Red Ginseng Attenuates Aβ-Induced Mitochondrial Dysfunction and Aβ-mediated Pathology in an Animal Model of Alzheimer’s Disease

 , and
, and
Abstract
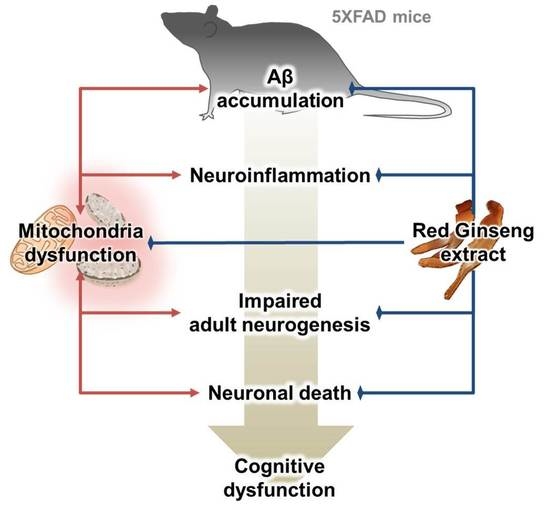
:
1. Introduction
2. Results
2.1. Cytotoxicity Evaluation of RGE in Hippocampal Neurons
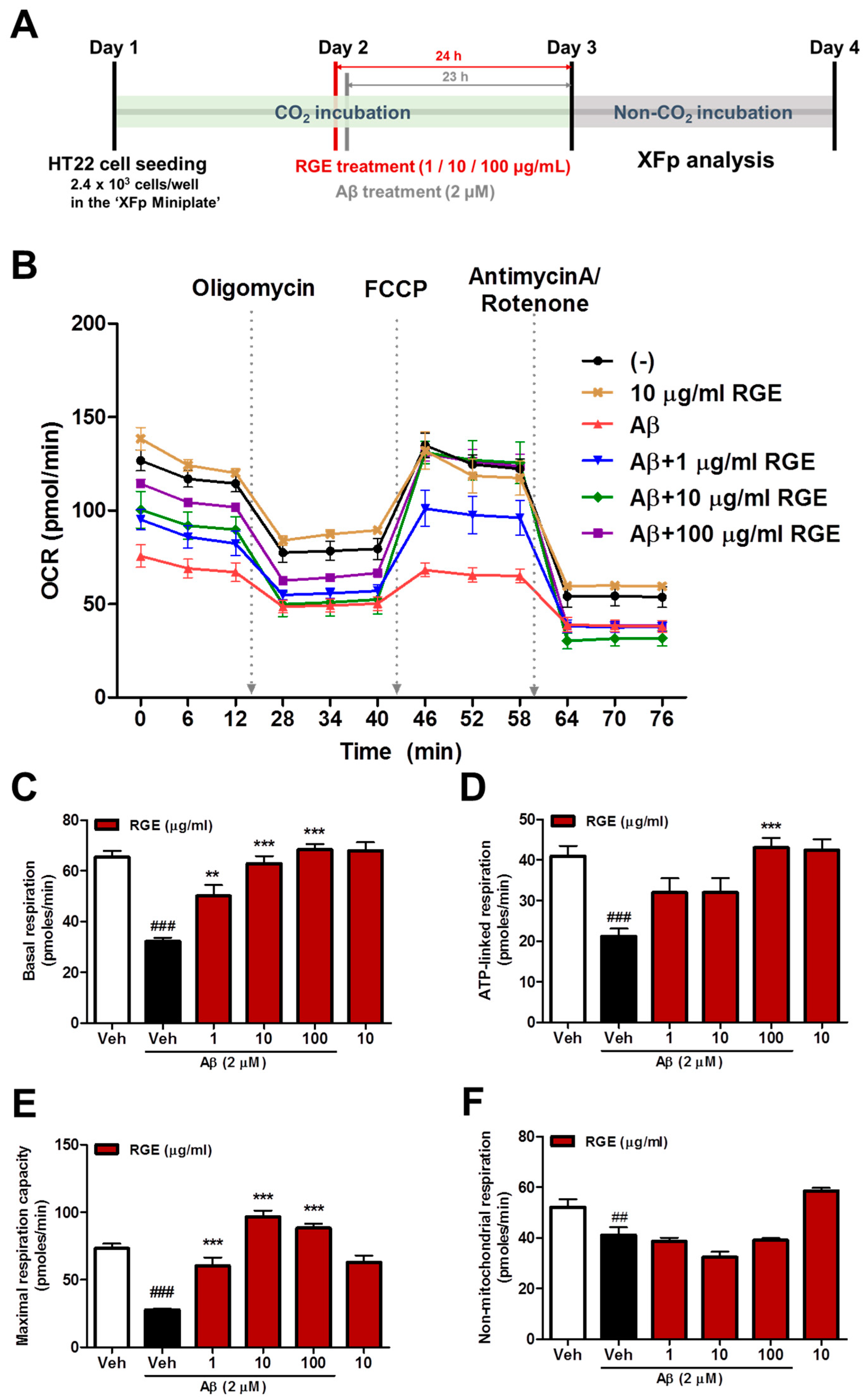
2.2. RGE Prevents Aβ-Induced Mitochondrial Dysfunction in HT22 Cells
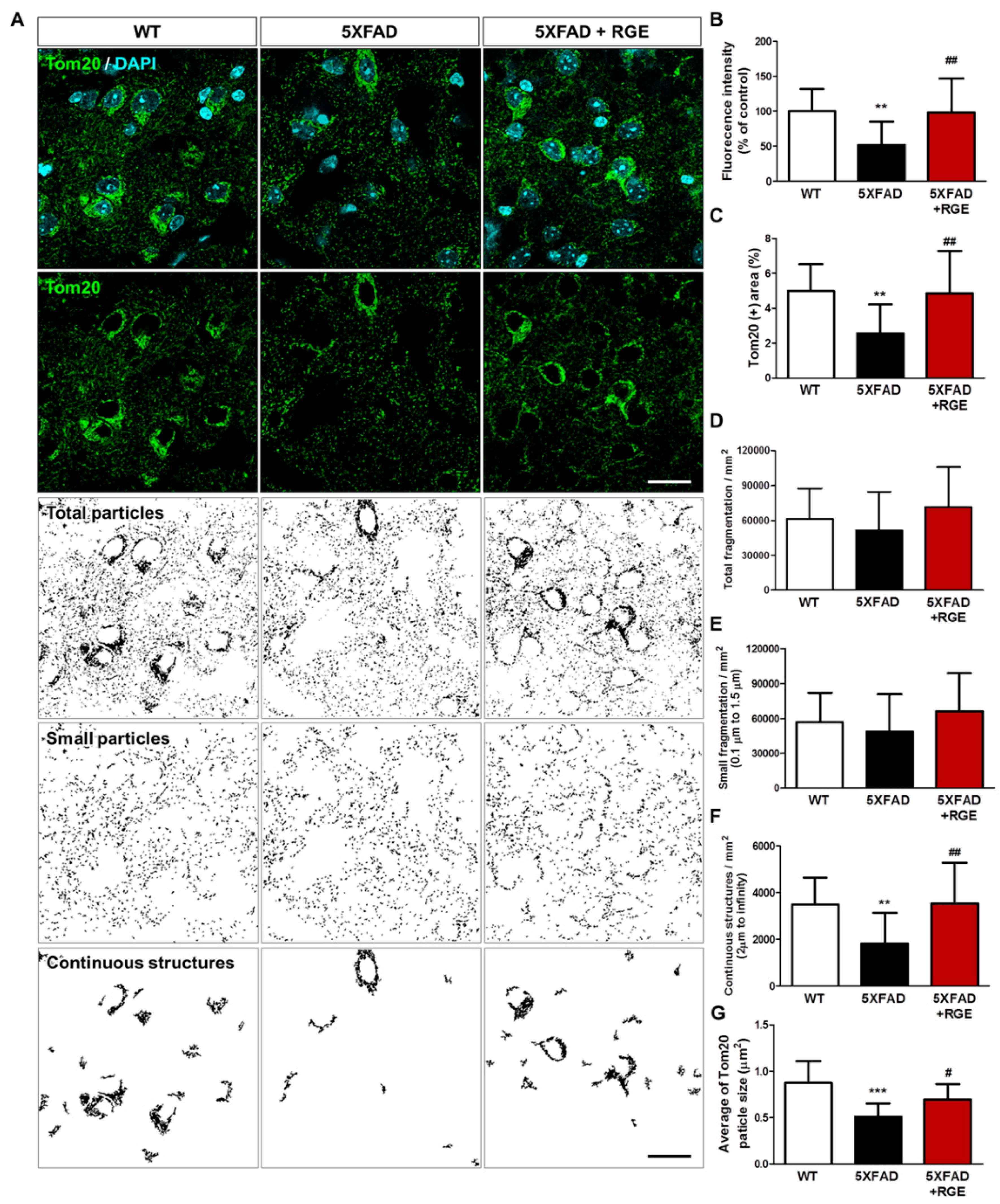
2.3. RGE Alleviates Abnormal Mitochondrial Dynamics in Aβ-Overexpressing Transgenic Mice
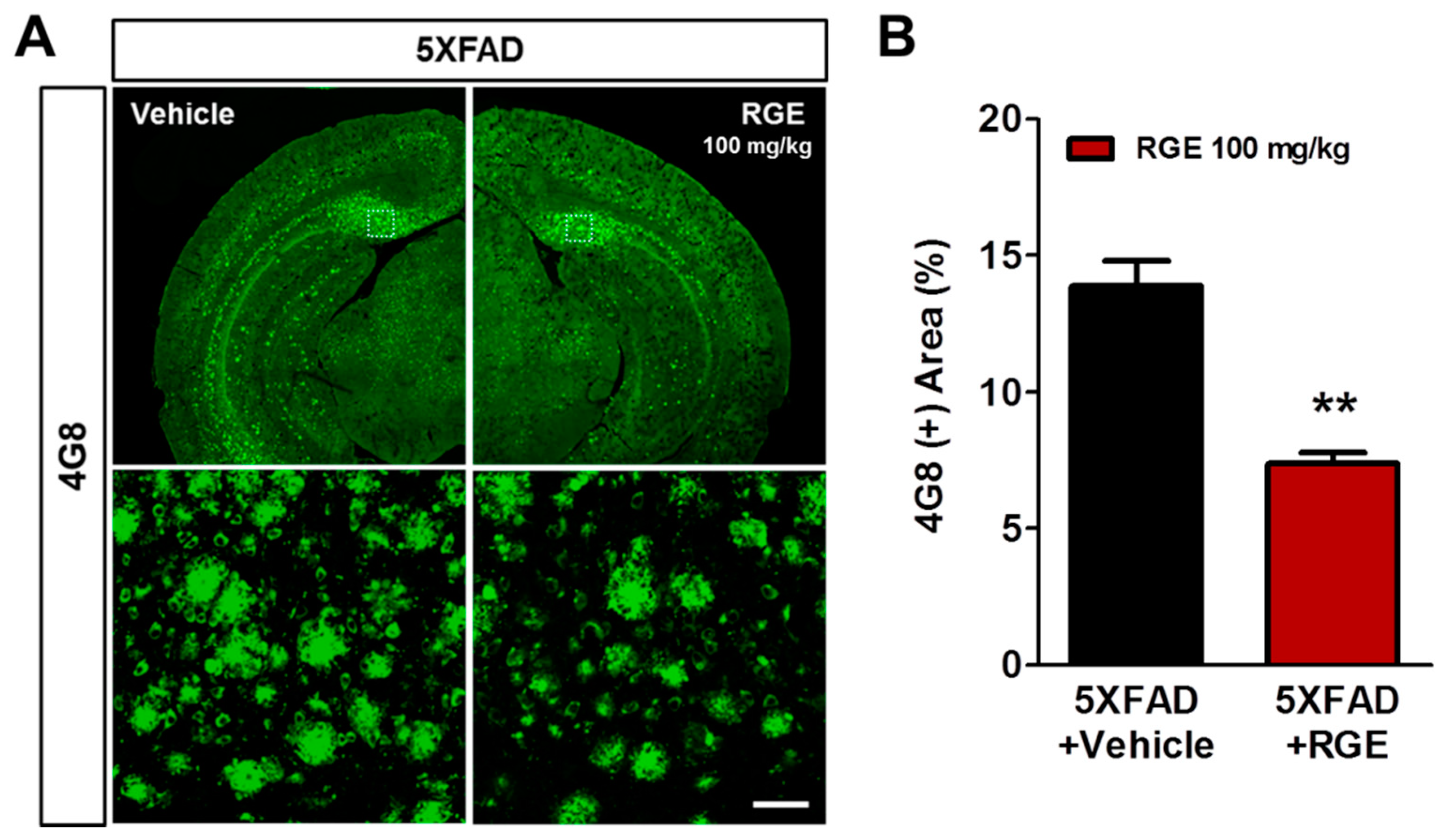
2.4. RGE Reduces Aβ Deposits in the Subiculum of 5XFAD Mice
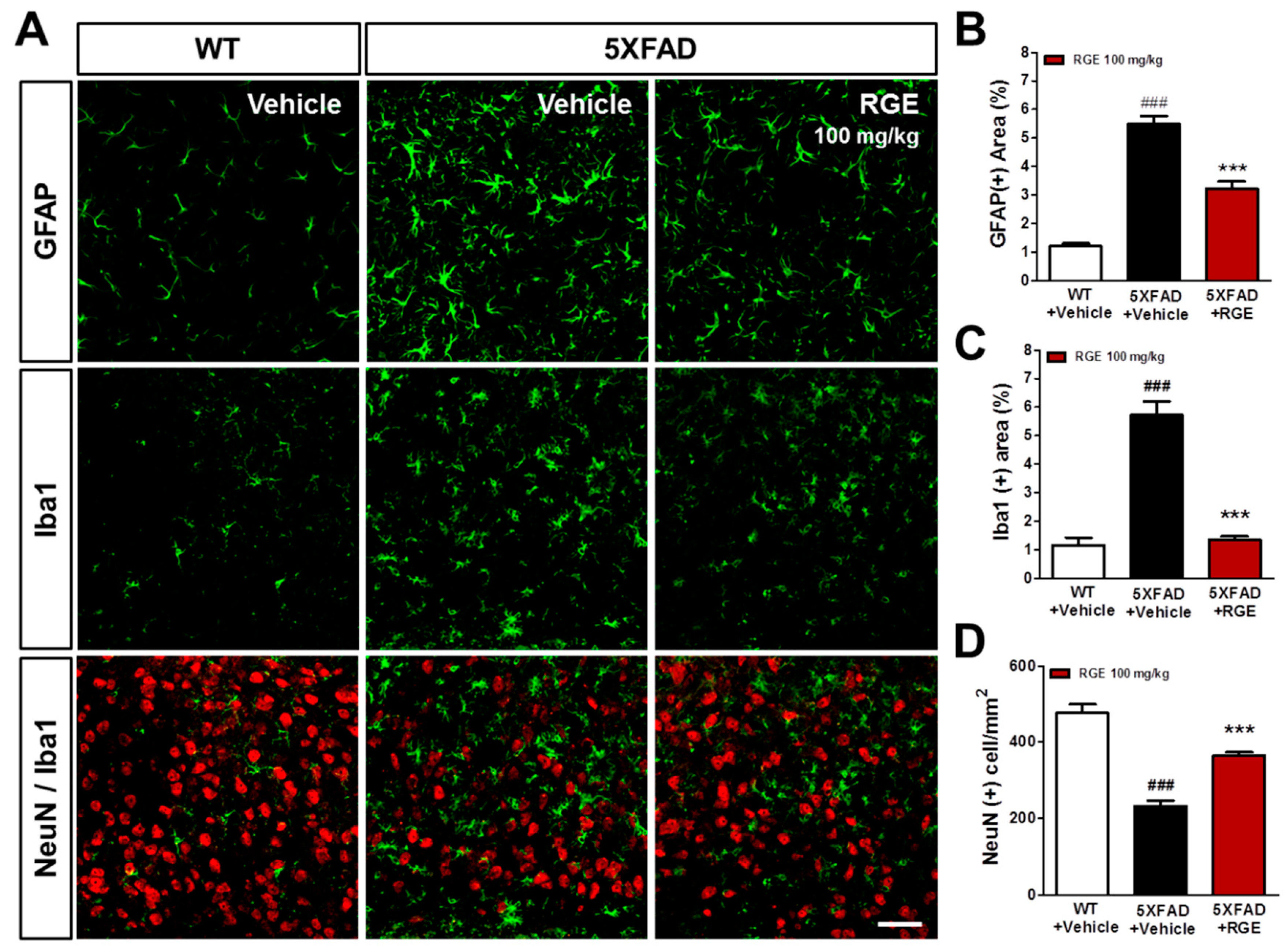
2.5. RGE Attenuates Neuroinflammation and Neuronal Death in the Subiculum of 5XFAD Mice
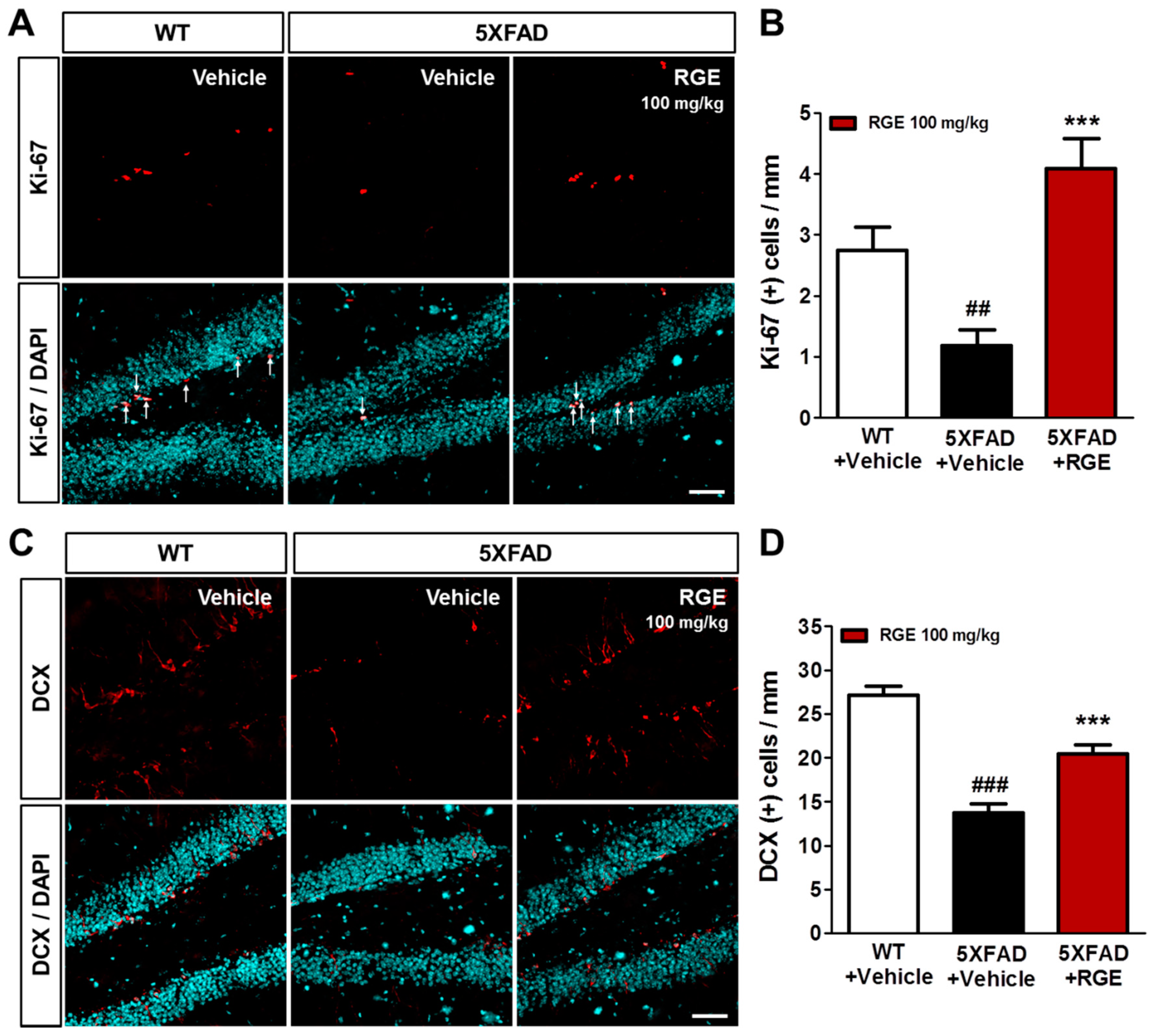
2.6. RGE Improves Impaired Hippocampal Adult Neurogenesis in 5XFAD Mice
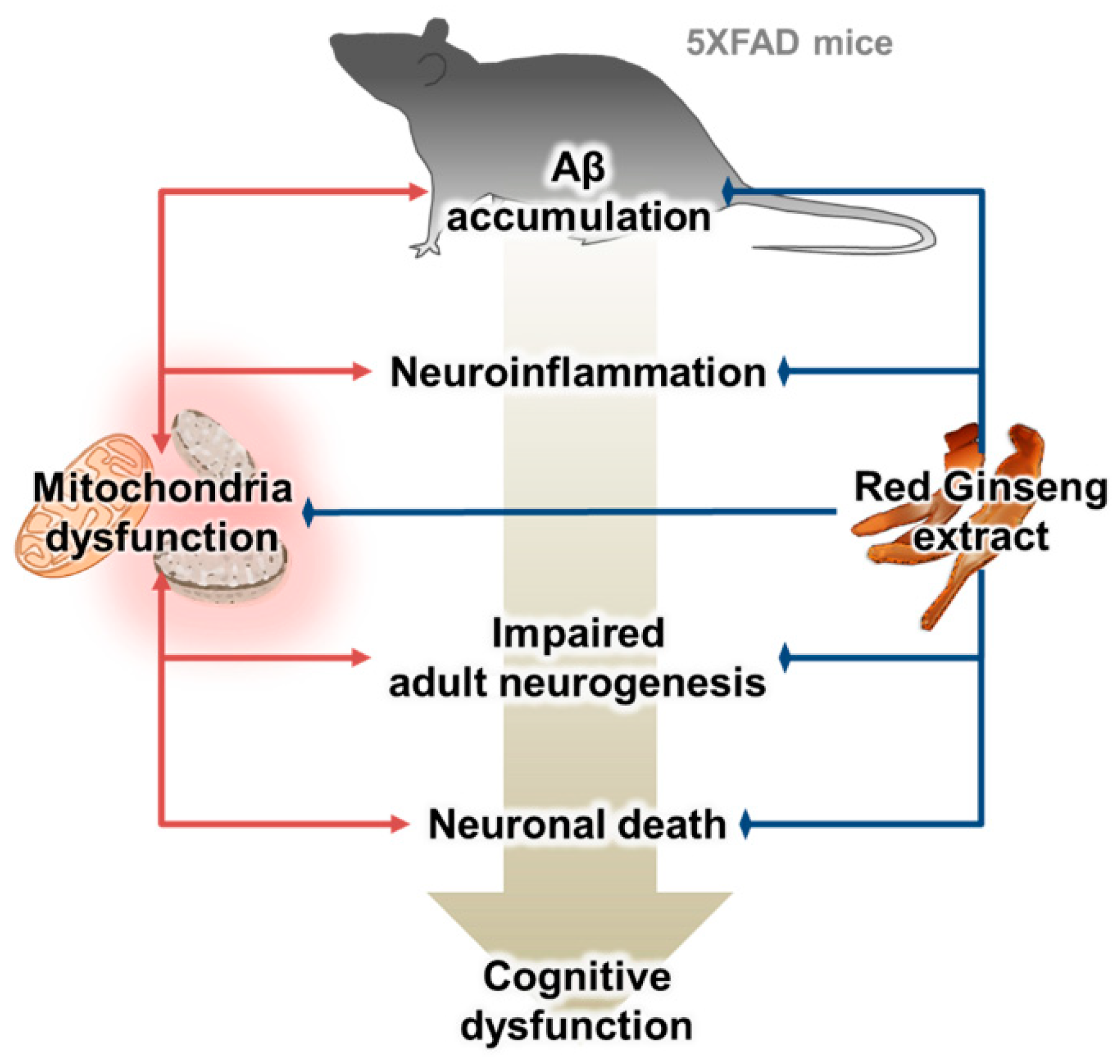
3. Discussion
4. Materials and Methods
4.1. Preparation and Characterization of RGE
4.2. Culture of HT22 Cell Line
4.3. Cell Viability Assay
4.4. Measurements of OCR
4.5. Animals and Administration
4.6. Preparation of Brain Tissue
4.7. Immunofluorescence Labeling
4.8. Image Acquisition and Analysis
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Aβ | amyloid beta |
| AChE | acetylcholinesterase |
| AD | Alzheimer’s disease |
| AFG | arginine-fructose-glucose |
| AP | acidic polysaccharide |
| APP | amyloid precursor protein |
| COX | cytochrome c oxidase |
| DAPI | 4′,6-diamidino-2-phenylindole |
| DCX | doublecortin |
| DG | dentate gyrus |
| DMEM | Dulbecco’s modified Eagle’s medium |
| GFAP | glial fibrillary acidic protein |
| Iba-1 | ionized calcium-binding adapter molecule 1 |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| NeuN | neuronal nuclei |
| OCR | oxygen consumption rate |
| PBS | phosphate-buffered saline |
| PD | Parkinson’s disease |
| PFA | paraformaldehyde |
| PG | Panax ginseng Meyer |
| RG | red ginseng |
References
- Winblad, B.; Amouyel, P.; Andrieu, S.; Ballard, C.; Brayne, C.; Brodaty, H.; Cedazo-Minguez, A.; Dubois, B.; Edvardsson, D.; Feldman, H.; et al. Defeating Alzheimer’s disease and other dementias: A priority for European science and society. Lancet Neurol. 2016, 15, 455–532. [Google Scholar] [CrossRef]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Moon, M.; Hong, H.S.; Nam, D.W.; Baik, S.H.; Song, H.; Kook, S.Y.; Kim, Y.S.; Lee, J.; Mook-Jung, I. Intracellular amyloid-beta accumulation in calcium-binding protein-deficient neurons leads to amyloid-beta plaque formation in animal model of Alzheimer’s disease. J. Alzheimer’s. Dis. 2012, 29, 615–628. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Alzheimer’s disease: A central role for amyloid. J. Neuropathol. Exp. Neurol. 1994, 53, 438–447. [Google Scholar] [CrossRef] [PubMed]
- Thinakaran, G.; Koo, E.H. Amyloid precursor protein trafficking, processing, and function. J. Biol. Chem. 2008, 283, 29615–29619. [Google Scholar] [CrossRef] [PubMed]
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef] [PubMed]
- McGeer, E.G.; McGeer, P.L. Neuroinflammation in Alzheimer’s disease and mild cognitive impairment: A field in its infancy. J. Alzheimer’s Dis. 2010, 19, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Mu, Y.; Gage, F.H. Adult hippocampal neurogenesis and its role in Alzheimer’s disease. Mol. Neurodegener. 2011, 6, 85. [Google Scholar] [CrossRef]
