β-Glucose-1,6-Bisphosphate Stabilizes Pathological Phophomannomutase2 Mutants In Vitro and Represents a Lead Compound to Develop Pharmacological Chaperones for the Most Common Disorder of Glycosylation, PMM2-CDG

 ,
, 
 and
and 
Abstract
:1. Introduction
- hexose = mannose, (1) + (2) = phosphomannomutase activity
- hexose = glucose, (1) + (2) = phosphoglucomutase activity
- hexose = mannose or glucose, (1) + (3) = phosphatase activity
2. Results
2.1. Synthesis and 31P-NMR Characterization of βG16P
2.2. βG16P Binds PMM2 and Induces a Conformational Change
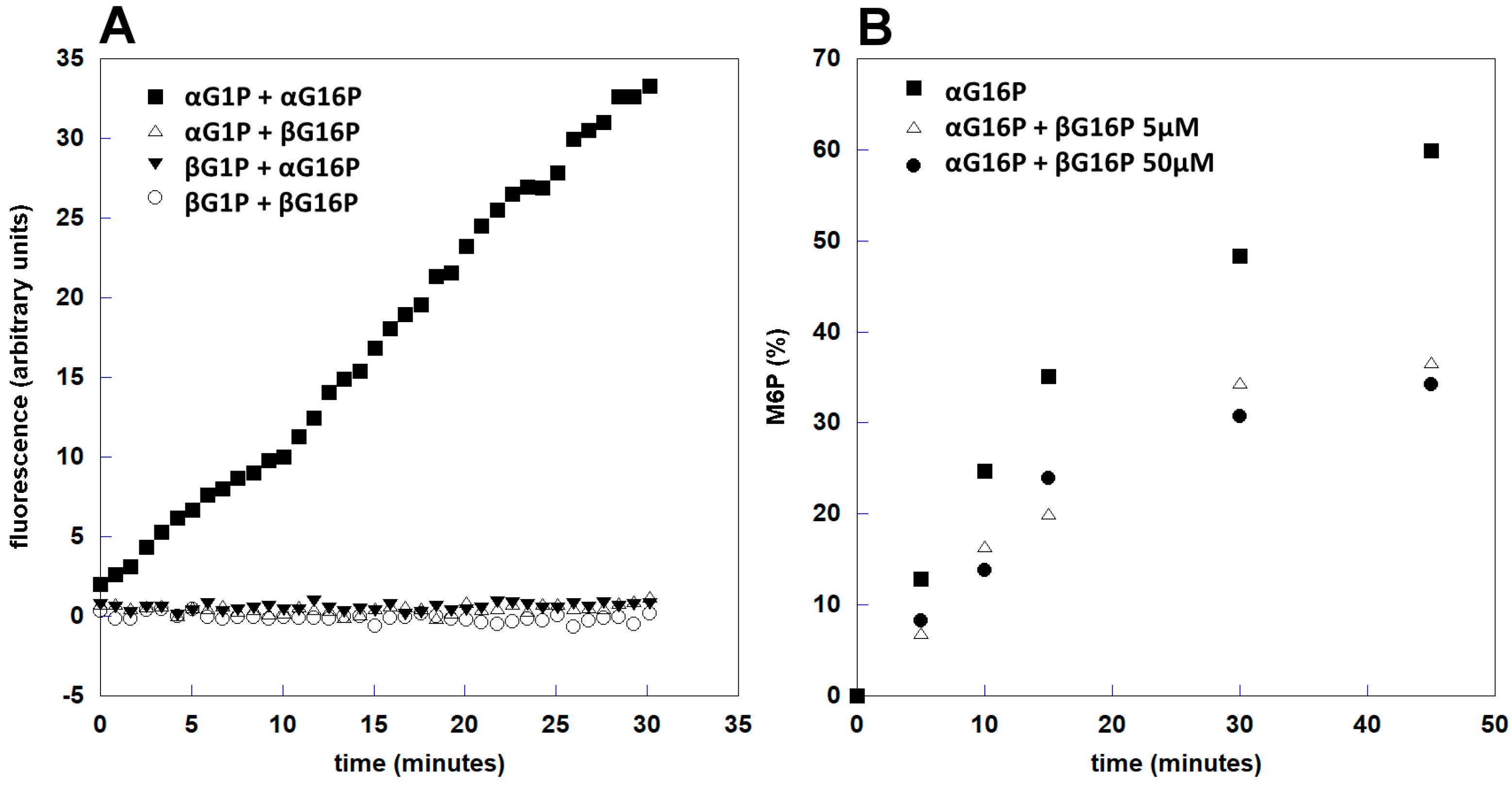
2.3. βG16P Inhibits PMM2
2.4. βG16P Stabilizes PMM2
2.5. βG16P is a Poor Substrate of PMM1
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Synthesis of α-Glucose-1-Phosphate and β-Glucose-1,6-Bisphosphate
