Glucosylsphingosine (lyso-Gb1) as a Biomarker for Monitoring Treated and Untreated Children with Gaucher Disease


 and
and 
Abstract
:1. Introduction
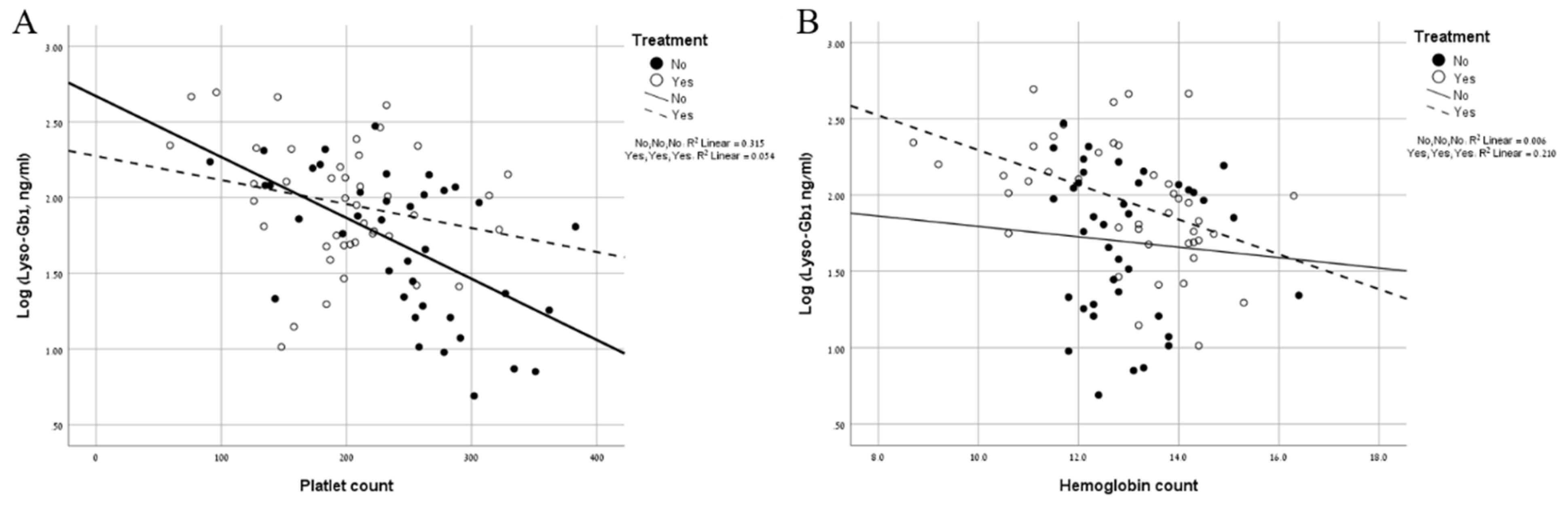
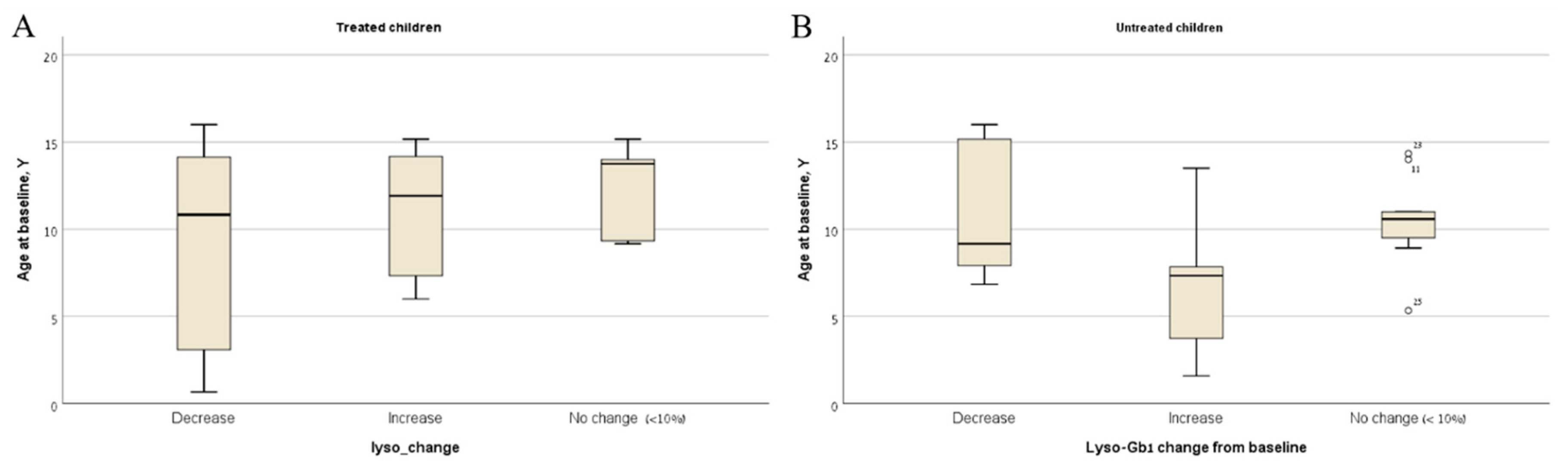
2. Results
3. Discussion
4. Material and Methods
4.1. Patients Samples
4.2. Statistical Methods
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Revel-Vilk, S.; Szer, J.; Mehta, A.; Zimran, A. How we manage Gaucher Disease in the era of choices. Br. J. Haematol. 2018, 182, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Gary, S.E.; Ryan, E.; Steward, A.M.; Sidransky, E. Recent advances in the diagnosis and management of Gaucher disease. Expert Rev. Endocrinol. Metab. 2018, 13, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Zimran, A.; Elstein, D. Chapter 72: Gaucher disease and related Lysosomal Storage Diseases. In Williams’ Hematology; Kaushansky, K., Lichtman, M.A., Prchal, J.T., Levi, M.M., Press, O.W., Burns, L.J., Caligiuri, M.L., Eds.; McGraw-Hill: New York, NY, USA, 2016. [Google Scholar]
- Barton, N.W.; Brady, R.O.; Dambrosia, J.M.; Di Bisceglie, A.M.; Doppelt, S.H.; Hill, S.C.; Mankin, H.J.; Murray, G.J.; Parker, R.I.; Argoff, C.E.; et al. Replacement therapy for inherited enzyme deficiency--macrophage-targeted glucocerebrosidase for Gaucher's disease. N. Engl. J. Med. 1991, 324, 1464–1470. [Google Scholar] [CrossRef] [PubMed]
- Andersson, H.; Kaplan, P.; Kacena, K.; Yee, J. Eight-year clinical outcomes of long-term enzyme replacement therapy for 884 children with Gaucher disease type 1. Pediatrics 2008, 122, 1182–1190. [Google Scholar] [CrossRef] [PubMed]
- Elstein, D.; Altarescu, G.; Abrahamov, A.; Zimran, A. Children with type 1 Gaucher disease: Changing profiles in the 21st century. Blood Cells Mol. Dis. 2018, 68, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.C.; Bier, L.; Overbey, J.R.; Cohen-Pfeffer, J.; Desai, K.; Desnick, R.J.; Balwani, M. Early manifestations of type 1 Gaucher disease in presymptomatic children diagnosed after parental carrier screening. Genet. Med. 