- Swerdlow, R.H. Mitochondria and Mitochondrial Cascades in Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 62, 1403–1416. [Google Scholar] [CrossRef]
- Byun, J.; Son, S.M.; Cha, M.Y.; Shong, M.; Hwang, Y.J.; Kim, Y.; Ryu, H.; Moon, M.; Kim, K.S.; Mook-Jung, I. CR6-interacting factor 1 is a key regulator in Abeta-induced mitochondrial disruption and pathogenesis of Alzheimer’s disease. Cell Death Differ. 2015, 22, 959–973. [Google Scholar] [CrossRef]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H.; Beal, M.F. Amyloid beta, mitochondrial dysfunction and synaptic damage: Implications for cognitive decline in aging and Alzheimer’s disease. Trends Mol. Med. 2008, 14, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H.; McWeeney, S.; Park, B.S.; Manczak, M.; Gutala, R.V.; Partovi, D.; Jung, Y.; Yau, V.; Searles, R.; Mori, M.; et al. Gene expression profiles of transcripts in amyloid precursor protein transgenic mice: Up-regulation of mitochondrial metabolism and apoptotic genes is an early cellular change in Alzheimer’s disease. Hum. Mol. Genet. 2004, 13, 1225–1240. [Google Scholar] [CrossRef] [PubMed]
- Sirk, D.; Zhu, Z.; Wadia, J.S.; Shulyakova, N.; Phan, N.; Fong, J.; Mills, L.R. Chronic exposure to sub-lethal beta-amyloid (Abeta) inhibits the import of nuclear-encoded proteins to mitochondria in differentiated PC12 cells. J. Neurochem. 2007, 103, 1989–2003. [Google Scholar] [CrossRef]
- Barsoum, M.J.; Yuan, H.; Gerencser, A.A.; Liot, G.; Kushnareva, Y.; Graber, S.; Kovacs, I.; Lee, W.D.; Waggoner, J.; Cui, J.; et al. Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. EMBO J. 2006, 25, 3900–3911. [Google Scholar] [CrossRef] [PubMed]
- Casley, C.S.; Canevari, L.; Land, J.M.; Clark, J.B.; Sharpe, M.A. Beta-amyloid inhibits integrated mitochondrial respiration and key enzyme activities. J. Neurochem. 2002, 80, 91–100. [Google Scholar] [CrossRef]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox. Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef] [PubMed]
- Pfanner, N.; Warscheid, B.; Wiedemann, N. Mitochondrial proteins: From biogenesis to functional networks. Nat. Rev. Mol. Cell Biol. 2019, 20, 267–284. [Google Scholar] [CrossRef]
- Jeon, S.G.; Song, E.J.; Lee, D.; Park, J.; Nam, Y.; Kim, J.I.; Moon, M. Traditional Oriental Medicines and Alzheimer’s Disease. Aging Dis. 2019, 10, 307–328. [Google Scholar] [CrossRef]
- Kim, K.H.; Lee, D.; Lee, H.L.; Kim, C.E.; Jung, K.; Kang, K.S. Beneficial effects of Panax ginseng for the treatment and prevention of neurodegenerative diseases: Past findings and future directions. J. Ginseng Res. 2018, 42, 239–247. [Google Scholar] [CrossRef]
- Yeo, H.B.; Yoon, H.K.; Lee, H.J.; Kang, S.G.; Jung, K.Y.; Kim, L. Effects of Korean Red Ginseng on Cognitive and Motor Function: A Double-blind, Randomized, Placebo-controlled Trial. J. Ginseng Res. 2012, 36, 190–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.; Oh, S. Administration of red ginseng ameliorates memory decline in aged mice. J. Ginseng Res. 2015, 39, 250–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, Y.M.; Nishijo, H.; Uwano, T.; Tamura, R.; Kawanishi, K.; Ono, T. Red ginseng ameliorated place navigation deficits in young rats with hippocampal lesions and aged rats. Physiol. Behav. 2000, 69, 511–525. [Google Scholar] [CrossRef]
