, J.M.; Tedgui, A. Elevated levels of shed membrane microparticles with procoagulant potential in the peripheral circulating blood of patients with acute coronary syndromes. Circulation 2000, 101, 841–843. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, S.; Sakata, Y.; Suna, S.; Nakatani, D.; Usami, M.; Hara, M.; Kitamura, T.; Hamasaki, T.; Nanto, S.; Kawahara, Y.; et al. Circulating p53-responsive microRNAs are predictive indicators of heart failure after acute myocardial infarction. Circ. Res. 2013, 113, 322–326. [Google Scholar] [CrossRef] [PubMed]
- Montoro-Garcia, S.; Shantsila, E.; Tapp, L.D.; Lopez-Cuenca, A.; Romero, A.I.; Hernandez-Romero, D.; Orenes-Pinero, E.; Manzano-Fernandez, S.; Valdes, M.; Marin, F.; et al. Small-size circulating microparticles in acute coronary syndromes: Relevance to fibrinolytic status, reparative markers and outcomes. Atherosclerosis 2013, 227, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Morel, O.; Pereira, B.; Averous, G.; Faure, A.; Jesel, L.; Germain, P.; Grunebaum, L.; Ohlmann, P.; Freyssinet, J.M.; Bareiss, P.; et al. Increased levels of procoagulant tissue factor-bearing microparticles within the occluded coronary artery of patients with ST-segment elevation myocardial infarction: Role of endothelial damage and leukocyte activation. Atherosclerosis 2009, 204, 636–641. [Google Scholar] [CrossRef] [PubMed]
- Rectenwald, J.E.; Myers, D.D., Jr.; Hawley, A.E.; Longo, C.; Henke, P.K.; Guire, K.E.; Schmaier, A.H.; Wakefield, T.W. D-dimer, P-selectin, and microparticles: Novel markers to predict deep venous thrombosis. A pilot study. Thromb. Haemost. 2005, 94, 1312–1317. [Google Scholar] [CrossRef]
- Skeppholm, M.; Mobarrez, F.; Malmqvist, K.; Wallen, H. Platelet-derived microparticles during and after acute coronary syndrome. Thromb. Haemost. 2012, 107, 1122–1129. [Google Scholar] [CrossRef]
- Andaloussi, S.E.; Mager, I.; Breakefield, X.O.; Wood, M.J. Extracellular vesicles: Biology and emerging therapeutic opportunities. Nat. Rev. Drug Discov. 2013, 12, 347–357. [Google Scholar] [CrossRef]
- Suades, R.; Padro, T.; Crespo, J.; Ramaiola, I.; Martin-Yuste, V.; Sabate, M.; Sans-Rosello, J.; Sionis, A.; Badimon, L. Circulating microparticle signature in coronary and peripheral blood of ST elevation myocardial infarction patients in relation to pain-to-PCI elapsed time. Int. J. Cardiol. 2016, 202, 378–387. [Google Scholar] [CrossRef]
- Stepien, E.; Stankiewicz, E.; Zalewski, J.; Godlewski, J.; Zmudka, K.; Wybranska, I. Number of microparticles generated during acute myocardial infarction and stable angina correlates with platelet activation. Arch. Med. Res. 2012, 43, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Chiva-Blanch, G.; Bratseth, V.; Ritschel, V.; Andersen, G.O.; Halvorsen, S.; Eritsland, J.; Arnesen, H.; Badimon, L.; Seljeflot, I. Monocyte-derived circulating microparticles (CD14(+), CD14(+)/CD11b(+) and CD14(+)/CD142(+)) are related to long-term prognosis for cardiovascular mortality in STEMI patients. Int. J. Cardiol. 2017, 227, 876–881. [Google Scholar] [CrossRef] [PubMed]
- Sinning, J.M.; Losch, J.; Walenta, K.; Bohm, M.; Nickenig, G.; Werner, N. Circulating CD31+/Annexin V+ microparticles correlate with cardiovascular outcomes. Eur. Heart J. 2011, 32, 2034–2041. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.; Sorensson, P.; Saleh, N.; Arheden, H.; Ryden, L.; Pernow, J. Circulating endothelial and platelet derived microparticles reflect the size of myocardium at risk in patients with ST-elevation myocardial infarction. Atherosclerosis 2012, 221, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Schiro, A.; Wilkinson, F.L.; Weston, R.; Smyth, J.V.; Serracino-Inglott, F.; Alexander, M.Y. Elevated levels of endothelial-derived microparticles, and serum CXCL9 and SCGF-beta are associated with unstable asymptomatic carotid plaques. Sci. Rep. 2015, 5, 16658. [Google Scholar] [CrossRef] [PubMed]