4.3. 31P-NMR Spectroscopy
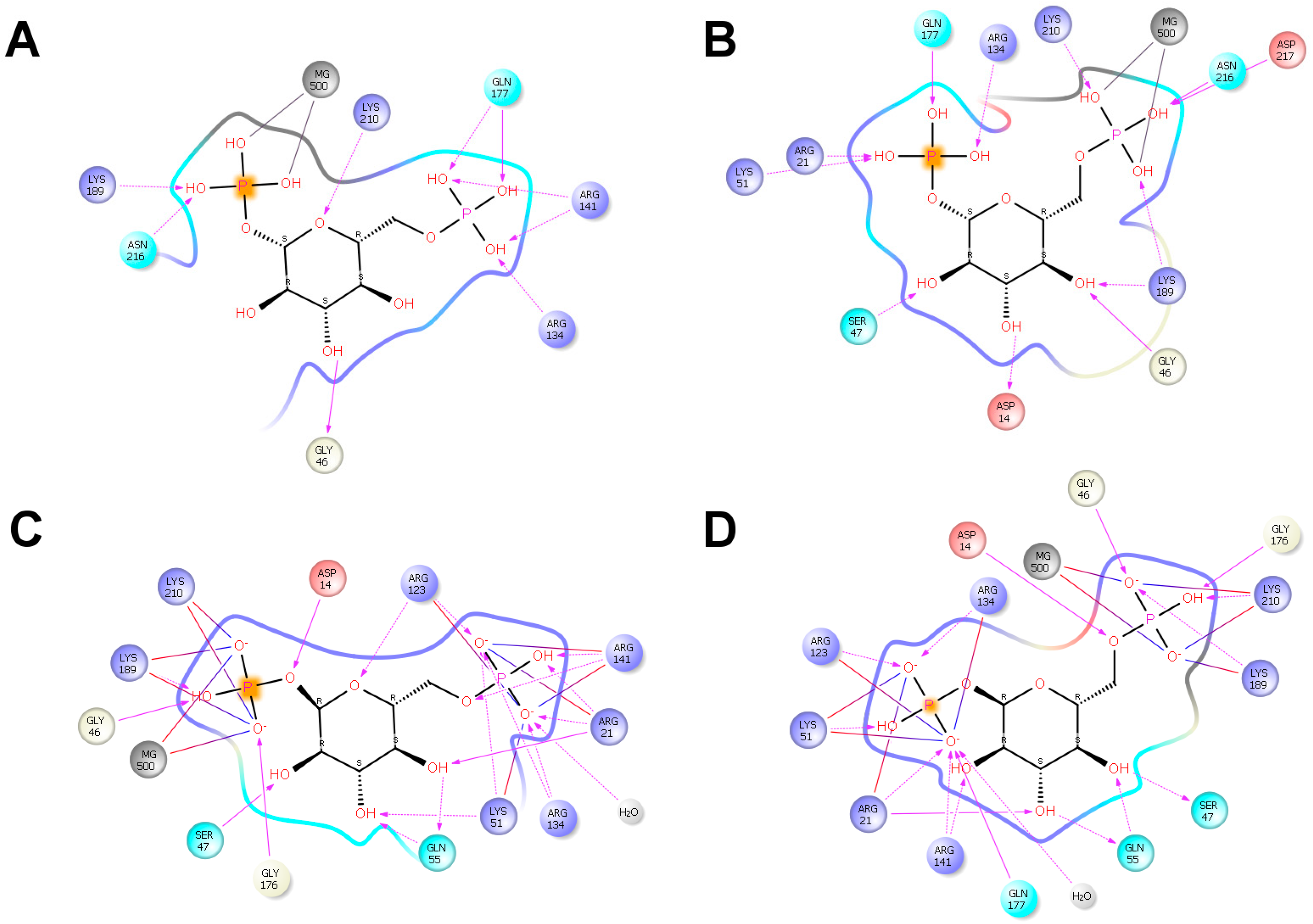
4.4. Docking and Structure Analysis
- (a)
- The global search was performed by combining a long (6 Å) and a short (1.5 Å) ligand perturbation steps, with a 75%/25% probability, respectively. Rotations were kept in the [0°–90°] range. A randomly chosen search direction was kept for two Monte Carlo steps, allowing a more complete exploration of the entire protein surface. No information about the bound structure was used to drive the search. Anisotropic normale mode perturbation included the lowest six modes, with maximum displacements of the alpha carbon of 1 Å. Within the lowest six modes, a randomly chosen mode was kept for six steps to facilitate large conformational exploration.