- Epperly, T.; Dunay, M.A.; Boice, J.L. Alzheimer Disease: Pharmacologic and Nonpharmacologic Therapies for Cognitive and Functional Symptoms. Am. Fam. Physician 2017, 95, 771–778. [Google Scholar] [PubMed]
- Heo, J.H.; Lee, S.T.; Chu, K.; Oh, M.J.; Park, H.J.; Shim, J.Y.; Kim, M. An open-label trial of Korean red ginseng as an adjuvant treatment for cognitive impairment in patients with Alzheimer’s disease. Eur. J. Neurol. 2008, 15, 865–868. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.H.; Lee, S.T.; Oh, M.J.; Park, H.J.; Shim, J.Y.; Chu, K.; Kim, M. Improvement of cognitive deficit in Alzheimer’s disease patients by long term treatment with korean red ginseng. J. Ginseng Res. 2011, 35, 457–461. [Google Scholar] [CrossRef]
- Heo, J.H.; Park, M.H.; Lee, J.H. Effect of Korean Red Ginseng on Cognitive Function and Quantitative EEG in Patients with Alzheimer’s Disease: A Preliminary Study. J. Altern. Complement. Med. 2016, 22, 280–285. [Google Scholar] [CrossRef]
- Dong, G.Z.; Jang, E.J.; Kang, S.H.; Cho, I.J.; Park, S.D.; Kim, S.C.; Kim, Y.W. Red ginseng abrogates oxidative stress via mitochondria protection mediated by LKB1-AMPK pathway. BMC Complement. Altern. Med. 2013, 13, 64. [Google Scholar] [CrossRef]
- Ryu, S.; Jeon, H.; Koo, S.; Kim, S. Korean Red Ginseng Enhances Neurogenesis in the Subventricular Zone of 1-Methyl-4-Phenyl-1, 2, 3, 6-Tetrahydropyridine-Treated Mice. Front. Aging Neurosci. 2018, 10, 355. [Google Scholar] [CrossRef]
- Park, J.K.; Shim, J.Y.; Cho, A.R.; Cho, M.R.; Lee, Y.J. Korean Red Ginseng Protects Against Mitochondrial Damage and Intracellular Inflammation in an Animal Model of Type 2 Diabetes Mellitus. J. Med. Food 2018, 21, 544–550. [Google Scholar] [CrossRef]
- Kong, D.; Tian, X.; Li, Y.; Zhang, S.; Cheng, Y.; Huo, L.; Ma, H.; Yang, Z.; Ren, L.; Zhang, M.; et al. Revealing the Inhibitory Effect of Ginseng on Mitochondrial Respiration through Synaptosomal Proteomics. Proteomics 2018, 18, e1700354. [Google Scholar] [CrossRef] [PubMed]
- Burte, F.; Carelli, V.; Chinnery, P.F.; Yu-Wai-Man, P. Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat. Rev. Neurol. 2015, 11, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Nakamura, K.; Iijima, M.; Sesaki, H. Mitochondrial dynamics in neurodegeneration. Trends Cell Biol. 2013, 23, 64–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Grande, A.M.; Robinson, J.K.; Previti, M.L.; Vasek, M.; Davis, J.; Van Nostrand, W.E. Early-onset subicular microvascular amyloid and neuroinflammation correlate with behavioral deficits in vasculotropic mutant amyloid beta-protein precursor transgenic mice. Neuroscience 2007, 146, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Nagele, R.G.; Wegiel, J.; Venkataraman, V.; Imaki, H.; Wang, K.C.; Wegiel, J. Contribution of glial cells to the development of amyloid plaques in Alzheimer’s disease. Neurobiol. Aging 2004, 25, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L.; et al. Inflammation and Alzheimer’s disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar] [CrossRef]