- Berezin, A.E.; Kremzer, A.A.; Martovitskaya, Y.V.; Samura, T.A.; Berezina, T.A.; Zulli, A.; Klimas, J.; Kruzliak, P. The utility of biomarker risk prediction score in patients with chronic heart failure. Int. J. Clin. Exp. Med. 2015, 8, 18255–18264. [Google Scholar] [CrossRef]
- Nozaki, T.; Sugiyama, S.; Sugamura, K.; Ohba, K.; Matsuzawa, Y.; Konishi, M.; Matsubara, J.; Akiyama, E.; Sumida, H.; Matsui, K.; et al. Prognostic value of endothelial microparticles in patients with heart failure. Eur. J. Heart Fail. 2010, 12, 1223–1228. [Google Scholar] [CrossRef]
- Matsumoto, N.; Nomura, S.; Kamihata, H.; Kimura, Y.; Iwasaka, T. Increased level of oxidized LDL-dependent monocyte-derived microparticles in acute coronary syndrome. Thromb. Haemost. 2004, 91, 146–154. [Google Scholar] [CrossRef]
- Christersson, C.; Thulin, A.; Siegbahn, A. Microparticles during long-term follow-up after acute myocardial infarction. Association to atherosclerotic burden and risk of cardiovascular events. Thromb. Haemost. 2017, 117, 1571–1581. [Google Scholar] [CrossRef]
- Thygesen, K.; Alpert, J.S.; Jaffe, A.S.; Simoons, M.L.; Chaitman, B.R.; White, H.D.; Katus, H.A.; Apple, F.S.; Lindahl, B.; Morrow, D.A.; et al. Third universal definition of myocardial infarction. J. Am. Coll. Cardiol. 2012, 60, 1581–1598. [Google Scholar] [CrossRef]
- Sun, C.; Zhao, W.B.; Chen, Y.; Hu, H.Y. Higher Plasma Concentrations of Platelet Microparticles in Patients With Acute Coronary Syndrome: A Systematic Review and Meta-analysis. Can. J. Cardiol. 2016, 32, 1321–1325. [Google Scholar] [CrossRef] [PubMed]
- Behan, M.W.; Fox, S.C.; Heptinstall, S.; Storey, R.F. Inhibitory effects of P2Y12 receptor antagonists on TRAP-induced platelet aggregation, procoagulant activity, microparticle formation and intracellular calcium responses in patients with acute coronary syndromes. Platelets 2005, 16, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Judge, H.M.; Buckland, R.J.; Sugidachi, A.; Jakubowski, J.A.; Storey, R.F. The active metabolite of prasugrel effectively blocks the platelet P2Y12 receptor and inhibits procoagulant and pro-inflammatory platelet responses. Platelets 2008, 19, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Franca, C.N.; Pinheiro, L.F.; Izar, M.C.; Brunialti, M.K.; Salomao, R.; Bianco, H.T.; Kasmas, S.H.; Barbosa, S.P.; de Nucci, G.; Fonseca, F.A. Endothelial progenitor cell mobilization and platelet microparticle release are influenced by clopidogrel plasma levels in stable coronary artery disease. Circ. J. 2012, 76, 729–736. [Google Scholar] [CrossRef] [PubMed]
- Biasucci, L.M.; Porto, I.; De Maria, G.L.; Di Vito, L.; Burzotta, F.; Tritarelli, A.; Gustapane, M.; Camaioni, C.; Cautilli, G.; Crea, F. Levels of Platelet-Derived Microparticles Are Related to Angiographic No-Reflow Phenomenon in Patients With ST-Elevation Myocardial Infarction Undergoing Primary-PCI. Circulation 2018, 122, A15198. [Google Scholar]