2017, 19, 652–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rolfs, A.; Giese, A.K.; Grittner, U.; Mascher, D.; Elstein, D.; Zimran, A.; Bottcher, T.; Lukas, J.; Hubner, R.; Golnitz, U.; et al. Glucosylsphingosine is a highly sensitive and specific biomarker for primary diagnostic and follow-up monitoring in Gaucher disease in a non-Jewish, Caucasian cohort of Gaucher disease patients. PLoS ONE 2013, 8, e79732. [Google Scholar] [CrossRef] [PubMed]
- Elstein, D.; Mellgard, B.; Dinh, Q.; Lan, L.; Qiu, Y.; Cozma, C.; Eichler, S.; Bottcher, T.; Zimran, A. Reductions in glucosylsphingosine (lyso-Gb1) in treatment-naive and previously treated patients receiving velaglucerase alfa for type 1 Gaucher disease: Data from phase 3 clinical trials. Mol. Genet. Metab. 2017, 122, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Murugesan, V.; Chuang, W.L.; Liu, J.; Lischuk, A.; Kacena, K.; Lin, H.; Pastores, G.M.; Yang, R.; Keutzer, J.; Zhang, K.; et al. Glucosylsphingosine is a key biomarker of Gaucher disease. Am. J. Hematol. 2016, 91, 1082–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidson, B.A.; Hassan, S.; Garcia, E.J.; Tayebi, N.; Sidransky, E. Exploring genetic modifiers of Gaucher disease: The next horizon. Hum. Mutat. 2018, 39, 1739–1751. [Google Scholar] [CrossRef] [PubMed]
- Dekker, N.; van Dussen, L.; Hollak, C.E.; Overkleeft, H.; Scheij, S.; Ghauharali, K.; van Breemen, M.J.; Ferraz, M.J.; Groener, J.E.; Maas, M.; et al. Elevated plasma glucosylsphingosine in Gaucher disease: Relation to phenotype, storage cell markers, and therapeutic response. Blood 2011, 118, e118–e127. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, G.A.; Kacena, K.; Cole, J.A.; Hollak, C.E.; Zhang, L.; Yee, J.; Mistry, P.K.; Zimran, A.; Charrow, J.; vom Dahl, S. Dose-response relationships for enzyme replacement therapy with imiglucerase/alglucerase in patients with Gaucher disease type 1. Genet. Med. 2009, 11, 92–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arkadir, D.; Dinur, T.; Revel-Vilk, S.; Becker Cohen, M.; Cozma, C.; Hovakimyan, M.; Eichler, S.; Rolfs, A.; Zimran, A. Glucosylsphingosine is a reliable response biomarker in Gaucher disease. Am. J. Hematol. 2018, 93, E140–E142. [Google Scholar] [CrossRef] [PubMed]