- Regen, F.; Hellmann-Regen, J.; Costantini, E.; Reale, M. Neuroinflammation and Alzheimer’s Disease: Implications for Microglial Activation. Curr. Alzheimer Res. 2017, 14, 1140–1148. [Google Scholar] [CrossRef]
- Di Filippo, M.; Chiasserini, D.; Tozzi, A.; Picconi, B.; Calabresi, P. Mitochondria and the link between neuroinflammation and neurodegeneration. J. Alzheimer’s Dis. 2010, 20 (Suppl. 2), S369–S379. [Google Scholar] [CrossRef]
- Sahay, A.; Scobie, K.N.; Hill, A.S.; O’Carroll, C.M.; Kheirbek, M.A.; Burghardt, N.S.; Fenton, A.A.; Dranovsky, A.; Hen, R. Increasing adult hippocampal neurogenesis is sufficient to improve pattern separation. Nature 2011, 472, 466–470. [Google Scholar] [CrossRef] [Green Version]
- Deng, W.; Aimone, J.B.; Gage, F.H. New neurons and new memories: How does adult hippocampal neurogenesis affect learning and memory? Nat. Rev. Neurosci. 2010, 11, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Toda, T.; Gage, F.H. Review: Adult neurogenesis contributes to hippocampal plasticity. Cell Tissue Res. 2018, 373, 693–709. [Google Scholar] [CrossRef] [PubMed]
- Gage, F.H. Adult neurogenesis in mammals. Science 2019, 364, 827–828. [Google Scholar] [CrossRef] [PubMed]
- Moon, M.; Cha, M.Y.; Mook-Jung, I. Impaired hippocampal neurogenesis and its enhancement with ghrelin in 5XFAD mice. J. Alzheimer’s Dis. 2014, 41, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Jimenez, E.P.; Flor-Garcia, M.; Terreros-Roncal, J.; Rabano, A.; Cafini, F.; Pallas-Bazarra, N.; Avila, J.; Llorens-Martin, M. Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer’s disease. Nat. Med. 2019, 25, 554–560. [Google Scholar] [CrossRef] [PubMed]
- Khacho, M.; Harris, R.; Slack, R.S. Mitochondria as central regulators of neural stem cell fate and cognitive function. Nat. Rev. Neurosci. 2019, 20, 34–48. [Google Scholar] [CrossRef]
- Richetin, K.; Moulis, M.; Millet, A.; Arrazola, M.S.; Andraini, T.; Hua, J.; Davezac, N.; Roybon, L.; Belenguer, P.; Miquel, M.C.; et al. Amplifying mitochondrial function rescues adult neurogenesis in a mouse model of Alzheimer’s disease. Neurobiol. Dis. 2017, 102, 113–124. [Google Scholar] [CrossRef]
- Jin, K.; Peel, A.L.; Mao, X.O.; Xie, L.; Cottrell, B.A.; Henshall, D.C.; Greenberg, D.A. Increased hippocampal neurogenesis in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 343–347. [Google Scholar] [CrossRef]
- Lee, S.T.; Chu, K.; Sim, J.Y.; Heo, J.H.; Kim, M. Panax ginseng enhances cognitive performance in Alzheimer disease. Alzheimer Dis. Assoc. Disord. 2008, 22, 222–226. [Google Scholar] [CrossRef]
- Pagani, L.; Eckert, A. Amyloid-Beta interaction with mitochondria. Int. J. Alzheimer’s Dis. 2011, 2011, 925050. [Google Scholar] [CrossRef]
- Sorrentino, V.; Romani, M.; Mouchiroud, L.; Beck, J.S.; Zhang, H.; D’Amico, D.; Moullan, N.; Potenza, F.; Schmid, A.W.; Rietsch, S.; et al. Enhancing mitochondrial proteostasis reduces amyloid-beta proteotoxicity. Nature 2017, 552, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Chan, D.C. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat. Rev. Mol. Cell. Biol. 2014, 15, 634–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Su, B.; Lee, H.G.; Li, X.; Perry, G.; Smith, M.A.; Zhu, X. Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J. Neurosci. 2009, 29, 9090–9103. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Jung, S.W.; Kim, S.Y.; Cho, I.H.; Kim, H.C.; Rhim, H.; Kim, M.; Nah, S.Y. Panax ginseng as an adjuvant treatment for Alzheimer’s disease. J. Ginseng Res. 2018, 42, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Kwan, K.K.L.; Leung, K.W.; Yao, P.; Wang, H.; Dong, T.T.; Tsim, K.W.K. Ginseng extracts modulate mitochondrial bioenergetics of live cardiomyoblasts: A functional comparison of different extraction solvents. J. Ginseng Res. 2018, in press. [Google Scholar] [CrossRef]
- Nah, S.Y. Ginseng ginsenoside pharmacology in the nervous system: Involvement in the regulation of ion channels and receptors. Front. Physiol. 2014, 5, 98. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.H.; Shin, T.J.; Choi, S.H.; Cho, H.J.; Lee, B.H.; Pyo, M.K.; Lee, J.H.; Kang, J.; Kim, H.J.; Park, C.W.; et al. Gintonin, newly identified compounds from ginseng, is novel lysophosphatidic acids-protein complexes and activates G protein-coupled lysophosphatidic acid receptors with high affinity. Mol. Cells. 2012, 33, 151–162. [Google Scholar] [CrossRef] [Green Version]
- Nah, S.Y.; Kim, D.H.; Rhim, H. Ginsenosides: Are any of them candidates for drugs acting on the central nervous system? CNS Drug Rev. 2007, 13, 381–404. [Google Scholar] [CrossRef]
- Huang, T.; Fang, F.; Chen, L.; Zhu, Y.; Zhang, J.; Chen, X.; Yan, S.S. Ginsenoside Rg1 attenuates oligomeric Abeta (1–42)-induced mitochondrial dysfunction. Curr. Alzheimer Res. 2012, 9, 388–395. [Google Scholar] [CrossRef]
- Zhou, J.S.; Wang, J.F.; He, B.R.; Cui, Y.S.; Fang, X.Y.; Ni, J.L.; Chen, J.; Wang, K.Z. Ginsenoside Rd attenuates mitochondrial permeability transition and cytochrome C release in isolated spinal cord mitochondria: Involvement of kinase-mediated pathways. Int. J. Mol. Sci. 2014, 15, 9859–9877. [Google Scholar] [CrossRef]
- Sun, M.; Huang, C.; Wang, C.; Zheng, J.; Zhang, P.; Xu, Y.; Chen, H.; Shen, W. Ginsenoside Rg3 improves cardiac mitochondrial population quality: Mimetic exercise training. Biochem. Biophys. Res. Commun. 2013, 441, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Hong, Y.; Tran, Q.; Cho, H.; Kim, M.; Kim, C.; Kwon, S.H.; Park, S.; Park, J.; Park, J. A new role for the ginsenoside RG3 in antiaging via mitochondria function in ultraviolet-irradiated human dermal fibroblasts. J. Ginseng Res. 2018, in press. [Google Scholar] [CrossRef]
- Li, X.T.; Chen, R.; Jin, L.M.; Chen, H.Y. Regulation on energy metabolism and protection on mitochondria of Panax ginseng polysaccharide. Am. J. Chin. Med. 2009, 37, 1139–1152. [Google Scholar] [CrossRef]
- Yan, X.; Hu, G.; Yan, W.; Chen, T.; Yang, F.; Zhang, X.; Zhao, G.; Liu, J. Ginsenoside Rd promotes non-amyloidogenic pathway of amyloid precursor protein processing by regulating phosphorylation of estrogen receptor alpha. Life Sci. 2017, 168, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Choi, R.J.; Roy, A.; Jung, H.J.; Ali, M.Y.; Min, B.S.; Park, C.H.; Yokozawa, T.; Fan, T.P.; Choi, J.S.; Jung, H.A. BACE1 molecular docking and anti-Alzheimer’s disease activities of ginsenosides. J. Ethnopharmacol. 2016, 190, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.; Chen, X.; Huang, T.; Lue, L.F.; Luddy, J.S.; Yan, S.S. Multi-faced neuroprotective effects of Ginsenoside Rg1 in an Alzheimer mouse model. Biochim. Biophys. Acta 2012, 1822, 286–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, G.; Su, P.; Zhang, S.; Guo, L.; Zhang, H.; Liang, Y.; Qin, C.; Zhang, W. Ginsenoside Re reduces Abeta production by activating PPARgamma to inhibit BACE1 in N2a/APP695 cells. Eur. J. Pharm. 2016, 793, 101–108. [Google Scholar] [CrossRef]