- Gasecka, A.; Nieuwland, R.; Budnik, M.; Dignat-George, F.; Eyileten, C.; Harrison, P.; Huczek, Z.; Kaplon-Cieslicka, A.; Lacroix, R.; Opolski, G.; et al. Randomized controlled trial protocol to investigate the antiplatelet therapy effect on extracellular vesicles (AFFECT EV) in acute myocardial infarction. Platelets 2018, 29, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Kafian, S.; Mobarrez, F.; Wallen, H.; Samad, B. Association between platelet reactivity and circulating platelet-derived microvesicles in patients with acute coronary syndrome. Platelets 2015, 26, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Bulut, D.; Becker, V.; Mugge, A. Acetylsalicylate reduces endothelial and platelet-derived microparticles in patients with coronary artery disease. Can. J. Physiol. Pharmacol. 2011, 89, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Giacomazzi, A.; Degan, M.; Calabria, S.; Meneguzzi, A.; Minuz, P. Antiplatelet Agents Inhibit the Generation of Platelet-Derived Microparticles. Front. Pharmacol. 2016, 7, 314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiva-Blanch, G.; Suades, R.; Padro, T.; Vilahur, G.; Pena, E.; Ybarra, J.; Pou, J.M.; Badimon, L. Microparticle Shedding by Erythrocytes, Monocytes and Vascular Smooth Muscular Cells Is Reduced by Aspirin in Diabetic Patients. Rev. Esp. Cardiol. 2016, 69, 672–680. [Google Scholar] [CrossRef] [PubMed]
- Duarte, R.C.; Goncalves, L.H.; Campos, F.M.; Filho, O.A.; Alves, M.T.; Fernandes, A.P.; Borges, K.B.; Dusse, L.M.; Faria, M.C.; Goncalves, G.S.; et al. Effect of acetylsalicylic acid on platelet activation and oxidative profile in a set of Brazilian patients with type 2 diabetes mellitus. Blood Coagul. Fibrinolysis 2015, 26, 123–130. [Google Scholar] [CrossRef]
- Lubsczyk, B.; Kollars, M.; Hron, G.; Kyrle, P.A.; Weltermann, A.; Gartner, V. Low dose acetylsalicylic acid and shedding of microparticles in vivo in humans. Eur. J. Clin. Investig. 2010, 40, 477–482. [Google Scholar] [CrossRef]
- Shirafuji, T.; Hamaguchi, H.; Kanda, F. Measurement of platelet-derived microparticle levels in the chronic phase of cerebral infarction using an enzyme-linked immunosorbent assay. Kobe J. Med. Sci. 2008, 54, 55–61. [Google Scholar]
- Morel, O.; Hugel, B.; Jesel, L.; Mallat, Z.; Lanza, F.; Douchet, M.P.; Zupan, M.; Chauvin, M.; Cazenave, J.P.; Tedgui, A.; et al. Circulating procoagulant microparticles and soluble GPV in myocardial infarction treated by primary percutaneous transluminal coronary angioplasty. A possible role for GPIIb-IIIa antagonists. J. Thromb. Haemost. 2004, 2, 1118–1126. [Google Scholar] [CrossRef]
- Velez, P.; Parguina, A.F.; Ocaranza-Sanchez, R.; Grigorian-Shamagian, L.; Rosa, I.; Alonso-Orgaz, S.; de la Cuesta, F.; Guitian, E.; Moreu, J.; Barderas, M.G.; et al. Identification of a circulating microvesicle protein network involved in ST-elevation myocardial infarction. Thromb. Haemost. 2014, 112, 716–726. [Google Scholar] [CrossRef] [PubMed]
- Cheow, E.S.; Cheng, W.C.; Lee, C.N.; de Kleijn, D.; Sorokin, V.; Sze, S.K. Plasma-derived Extracellular Vesicles Contain Predictive Biomarkers and Potential Therapeutic Targets for Myocardial Ischemic (MI) Injury. Mol. Cell. Proteom. 2016, 15, 2628–2640. [Google Scholar] [CrossRef] [Green Version]
- De Hoog, V.C.; Timmers, L.; Schoneveld, A.H.; Wang, J.W.; van de Weg, S.M.; Sze, S.K.; van Keulen, J.K.; Hoes, A.W.; den Ruijter, H.M.; de Kleijn, D.P.; et al. Serum extracellular vesicle protein levels are associated with acute coronary syndrome. Eur. Heart J. Acute Cardiovasc. Care 2013, 2, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Huisse, M.G.; Lanoy, E.; Tcheche, D.; Feldman, L.J.; Bezeaud, A.; Angles-Cano, E.; Mary-Krause, M.; de Prost, D.; Guillin, M.C.; Steg, P.G. Prothrombotic markers and early spontaneous recanalization in ST-segment elevation myocardial infarction. Thromb. Haemost. 2007, 98, 420–426. [Google Scholar] [PubMed]