- Mistry, P.K.; Lukina, E.; Ben Turkia, H.; Shankar, S.P.; Baris, H.; Ghosn, M.; Mehta, A.; Packman, S.; Pastores, G.; Petakov, M.; et al. Outcomes after 18 months of eliglustat therapy in treatment-naive adults with Gaucher disease type 1: The phase 3 ENGAGE trial. Am. J. Hematol. 2017, 92, 1170–1176. [Google Scholar] [CrossRef] [PubMed]
- Brautbar, A.; Abrahamov, A.; Hadas-Halpern, I.; Elstein, D.; Zimran, A. Gaucher disease in Arab patients at an Israeli referral clinic. Isr. Med. Assoc. J. 2008, 10, 600–602. [Google Scholar] [PubMed]
- Abrahamov, A.; Elstein, D.; Gross-Tsur, V.; Farber, B.; Glaser, Y.; Hadas-Halpern, I.; Ronen, S.; Tafakjdi, M.; Horowitz, M.; Zimran, A. Gaucher’s disease variant characterised by progressive calcification of heart valves and unique genotype. Lancet 1995, 346, 1000–1003. [Google Scholar] [CrossRef]
- Mistry, P.K. Genotype/phenotype correlations in Gaucher’s disease. Lancet 1995, 346, 982–983. [Google Scholar] [CrossRef]
- Zimran, A.; Gross, E.; West, C.; Sorge, J.; Kubitz, M.; Beutler, E. Prediction of severity of Gaucher’s disease by identification of mutations at DNA level. Lancet 1989, 2, 349–352. [Google Scholar] [CrossRef]
- Yetter, E.M.; Acosta, K.B.; Olson, M.C.; Blundell, K. Estimating Splenic Volume: Sonographic Measurements Correlated with Helical CT Determination. Am. J. Roentgenol. 2003, 181, 1615–1620. [Google Scholar] [CrossRef] [PubMed]
- Elstein, D.; Hadas-Halpern, I.; Azuri, Y.; Abrahamov, A.; Bar-Ziv, Y.; Zimran, A. Accuracy of ultrasonography in assessing spleen and liver size in patients with Gaucher disease: Comparison to computed tomographic measurements. J. Ultrasound Med. 1997, 16, 209–211. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Total | Mild Type 1 | Severe Type 1 | Type 3 | |
|---|---|---|---|---|
| N | 81 | 35 | 34 | 12 |
| Age, years* | 11 (1–18) | 11 (4–18) | 12 (2–16) | 9.5 (1–18) |