- Hwang, S.H.; Shin, E.J.; Shin, T.J.; Lee, B.H.; Choi, S.H.; Kang, J.; Kim, H.J.; Kwon, S.H.; Jang, C.G.; Lee, J.H.; et al. Gintonin, a ginseng-derived lysophosphatidic acid receptor ligand, attenuates Alzheimer’s disease-related neuropathies: Involvement of non-amyloidogenic processing. J. Alzheimer’s Dis. 2012, 31, 207–223. [Google Scholar] [CrossRef]
- Sun, S.; Qi, L.W.; Du, G.J.; Mehendale, S.R.; Wang, C.Z.; Yuan, C.S. Red notoginseng: Higher ginsenoside content and stronger anticancer potential than Asian and American ginseng. Food Chem. 2011, 125, 1299–1305. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Liu, J.; Yan, X.; Qin, K.; Shi, M.; Lin, T.; Zhu, Y.; Kang, T.; Zhao, G. Protective effects of ginsenoside Rd against okadaic acid-induced neurotoxicity in vivo and in vitro. J. Ethnopharmacol. 2011, 138, 135–141. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, J.; Zhang, Z.; Bi, P.; Qi, Z.; Zhang, C. Anti-neuroinflammation effect of ginsenoside Rbl in a rat model of Alzheimer disease. Neurosci. Lett. 2011, 487, 70–72. [Google Scholar] [CrossRef]
- Wilkins, H.M.; Swerdlow, R.H. Relationships between Mitochondria and Neuroinflammation: Implications for Alzheimer’s Disease. Curr. Top. Med. Chem. 2016, 16, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.W.; Zhang, X.; Huang, W.J. Role of neuroinflammation in neurodegenerative diseases (Review). Mol. Med. Rep. 2016, 13, 3391–3396. [Google Scholar] [CrossRef] [Green Version]
- Kempermann, G.; Wiskott, L.; Gage, F.H. Functional significance of adult neurogenesis. Curr. Opin. Neurobiol. 2004, 14, 186–191. [Google Scholar] [CrossRef]
- Lazarov, O.; Mattson, M.P.; Peterson, D.A.; Pimplikar, S.W.; van Praag, H. When neurogenesis encounters aging and disease. Trends Neurosci. 2010, 33, 569–579. [Google Scholar] [CrossRef] [Green Version]
- Devi, L.; Ohno, M. Genetic reductions of beta-site amyloid precursor protein-cleaving enzyme 1 and amyloid-beta ameliorate impairment of conditioned taste aversion memory in 5XFAD Alzheimer’s disease model mice. Eur. J. Neurosci. 2010, 31, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.L.; Morstorf, T.; Zhong, K. Alzheimer’s disease drug-development pipeline: Few candidates, frequent failures. Alzheimer’s Res. 2014, 6, 37. [Google Scholar] [CrossRef] [PubMed]
- Nordberg, A.; Svensson, A.L. Cholinesterase inhibitors in the treatment of Alzheimer’s disease: A comparison of tolerability and pharmacology. Drug Saf. 1998, 19, 465–480. [Google Scholar] [CrossRef]
- Hogan, D.B. Long-term efficacy and toxicity of cholinesterase inhibitors in the treatment of Alzheimer disease. Can. J. Psychiatry 2014, 59, 618–623. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.N.; Yan, Y.H.; Zhu, T.L.; Ma, B.K.; Fan, H.R.; Liu, Y.M.; Li, W.G.; Li, F. Long-Term NMDAR Antagonism Correlates Weight Loss With Less Eating. Front. Psychiatry 2019, 10, 15. [Google Scholar] [CrossRef]