- Switonska, M.; Slomka, A.; Sinkiewicz, W.; Zekanowska, E. Tissue-factor-bearing microparticles (MPs-TF) in patients with acute ischaemic stroke: The influence of stroke treatment on MPs-TF generation. Eur. J. Neurol. 2015, 22, 395–401. [Google Scholar] [CrossRef]
- Keuren, J.F.; Jie, K.S.; Leers, M.P. Increased expression of TF on monocytes, but decreased numbers of TF bearing microparticles in blood from patients with acute myocardial infarction. Eur. J. Haematol. 2009, 83, 387–388. [Google Scholar] [CrossRef]
- Maly, M.; Hrachovinova, I.; Tomasov, P.; Salaj, P.; Hajek, P.; Veselka, J. Patients with acute coronary syndromes have low tissue factor activity and microparticle count, but normal concentration of tissue factor antigen in platelet free plasma: A pilot study. Eur. J. Haematol. 2009, 82, 148–153. [Google Scholar] [CrossRef] [PubMed]
- Bi, S.; Wang, C.; Jin, Y.; Lv, Z.; Xing, X.; Lu, Q. Correlation between serum exosome derived miR-208a and acute coronary syndrome. Int. J. Clin. Exp. Med. 2015, 8, 4275–4280. [Google Scholar] [PubMed]
- Kuwabara, Y.; Ono, K.; Horie, T.; Nishi, H.; Nagao, K.; Kinoshita, M.; Watanabe, S.; Baba, O.; Kojima, Y.; Shizuta, S.; et al. Increased microRNA-1 and microRNA-133a levels in serum of patients with cardiovascular disease indicate myocardial damage. Circ. Cardiovasc. Genet. 2011, 4, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Jansen, F.; Yang, X.; Proebsting, S.; Hoelscher, M.; Przybilla, D.; Baumann, K.; Schmitz, T.; Dolf, A.; Endl, E.; Franklin, B.S.; et al. MicroRNA expression in circulating microvesicles predicts cardiovascular events in patients with coronary artery disease. J. Am. Heart Assoc. 2014, 3, e001249. [Google Scholar] [CrossRef] [PubMed]
- Dolz, S.; Gorriz, D.; Tembl, J.I.; Sanchez, D.; Fortea, G.; Parkhutik, V.; Lago, A. Circulating MicroRNAs as Novel Biomarkers of Stenosis Progression in Asymptomatic Carotid Stenosis. Stroke 2017, 48, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Emanueli, C.; Shearn, A.I.; Laftah, A.; Fiorentino, F.; Reeves, B.C.; Beltrami, C.; Mumford, A.; Clayton, A.; Gurney, M.; Shantikumar, S.; et al. Coronary Artery-Bypass-Graft Surgery Increases the Plasma Concentration of Exosomes Carrying a Cargo of Cardiac MicroRNAs: An Example of Exosome Trafficking Out of the Human Heart with Potential for Cardiac Biomarker Discovery. PLoS ONE 2016, 11, e0154274. [Google Scholar] [CrossRef]
- Ye, R.; Ye, C.; Huang, Y.; Liu, L.; Wang, S. Circulating tissue factor positive microparticles in patients with acute recurrent deep venous thrombosis. Thromb. Res. 2012, 130, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Elyamany, G.; Alzahrani, A.M.; Bukhary, E. Cancer-associated thrombosis: An overview. Clin. Med. Insights Oncol. 2014, 8, 129–137. [Google Scholar] [CrossRef]
- Falanga, A.; Consonni, R.; Marchetti, M.; Locatelli, G.; Garattini, E.; Passerini, C.G.; Gordon, S.G.; Barbui, T. Cancer procoagulant and tissue factor are differently modulated by all-trans-retinoic acid in acute promyelocytic leukemia cells. Blood 1998, 92, 143–151. [Google Scholar]
- Khorana, A.A.; Francis, C.W.; Culakova, E.; Kuderer, N.M.; Lyman, G.H. Frequency, risk factors, and trends for venous thromboembolism among hospitalized cancer patients. Cancer 2007, 110, 2339–2346. [Google Scholar] [CrossRef]
- Young, A.; Chapman, O.; Connor, C.; Poole, C.; Rose, P.; Kakkar, A.K. Thrombosis and cancer. Nat. Rev. Clin. Oncol. 2012, 9, 437–449. [Google Scholar] [CrossRef] [PubMed]
- Bidot, L.; Jy, W.; Bidot, C., Jr.; Jimenez, J.J.; Fontana, V.; Horstman, L.L.; Ahn, Y.S. Microparticle-mediated thrombin generation assay: Increased activity in patients with recurrent thrombosis. J. Thromb. Haemost. 2008, 6, 913–919. [Google Scholar] [CrossRef] [PubMed]