- (b)
- The local search used translations of only 0.5 A and rotations in the [0°–180°] range. Furthermore, to keep the ligand in the active site, random search direction was maintained to only one iteration.
4.5. Protein Expression and Purification
4.6. Limited Proteolysis
4.7. Enzyme Assay by Fluorescence Spectroscopy
4.8. Enzyme Assay by 31P-NMR
4.9. Thermal Shift Assay
4.10. Miscellaneous
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CP | creatine phosphate |
| DTT | dithiothreitol |
| αG1P | α-glucose-1-phosphate |
| αG16P | α-glucose-1,6-bisphosphate |
| βG16P | β-glucose-1,6-bisphosphate |
| IMP | inosine monophosphate |
| αM1P | α-mannose-1-phosphate |
| M6P | mannose-6-phosphate |
| PC | pharmacological chaperone |
References
- Pirard, M.; Achouri, Y.; Collet, J.F.; Schollen, E.; Matthijs, G.; Van Schaftingen, E. Kinetic properties and tissular distribution of mammalian phosphomannomutase isozymes. Biochem. J. 1999, 339, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Veiga-da-Cunha, M.; Vleugels, W.; Maliekal, P.; Matthijs, G.; Van Schaftingen, E. Mammalian phosphomannomutase PMM1 is the brain IMP-sensitive glucose-1,6-bisphosphatase. J. Biol. Chem. 2008, 283, 33988–33993. [Google Scholar] [CrossRef] [PubMed]
- Citro, V.; Cimmaruta, C.; Liguori, L.; Viscido, G.; Cubellis, M.V.; Andreotti, G. A mutant of phosphomannomutase1 retains full enzymatic activity, but is not activated by IMP: Possible implications for the disease PMM2-CDG. PLoS ONE 2017, 12, e0189629. [Google Scholar] [CrossRef] [PubMed]
- Van Schaftingen, E.; Jaeken, J. Phosphomannomutase deficiency is a cause of carbohydrate-deficient glycoprotein syndrome type I. FEBS Lett. 1995, 377, 318–320. [Google Scholar] [PubMed] [Green Version]
- Kjaergaard, S.; Skovby, F.; Schwartz, M. Carbohydrate-deficient glycoprotein syndrome type 1A: Expression and characterisation of wild type and mutant PMM2 in E. coli. Eur. J. Hum. Genet. 1999, 7, 884–888. [Google Scholar] [CrossRef] [PubMed]
- Matthijs, G.; Schollen, E.; Bjursell, C.; Erlandson, A.; Freeze, H.; Imtiaz, F.; Kjaergaard, S.; Martinsson, T.; Schwartz, M.; Seta, N.; et al. Mutations in PMM2 that cause congenital disorders of glycosylation, type Ia (CDG-Ia). Hum. Mutat. 2000, 16, 386–394. [Google Scholar] [CrossRef]
- Pirard, M.; Matthijs, G.; Heykants, L.; Schollen, E.; Grunewald, S.; Jaeken, J.; van Schaftingen, E. Effect of mutations found in carbohydrate-deficient glycoprotein syndrome type IA on the activity of phosphomannomutase 2. FEBS Lett. 1999, 452, 319–322. [Google Scholar] [CrossRef]
- Kjaergaard, S.; Skovby, F.; Schwartz, M. Absence of homozygosity for predominant mutations in PMM2 in Danish patients with carbohydrate-deficient glycoprotein syndrome type 1. Eur. J. Hum. Genet. 1998, 6, 331–336. [Google Scholar] [CrossRef]
- Matthijs, G.; Schollen, E.; Van Schaftingen, E.; Cassiman, J.J.; Jaeken, J. Lack of homozygotes for the most frequent disease allele in carbohydrate-deficient glycoprotein syndrome type 1A. Am. J. Hum. Genet. 1998, 62, 542–550. [Google Scholar] [CrossRef]
- Andreotti, G.; Cabeza de Vaca, I.; Poziello, A.; Monti, M.C.; Guallar, V.; Cubellis, M.V. Conformational response to ligand binding in phosphomannomutase2: Insights into inborn glycosylation disorder. J. Biol. Chem. 2014, 289, 34900–34910. [Google Scholar] [CrossRef]