- Galimberti, D.; Scarpini, E. Disease-modifying treatments for Alzheimer’s disease. Adv. Neurol. Disord. 2011, 4, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Shi, J.; Zhang, X.; Wang, Y. Herbal therapy: A new pathway for the treatment of Alzheimer’s disease. Alzheimer’s Res. 2010, 2, 30. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.T.; Zheng, X.W.; Chen, S.; Shan, C.S.; Xu, Q.Q.; Zhu, J.Z.; Bao, X.Y.; Lin, Y.; Zheng, G.Q.; Wang, Y. Chinese herbal medicine for Alzheimer’s disease: Clinical evidence and possible mechanism of neurogenesis. Biochem. Pharm. 2017, 141, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Shen, L.H.; Zhang, J.T. Anti-amnestic and anti-aging effects of ginsenoside Rg1 and Rb1 and its mechanism of action. Acta Pharm. Sin. 2005, 26, 143–149. [Google Scholar] [CrossRef]
- Choi, K.T. Botanical characteristics, pharmacological effects and medicinal components of Korean Panax ginseng C A Meyer. Acta Pharm. Sin. 2008, 29, 1109–1118. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Mo, E.J.; Choi, J.E.; Jo, Y.H.; Jang, H.; Jeong, J.Y.; Jin, Q.; Chung, H.N.; Hwang, B.Y.; Lee, M.K. Effect of Korean Red Ginseng extraction conditions on antioxidant activity, extraction yield, and ginsenoside Rg1 and phenolic content: Optimization using response surface methodology. J. Ginseng Res. 2016, 40, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Rasoanaivo, P.; Wright, C.W.; Willcox, M.L.; Gilbert, B. Whole plant extracts versus single compounds for the treatment of malaria: Synergy and positive interactions. Malar. J. 2011, 10 (Suppl. 1), S4. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Malinovska, L.; Saha, S.; Wang, J.; Alberti, S.; Krishnan, Y.; Hyman, A.A. ATP as a biological hydrotrope. Science 2017, 356, 753–756. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, S.M.; Santana, I.; Swerdlow, R.H.; Oliveira, C.R. Mitochondria dysfunction of Alzheimer’s disease cybrids enhances Abeta toxicity. J. Neurochem. 2004, 89, 1417–1426. [Google Scholar] [CrossRef] [PubMed]
- Naval, M.V.; Gomez-Serranillos, M.P.; Carretero, M.E.; Villar, A.M. Neuroprotective effect of a ginseng (Panax ginseng) root extract on astrocytes primary culture. J. Ethnopharmacol. 2007, 112, 262–270. [Google Scholar] [CrossRef]
- Seo, J.Y.; Ju, S.H.; Oh, J.; Lee, S.K.; Kim, J.S. Neuroprotective and Cognition-Enhancing Effects of Compound K Isolated from Red Ginseng. J. Agric. Food Chem. 2016, 64, 2855–2864. [Google Scholar] [CrossRef] [PubMed]
- In, G.; Ahn, N.G.; Bae, B.S.; Lee, M.W.; Park, H.W.; Jang, K.H.; Cho, B.G.; Han, C.K.; Park, C.K.; Kwak, Y.S. In situ analysis of chemical components induced by steaming between fresh ginseng, steamed ginseng, and red ginseng. J. Ginseng Res. 2017, 41, 361–369. [Google Scholar] [CrossRef]
- Do, J.; Lee, H.; Lee, S.; Jang, J.; Lee, S.; Sung, H. Colorimetric determination of acidic polysaccharide from Panax ginseng, its extraction condition and stability. Korean J. Ginseng Sci. 1993, 17, 139–144. [Google Scholar]