- Bucciarelli, P.; Martinelli, I.; Artoni, A.; Passamonti, S.M.; Previtali, E.; Merati, G.; Tripodi, A.; Mannucci, P.M. Circulating microparticles and risk of venous thromboembolism. Thromb. Res. 2012, 129, 591–597. [Google Scholar] [CrossRef] [PubMed]
- Flores-Nascimento, M.C.; Beltrame, M.P.; De Paula, E.V.; Montalvao, S.L.; Pereira, F.G.; Orsi, F.L.; Lorand-Metze, I.; Annichino-Bizzacchi, J.M. Microparticles in deep venous thrombosis, antiphospholipid syndrome and Factor V Leiden. Platelets 2009, 20, 367–375. [Google Scholar] [CrossRef] [PubMed]
- Steppich, B.A.; Hassenpflug, M.; Braun, S.L.; Schomig, K.; von Beckerath, O.; von Beckerath, N.; Eckstein, H.H.; Ott, I. Circulating tissue factor and microparticles are not increased in patients with deep vein thrombosis. Vasa 2011, 40, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Campello, E.; Spiezia, L.; Radu, C.M.; Bulato, C.; Gavasso, S.; Tormene, D.; Woodhams, B.; Dalla Valle, F.; Simioni, P. Circulating microparticles and the risk of thrombosis in inherited deficiencies of antithrombin, protein C and protein S. Thromb. Haemost. 2016, 115, 81–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, E.; Ambrose, N.L.; Ahnstrom, J.; Kiprianos, A.P.; Stanford, M.R.; Eleftheriou, D.; Brogan, P.A.; Mason, J.C.; Johns, M.; Laffan, M.A.; et al. A low balance between microparticles expressing tissue factor pathway inhibitor and tissue factor is associated with thrombosis in Behcet’s Syndrome. Sci. Rep. 2016, 6, 38104. [Google Scholar] [CrossRef]
- Palkovits, J.; Novacek, G.; Kollars, M.; Hron, G.; Osterode, W.; Quehenberger, P.; Kyrle, P.A.; Vogelsang, H.; Reinisch, W.; Papay, P.; et al. Tissue factor exposing microparticles in inflammatory bowel disease. J. Crohns Colitis 2013, 7, 222–229. [Google Scholar] [CrossRef] [Green Version]
- Sellam, J.; Proulle, V.; Jungel, A.; Ittah, M.; Miceli Richard, C.; Gottenberg, J.E.; Toti, F.; Benessiano, J.; Gay, S.; Freyssinet, J.M.; et al. Increased levels of circulating microparticles in primary Sjogren’s syndrome, systemic lupus erythematosus and rheumatoid arthritis and relation with disease activity. Arthritis Res. Ther. 2009, 11, R156. [Google Scholar] [CrossRef]
- Nieuwland, R.; Berckmans, R.J.; McGregor, S.; Boing, A.N.; Romijn, F.P.; Westendorp, R.G.; Hack, C.E.; Sturk, A. Cellular origin and procoagulant properties of microparticles in meningococcal sepsis. Blood 2000, 95, 930–935. [Google Scholar]
- Campello, E.; Radu, C.M.; Duner, E.; Lombardi, A.M.; Spiezia, L.; Bendo, R.; Ferrari, S.; Simioni, P.; Fabris, F. Activated Platelet-Derived and Leukocyte-Derived Circulating Microparticles and the Risk of Thrombosis in Heparin-Induced Thrombocytopenia: A Role for PF4-Bearing Microparticles? Cytom. B Clin. Cytom. 2018, 94, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Fu, Y.; Xu, L.; Xiao, L.; Yue, Y.; Liu, S.; Huang, Q.; Li, S.; Li, Y. Diagnostic value of platelet-derived microparticles in pulmonary thromboembolism: A population-based study. Exp. Ther. Med. 2018, 16, 3099–3106. [Google Scholar] [CrossRef] [PubMed]
- Jamaly, S.; Basavaraj, M.G.; Starikova, I.; Olsen, R.; Braekkan, S.K.; Hansen, J.B. Elevated plasma levels of P-selectin glycoprotein ligand-1-positive microvesicles in patients with unprovoked venous thromboembolism. J. Thromb. Haemost. 2018. [Google Scholar] [CrossRef] [PubMed]