| Male, % | 38 (47%) | 16 (38%) | 18 (52%) | 4 (33%) |
| ERT* | 42 (51%) | 2 (5.7%) | 30 (88%) | 10 (83%) |
| Platelet count, ×103/mL | 214 (59–383) | 251 (134–383) | 209 (76–334) | 190 (59–322) |
| Hemoglobin, mg/dl | 12.9 (8.7–16.4) | 12.8(11.5–16.4) | 13.15 (9.2–15.3) | 12.4 (8.7–16.3) |
| Spleen (MN)* | 1.3 (0–16.7) | 1.3 (0.5–4.7) | 1.3 (0.6–10.3) | 3.4 (1.2–16.7) |
| Liver (MN)* | 1.4 (0.3–3.6) | 1.4 (1–2.3) | 1.3 (0.3–2.8) | 2.3 (1.3–3.5) |
| Lyso-Gb1 level, ng/mL* | 76.3 (4.9–495) | 64 (4.9–208) | 98 (7.3–495) | 100.4 (21.4–210) |
| Weight, Kg* | 37.8 (10.1–76) | 34.1 (14.7–70.9) | 49 (15.4–76) | 29.8 (10.1–52.2) |
| Pre-Treatment | Un-Treated | |
|---|---|---|
| N | 10 | 28 |
| Age, years | 5.5 (2–14) | 8.5 (1–16) |
| Male, % | 5 (55%) | 11 (37%) |
| Platelet count, ×103/mL | 82.5 (68–228) | 236.5 (117–339) |
| Hemoglobin, mg/dl | 11.1 (6.7–12.4) | 12.7 (11.1–15.7) |
| Spleen (MN)* | 3.9 (1.1–22.9) | 1.35 (0.5–5.2) |
| Liver (MN)* | 2.4 (1.2–4.5) | 1.7 (1–3) |
| Lyso-Gb1 level, ng/mL* | 262.5 (101–1270) | 61.45 (6.1–157) |
| Untreated | Treated, Pretreatment Baseline | ||
|---|---|---|---|
| Yes | No | ||
| N | 28 | 10 | 30 |
| Male | 11 | 5 | 16 |
| Age, years* | 12 (4–18) | 8.5 (3–18) | 16 (3–19) |
| Months of follow-up* | 31.85 ( 6.7–45) | 27.6 (6.7–44) | 28.75 (9.3–49.9) |
| Number of visits | 3 (2–6) | 4 (3-6) | 4 (2–9) |
| Unchanged** (n) | 9 (32%) | 1 (10%) | 5 (16%) |
| Increased (n) | 13 (46%) | 0 (0%) | 8 (26%) |
| Increase change* | 12 (1.29–128) | 67.4 (5.7–368) | |
| Decreased (n) | 6 (21%) | 9 (90%) | 17 (56%) |
| Decrease change* | 11.2 (4–50.4) | 143.6 (13–1207.7) | 32.7 (4.2–172) |
| Age (Y)* | Gender | Genotype | Mo. on Tx* | Dosa u/kg/mo* | Follow Up (mo) | Baseline Lyso- Gb1 | Change from Baseline | Possible Explanation | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Lyso-Gb1 | PLT | Hb | Spleen MN | Liver MN | ||||||||
| 9 | male | Severe GD1 | 72 | 36.7 | 140 | 79↑ | 9↓ | 0.5≈ | 0.5≈ | 0.4≈ | ||
| 9 | male | Severe GD1 | 81.2 | 52 | 23.6 | 168 | 18↑ | 31↓ | 0.4≈ | Weight gain** | ||