- Gui, Y.; Ryu, G.H. Effects of extrusion cooking on physicochemical properties of white and red ginseng (powder). J. Ginseng Res. 2014, 38, 146–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.W.; In, G.; Han, S.T.; Lee, M.W.; Kim, S.Y.; Kim, K.T.; Cho, B.G.; Han, G.H.; Chang, I.M. Simultaneous determination of 30 ginsenosides in Panax ginseng preparations using ultra performance liquid chromatography. J. Ginseng Res. 2013, 37, 457–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.G.; Kim, N.; Huh, E.; Lee, H.; Oh, M.H.; Park, J.D.; Pyo, M.K.; Oh, M.S. White Ginseng Protects Mouse Hippocampal Cells against Amyloid-Beta Oligomer Toxicity. Phytother. Res. 2017, 31, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Min, D.S.; Lee, C.W.; Song, K.H.; Kim, Y.S.; Kim, H.P. Ginsenosides from Korean Red Ginseng ameliorate lung inflammatory responses: Inhibition of the MAPKs/NF-kappaB/c-Fos pathways. J. Ginseng Res. 2018, 42, 476–484. [Google Scholar] [CrossRef]
- Park, H.-J.; Han, J.-M.; Kim, H.-G.; Choi, M.-K.; Lee, J.-S.; Son, C.-G. Anti-Myelosuppression Effects of Korean Red Ginseng in SD Rat Injected with 5-fluorouracil. J. Korean Med. 2012, 33, 47–55. [Google Scholar] [CrossRef]
- Paxinos, G.; Keith, B.J.; Franklin, M. Paxinos and Franklin’s the Mouse Brain in Stereotaxic Coordinates, 4th ed.; Elsevier Science: Amsterdam, The Netherlands, 2012. [Google Scholar]
- Kim, J.; Kim, S.H.; Lee, D.S.; Lee, D.J.; Chung, S.; Yang, H.O. Effects of fermented ginseng on memory impairment and beta-amyloid reduction in Alzheimer’s disease experimental models. J. Ginseng Res. 2013, 37, 100–107. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ginsenoside (mg/g) | AFG (mg/g) | AP (mg/g) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Rb1 | Rb2 | Rc | Rd | Re | Rf | Rg1 | Rg2s | Rg3r | Rg3s | Rh1 | ||
| 6.23 | 2.45 | 2.94 | 1.27 | 0.93 | 1.37 | 0.64 | 1.78 | 1.77 | 3.50 | 1.68 | 5.58 | 98.46 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shin, S.J.; Jeon, S.G.; Kim, J.-i.; Jeong, Y.-o.; Kim, S.; Park, Y.H.; Lee, S.-K.; Park, H.H.; Hong, S.B.; Oh, S.; et al. Red Ginseng Attenuates Aβ-Induced Mitochondrial Dysfunction and Aβ-mediated Pathology in an Animal Model of Alzheimer’s Disease. Int. J. Mol. Sci. 2019, 20, 3030. https://doi.org/10.3390/ijms20123030
Shin SJ, Jeon SG, Kim J-i, Jeong Y-o, Kim S, Park YH, Lee S-K, Park HH, Hong SB, Oh S, et al. Red Ginseng Attenuates Aβ-Induced Mitochondrial Dysfunction and Aβ-mediated Pathology in an Animal Model of Alzheimer’s Disease. International Journal of Molecular Sciences. 2019; 20(12):3030. https://doi.org/10.3390/ijms20123030
Chicago/Turabian StyleShin, Soo Jung, Seong Gak Jeon, Jin-il Kim, Yu-on Jeong, Sujin Kim, Yong Ho Park, Seong-Kyung Lee, Hyun Ha Park, Sang Bum Hong, Sua Oh, and et al. 2019. "Red Ginseng Attenuates Aβ-Induced Mitochondrial Dysfunction and Aβ-mediated Pathology in an Animal Model of Alzheimer’s Disease" International Journal of Molecular Sciences 20, no. 12: 3030. https://doi.org/10.3390/ijms20123030
APA StyleShin, S. J., Jeon, S. G., Kim, J. -i., Jeong, Y. -o., Kim, S., Park, Y. H., Lee, S. -K., Park, H. H., Hong, S. B., Oh, S., Hwang, J. -y., Kim, H. s., Park, H., Nam, Y., Lee, Y. Y., Kim, J. -J., Park, S. -H., Kim, J. -S., & Moon, M. (2019). Red Ginseng Attenuates Aβ-Induced Mitochondrial Dysfunction and Aβ-mediated Pathology in an Animal Model of Alzheimer’s Disease. International Journal of Molecular Sciences, 20(12), 3030. https://doi.org/10.3390/ijms20123030