- Falanga, A.; Russo, L.; Milesi, V.; Vignoli, A. Mechanisms and risk factors of thrombosis in cancer. Crit. Rev. Oncol. Hematol. 2017, 118, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Mitrugno, A.; Tormoen, G.W.; Kuhn, P.; McCarty, O.J. The prothrombotic activity of cancer cells in the circulation. Blood Rev. 2016, 30, 11–19. [Google Scholar] [CrossRef]
- Campello, E.; Spiezia, L.; Radu, C.M.; Bulato, C.; Castelli, M.; Gavasso, S.; Simioni, P. Endothelial, platelet, and tissue factor-bearing microparticles in cancer patients with and without venous thromboembolism. Thromb. Res. 2011, 127, 473–477. [Google Scholar] [CrossRef]
- Tesselaar, M.E.; Romijn, F.P.; van der Linden, I.K.; Bertina, R.M.; Osanto, S. Microparticle-associated tissue factor activity in cancer patients with and without thrombosis. J. Thromb. Haemost. 2009, 7, 1421–1423. [Google Scholar] [CrossRef]
- Van Es, N.; Hisada, Y.; Di Nisio, M.; Cesarman, G.; Kleinjan, A.; Mahe, I.; Otten, H.M.; Kamphuisen, P.W.; Berckmans, R.J.; Buller, H.R.; et al. Extracellular vesicles exposing tissue factor for the prediction of venous thromboembolism in patients with cancer: A prospective cohort study. Thromb. Res. 2018, 166, 54–59. [Google Scholar] [CrossRef]
- Van Es, N.; Louzada, M.; Carrier, M.; Tagalakis, V.; Gross, P.L.; Shivakumar, S.; Rodger, M.A.; Wells, P.S. Predicting the risk of recurrent venous thromboembolism in patients with cancer: A prospective cohort study. Thromb. Res. 2018, 163, 41–46. [Google Scholar] [CrossRef]
- Manly, D.A.; Wang, J.; Glover, S.L.; Kasthuri, R.; Liebman, H.A.; Key, N.S.; Mackman, N. Increased microparticle tissue factor activity in cancer patients with Venous Thromboembolism. Thromb. Res. 2010, 125, 511–512. [Google Scholar] [CrossRef] [Green Version]
- Del Conde, I.; Bharwani, L.D.; Dietzen, D.J.; Pendurthi, U.; Thiagarajan, P.; Lopez, J.A. Microvesicle-associated tissue factor and Trousseau’s syndrome. J. Thromb. Haemost. 2007, 5, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Tilley, R.E.; Holscher, T.; Belani, R.; Nieva, J.; Mackman, N. Tissue factor activity is increased in a combined platelet and microparticle sample from cancer patients. Thromb. Res. 2008, 122, 604–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Doormaal, F.; Kleinjan, A.; Berckmans, R.J.; Mackman, N.; Manly, D.; Kamphuisen, P.W.; Richel, D.J.; Buller, H.R.; Sturk, A.; Nieuwland, R. Coagulation activation and microparticle-associated coagulant activity in cancer patients. An exploratory prospective study. Thromb. Haemost. 2012, 108, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Auwerda, J.J.; Yuana, Y.; Osanto, S.; de Maat, M.P.; Sonneveld, P.; Bertina, R.M.; Leebeek, F.W. Microparticle-associated tissue factor activity and venous thrombosis in multiple myeloma. Thromb. Haemost. 2011, 105, 14–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gezelius, E.; Flou Kristensen, A.; Bendahl, P.O.; Hisada, Y.; Risom Kristensen, S.; Ek, L.; Bergman, B.; Wallberg, M.; Falkmer, U.; Mackman, N.; et al. Coagulation biomarkers and prediction of venous thromboembolism and survival in small cell lung cancer: A sub-study of RASTEN—A randomized trial with low molecular weight heparin. PLoS ONE 2018, 13, e0207387. [Google Scholar] [CrossRef] [PubMed]
- Thaler, J.; Ay, C.; Mackman, N.; Bertina, R.M.; Kaider, A.; Marosi, C.; Key, N.S.; Barcel, D.A.; Scheithauer, W.; Kornek, G.; et al. Microparticle-associated tissue factor activity, venous thromboembolism and mortality in pancreatic, gastric, colorectal and brain cancer patients. J. Thromb. Haemost. 2012, 10, 1363–1370. [Google Scholar] [CrossRef] [PubMed]