- Wang, Y.-J.; Di, X.-J.; Mu, T.-W. Using pharmacological chaperones to restore proteostasis. Pharmacol. Res. 2014, 83, 3–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, J.Q. A counterintuitive approach to treat enzyme deficiencies: Use of enzyme inhibitors for restoring mutant enzyme activity. Biol. Chem. 2008, 389, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Jorge-Finnigan, A.; Brasil, S.; Underhaug, J.; Ruiz-Sala, P.; Merinero, B.; Banerjee, R.; Desviat, L.R.; Ugarte, M.; Martinez, A.; Pérez, B. Pharmacological chaperones as a potential therapeutic option in methylmalonic aciduria cblB type. Hum. Mol. Genet. 2013, 22, 3680–3689. [Google Scholar] [CrossRef] [PubMed]
- Leidenheimer, N.J.; Ryder, K.G. Pharmacological chaperoning: A primer on mechanism and pharmacology. Pharmacol. Res. 2014, 83, 10–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Citro, V.; Pena-Garcia, J.; den-Haan, H.; Perez-Sanchez, H.; Del Prete, R.; Liguori, L.; Cimmaruta, C.; Lukas, J.; Cubellis, M.V.; Andreotti, G. Identification of an Allosteric Binding Site on Human Lysosomal Alpha-Galactosidase Opens the Way to New Pharmacological Chaperones for Fabry Disease. PLoS ONE 2016, 11, e0165463. [Google Scholar] [CrossRef] [PubMed]
- Jaeken, J.; Matthijs, G. Congenital disorders of glycosylation. Annu Rev. Genom. Hum. Genet. 2001, 2, 129–151. [Google Scholar] [CrossRef] [PubMed]
- Grunewald, S.; Schollen, E.; Van Schaftingen, E.; Jaeken, J.; Matthijs, G. High residual activity of PMM2 in patients’ fibroblasts: Possible pitfall in the diagnosis of CDG-Ia (phosphomannomutase deficiency). Am. J. Hum. Genet. 2001, 68, 347–354. [Google Scholar] [CrossRef]
- Andreotti, G.; Pedone, E.; Giordano, A.; Cubellis, M.V. Biochemical phenotype of a common disease-causing mutation and a possible therapeutic approach for the phosphomannomutase 2-associated disorder of glycosylation. Mol. Genet. Genom. Med. 2013, 1, 32–44. [Google Scholar] [CrossRef]
- Yuste-Checa, P.; Brasil, S.; Gamez, A.; Underhaug, J.; Desviat, L.R.; Ugarte, M.; Pérez-Cerdá, C.; Martinez, A.; Pérez, B. Pharmacological Chaperoning: A Potential Treatment for PMM2-CDG. Hum. Mutat. 2017, 38, 160–168. [Google Scholar] [CrossRef]
- Yuste-Checa, P.; Gamez, A.; Brasil, S.; Desviat, L.R.; Ugarte, M.; Perez-Cerda, C.; Pérez, B. The Effects of PMM2-CDG-Causing Mutations on the Folding, Activity, and Stability of the PMM2 Protein. Hum. Mutat. 2015, 36, 851–860. [Google Scholar] [CrossRef] [Green Version]
- Vega, A.I.; Perez-Cerda, C.; Abia, D.; Gamez, A.; Briones, P.; Artuch, R.; Desviat, L.R.; Ugarte, M.; Pérez, B. Expression analysis revealing destabilizing mutations in phosphomannomutase 2 deficiency (PMM2-CDG): Expression analysis of PMM2-CDG mutations. J. Inherit. Metab. Dis. 2011, 34, 929–939. [Google Scholar] [CrossRef] [PubMed]
- Andreotti, G.; Monti, M.C.; Citro, V.; Cubellis, M.V. Heterodimerization of Two Pathological Mutants Enhances the Activity of Human Phosphomannomutase2. PLoS ONE 2015, 10, e0139882. [Google Scholar] [CrossRef]
- Lao, J.P.; DiPrimio, N.; Prangley, M.; Sam, F.S.; Mast, J.D.; Perlstein, E.O. Yeast Models of Phosphomannomutase 2 Deficiency, A Congenital Disorder of Glycosylation. G3 (Bethesda) 2019, 9, 413–423. [Google Scholar] [CrossRef]