| 18 | male | Severe GD1 | 92.3 | 42 | 18.6 | 95 | 368↑ | 24↓ | 1.1↓ | 1.8↑ | 0.3≈ | Compliance |
| 18 | female | Severe GD1 | 130.9 | 35 | 36.9 | 281 | 180↑ | 18↓ | 0.5≈ | 0.6↓ | 0.1≈ | |
| 10 | male | Severe GD1 | 90.3 | 42 | 40.0 | 164 | 48↑ | 56↓ | 1.1↑ | 1.4↓ | 0.2≈ | |
| 14 | male | Severe GD1 | 113.7 | 114 | 38.3 | 45 | 6↑ | 4≈ | 1.3↓ | 6.4↑ | 1.2↓ | Weight gain** |
| 16 | male | Severe GD1 | 137.0 | 50 | 30.2 | 124 | 77↑ | 20↑ | 2.5↓ | 0.6↓ | 0.3≈ | Weight gain** |
| 17 | male | GD3 | 124.8 | 60 | 18.6 | 32 | 13↑ | 20↓ | 1.3↑ | 0.5↑ | 0.8↑ | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hurvitz, N.; Dinur, T.; Becker-Cohen, M.; Cozma, C.; Hovakimyan, M.; Oppermann, S.; Demuth, L.; Rolfs, A.; Abramov, A.; Zimran, A.; et al. Glucosylsphingosine (lyso-Gb1) as a Biomarker for Monitoring Treated and Untreated Children with Gaucher Disease. Int. J. Mol. Sci. 2019, 20, 3033. https://doi.org/10.3390/ijms20123033
Hurvitz N, Dinur T, Becker-Cohen M, Cozma C, Hovakimyan M, Oppermann S, Demuth L, Rolfs A, Abramov A, Zimran A, et al. Glucosylsphingosine (lyso-Gb1) as a Biomarker for Monitoring Treated and Untreated Children with Gaucher Disease. International Journal of Molecular Sciences. 2019; 20(12):3033. https://doi.org/10.3390/ijms20123033
Chicago/Turabian StyleHurvitz, Noa, Tama Dinur, Michal Becker-Cohen, Claudia Cozma, Marina Hovakimyan, Sebastian Oppermann, Laura Demuth, Arndt Rolfs, Aya Abramov, Ari Zimran, and et al. 2019. "Glucosylsphingosine (lyso-Gb1) as a Biomarker for Monitoring Treated and Untreated Children with Gaucher Disease" International Journal of Molecular Sciences 20, no. 12: 3033. https://doi.org/10.3390/ijms20123033
APA StyleHurvitz, N., Dinur, T., Becker-Cohen, M., Cozma, C., Hovakimyan, M., Oppermann, S., Demuth, L., Rolfs, A., Abramov, A., Zimran, A., & Revel-Vilk, S. (2019). Glucosylsphingosine (lyso-Gb1) as a Biomarker for Monitoring Treated and Untreated Children with Gaucher Disease. International Journal of Molecular Sciences, 20(12), 3033. https://doi.org/10.3390/ijms20123033