- Hisada, Y.; Alexander, W.; Kasthuri, R.; Voorhees, P.; Mobarrez, F.; Taylor, A.; McNamara, C.; Wallen, H.; Witkowski, M.; Key, N.S.; et al. Measurement of microparticle tissue factor activity in clinical samples: A summary of two tissue factor-dependent FXa generation assays. Thromb. Res. 2016, 139, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Liu, H.; Shen, Y.; Zhang, Y.; Pan, S. Platelet-derived microvesicles are involved in cardio-protective effects of remote preconditioning. Int. J. Clin. Exp. Pathol. 2015, 8, 10832–10839. [Google Scholar]
- Shan, L.Y.; Li, J.Z.; Zu, L.Y.; Niu, C.G.; Ferro, A.; Zhang, Y.D.; Zheng, L.M.; Ji, Y. Platelet-derived microparticles are implicated in remote ischemia conditioning in a rat model of cerebral infarction. CNS Neurosci. Ther. 2013, 19, 917–925. [Google Scholar] [CrossRef]
- Kervadec, A.; Bellamy, V.; El Harane, N.; Arakelian, L.; Vanneaux, V.; Cacciapuoti, I.; Nemetalla, H.; Perier, M.C.; Toeg, H.D.; Richart, A.; et al. Cardiovascular progenitor-derived extracellular vesicles recapitulate the beneficial effects of their parent cells in the treatment of chronic heart failure. J. Heart Lung Transpl. 2016, 35, 795–807. [Google Scholar] [CrossRef]
- Timmers, L.; Lim, S.K.; Hoefer, I.E.; Arslan, F.; Lai, R.C.; van Oorschot, A.A.; Goumans, M.J.; Strijder, C.; Sze, S.K.; Choo, A.; et al. Human mesenchymal stem cell-conditioned medium improves cardiac function following myocardial infarction. Stem Cell Res. 2011, 6, 206–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arslan, F.; Lai, R.C.; Smeets, M.B.; Akeroyd, L.; Choo, A.; Aguor, E.N.; Timmers, L.; van Rijen, H.V.; Doevendans, P.A.; Pasterkamp, G.; et al. Mesenchymal stem cell-derived exosomes increase ATP levels, decrease oxidative stress and activate PI3K/Akt pathway to enhance myocardial viability and prevent adverse remodeling after myocardial ischemia/reperfusion injury. Stem Cell Res. 2013, 10, 301–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hergenreider, E.; Heydt, S.; Treguer, K.; Boettger, T.; Horrevoets, A.J.; Zeiher, A.M.; Scheffer, M.P.; Frangakis, A.S.; Yin, X.; Mayr, M.; et al. Atheroprotective communication between endothelial cells and smooth muscle cells through miRNAs. Nat. Cell Biol. 2012, 14, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Zhang, Z.; Ma, T.; Yang, Z.; Zhang, J.; Liu, X.; Lu, D.; Shen, Z.; Yang, J.; Meng, Q. Endothelial progenitor cell-derived exosomes, loaded with miR-126, promoted deep vein thrombosis resolution and recanalization. Stem Cell Res. Ther. 2018, 9, 223. [Google Scholar] [CrossRef] [PubMed]
- Pawlowski, C.L.; Li, W.; Sun, M.; Ravichandran, K.; Hickman, D.; Kos, C.; Kaur, G.; Sen Gupta, A. Platelet microparticle-inspired clot-responsive nanomedicine for targeted fibrinolysis. Biomaterials 2017, 128, 94–108. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Type of EVs | Biogenesis | Isolation | Detection |
|---|---|---|---|
| Microvesicles (100 nm–1 µm) | Blebbing of plasma membrane | Ultracentrifugation, density gradients | AFM, EM, NTA, IF, FC, ELISA |
| Exosomes (30–100 nm) | Release by exocytosis of MVBs | Ultracentrifugation, immunopurification, density gradient, commercial kit, size exclusion chromatography | AFM, EM, NTA, RPS, DLS, WB, ELISA |
| Apoptotic body (500 nm–4 µm) | Blebbing of apoptotic cells | Centrifugation, filtration, FACS | IF, FC |
| MVs | Exosomes | |
|---|---|---|
| Platelets | GPIb, TF, CD31, CD36, CD62P, CD61, CD40L, vWF, fibrinogen, thrombospondin | miR126-3p, mi-R21, mi-223, miR-339, miR-328, miR-22, miR-185, miR-320b, GPIb, GPV, CXCL4, CXCL7, HMGB1 |
| Megakaryocytes | GPVI, CD42b | |
| Erythrocytes | Band 3, Actin, Hemoglobin A, CD55, CD59, Iron, Annexin A1, annexin A2, glut1 | |
| Leukocytes | ICAM-1, TF, PSGL-1, CD62L, C3, MMPs, inflammatory cytokines | miR-222, miR-155, miR-146a, miR-146b and miR-125a-5p, miR-21-5p |
| Endothelial cells | Annexin A1, annexin A2, actin, cofilin, calnexin, calreticulin, caveolin-1, thrombospondin, CD59, ICAM-1, α5β1, α2β1 | miR-214, miR-210, miR-126, miR-146a, MiR-206, ARF6, NCX1 |
| Type of EVs | Major Findings | Reference |
|---|---|---|
| Platelet-derived EVSs | PMPs induce fibrin deposition on atherosclerotic arteries. Increase platelet aggregation and adhesion to collagen. Shorten epinephrine/collagen closure and reduce clotting time | [142] |
| PMPs induce trans-activation of platelets by transferring arachidonic acid | [143] | |
| PMPs promote thrombin generation in a Factor XII-dependent fashion | [144] | |
| PMPs bind protein S and support the anticoagulant activity of activated protein C | [151] | |
| Platelet-exosomes inhibis platelet activation, endothelial mobility, inflammation and proatherothombotic cellular functions | [155,156] | |
| Erythrocytes-derived MVs | Induce coagulation through a factor XI- and factor XII-dependent mechanism | [144,146,147] |
| Bind protein S and support the anticoagulant activity of activated protein C | [150,151] | |
| Leukocyte-MVs | Sustain plasmin generation | [153] |
| Mast cell-exosome | Stimulate expression and activity of plasminogen activator inhibitor type 1 | [157] |
| Monocyte-MVs | Trigger coagulation predominantly via TF | [144,145] |
| Induce overexpression of TF and the reduction of TFPI and TM on endothelial cells | [148] | |
| Participate to fibrin generation and thrombus growth in vivo | [139] | |
| Macrophages-EVs | Modulate procoagulant activity and arterial thrombosis in vivo | [161] |
| EC-MVs | Stimulate TF expression and procoagulant activity in monocytic cell line | [149] |
| Enhance plasminogen activation, plasmin generation and fibrinolysis | [173] | |
| Bind to platelet CD36 and support thrombus formation in vivo | [160] | |
| Cancer cell-EV | Reduce bleeding time and time of vessel occlusion | [140] |
| Cancer cell-MVs enhanced blood coagulation and platelet aggregation | [141] | |
| Promote TF-dependent coagulation and thrombus formation in vivo | [166,167,168,169,170] | |
| Cancer cell-Exosomes accelerate venous thrombosis in vivo by inducing the release of NETs | [171,172] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zarà, M.; Guidetti, G.F.; Camera, M.; Canobbio, I.; Amadio, P.; Torti, M.; Tremoli, E.; Barbieri, S.S. Biology and Role of Extracellular Vesicles (EVs) in the Pathogenesis of Thrombosis. Int. J. Mol. Sci. 2019, 20, 2840. https://doi.org/10.3390/ijms20112840
Zarà M, Guidetti GF, Camera M, Canobbio I, Amadio P, Torti M, Tremoli E, Barbieri SS. Biology and Role of Extracellular Vesicles (EVs) in the Pathogenesis of Thrombosis. International Journal of Molecular Sciences. 2019; 20(11):2840. https://doi.org/10.3390/ijms20112840
Chicago/Turabian StyleZarà, Marta, Gianni Francesco Guidetti, Marina Camera, Ilaria Canobbio, Patrizia Amadio, Mauro Torti, Elena Tremoli, and Silvia Stella Barbieri. 2019. "Biology and Role of Extracellular Vesicles (EVs) in the Pathogenesis of Thrombosis" International Journal of Molecular Sciences 20, no. 11: 2840. https://doi.org/10.3390/ijms20112840
APA StyleZarà, M., Guidetti, G. F., Camera, M., Canobbio, I., Amadio, P., Torti, M., Tremoli, E., & Barbieri, S. S. (2019). Biology and Role of Extracellular Vesicles (EVs) in the Pathogenesis of Thrombosis. International Journal of Molecular Sciences, 20(11), 2840. https://doi.org/10.3390/ijms20112840