- Hay Mele, B.; Citro, V.; Andreotti, G.; Cubellis, M.V. Drug repositioning can accelerate discovery of pharmacological chaperones. Orphanet J. Rare Dis. 2015, 10, 55. [Google Scholar] [CrossRef]
- Iyer, S.; Murthy, K.; Parton, Z.; Tsang, H.; Sam, F.S.; DiPrimio, N.; Lao, J.; Perlstein, E.O. Repurposing the aldose reductase inhibitor and diabetic neuropathy drug epalrestat for the congenital disorder of glycosylation PMM2-CDG. bioRxiv 2019. [Google Scholar] [CrossRef]
- Borrelli, K.W.; Vitalis, A.; Alcantara, R.; Guallar, V. PELE: Protein energy landscape exploration. A novel Monte Carlo based technique. J. Chem. Theory Comput. 2005, 1, 1304–1311. [Google Scholar] [CrossRef]
- Freeze, H.H. Towards a therapy for phosphomannomutase 2 deficiency, the defect in CDG-Ia patients. Biochim. Biophys. Acta 2009, 1792, 835–840. [Google Scholar] [CrossRef] [Green Version]
- Brasil, S.; Pascoal, C.; Francisco, R.; Marques-da-Silva, D.; Andreotti, G.; Videira, P.A.; Morava, E.; Jaeken, J.; Dos Reis Ferreira, V. CDG Therapies: From Bench to Bedside. Int. J. Mol. Sci. 2018, 19, 1304. [Google Scholar] [CrossRef]
- Kjaergaard, S.; Kristiansson, B.; Stibler, H.; Freeze, H.H.; Schwartz, M.; Martinsson, T.; Skovby, F. Failure of short-term mannose therapy of patients with carbohydrate-deficient glycoprotein syndrome type 1A. Acta Paediatr. 1998, 87, 884–888. [Google Scholar] [CrossRef]
- Mayatepek, E.; Kohlmuller, D. Mannose supplementation in carbohydrate-deficient glycoprotein syndrome type I and phosphomannomutase deficiency. Eur. J. Pediatr. 1998, 157, 605–606. [Google Scholar] [CrossRef]
- Eklund, E.A.; Merbouh, N.; Ichikawa, M.; Nishikawa, A.; Clima, J.M.; Dorman, J.A.; Norberg, T.; Freeze, H.H. Hydrophobic Man-1-P derivatives correct abnormal glycosylation in Type I congenital disorder of glycosylation fibroblasts. Glycobiology 2005, 15, 1084–1093. [Google Scholar] [CrossRef] [Green Version]
- Rutschow, S.; Thiem, J.; Kranz, C.; Marquardt, T. Membrane-permeant derivatives of mannose-1-phosphate. Bioorg. Med. Chem. 2002, 10, 4043–4049. [Google Scholar] [CrossRef]
- Sharma, V.; Ichikawa, M.; He, P.; Scott, D.A.; Bravo, Y.; Dahl, R.; Ng, B.G.; Cosford, N.D.; Freeze, H.H. Phosphomannose isomerase inhibitors improve N-glycosylation in selected phosphomannomutase-deficient fibroblasts. J. Biol. Chem. 2011, 286, 39431–39438. [Google Scholar] [CrossRef]
- Shang, J.; Lehrman, M.A. Metformin-stimulated mannose transport in dermal fibroblasts. J. Biol. Chem. 2004, 279, 9703–9712. [Google Scholar] [CrossRef]
- Martinez-Monseny, A.F.; Bolasell, M.; Callejon-Poo, L.; Cuadras, D.; Freniche, V.; Itzep, D.C.; Gassiot, S.; Arango, P.; Casas-Alba, D.; de la Morena, E.; et al. AZATAX: Acetazolamide safety and efficacy in cerebellar syndrome in PMM2 congenital disorder of glycosylation (PMM2-CDG). Ann. Neurol. 2019, 85, 740–751. [Google Scholar] [CrossRef]
- Pandurangan, A.P.; Ochoa-Montano, B.; Ascher, D.B.; Blundell, T.L. SDM: A server for predicting effects of mutations on protein stability. Nucleic Acids Res. 2017, 45, W229–W235. [Google Scholar] [CrossRef]
- Levy, H.L.; Milanowski, A.; Chakrapani, A.; Cleary, M.; Lee, P.; Trefz, F.K.; Whitley, C.B.; Feillet, F.; Feigenbaum, A.S.; Bebchuk, J.D.; et al. Efficacy of sapropterin dihydrochloride (tetrahydrobiopterin, 6R-BH4) for reduction of phenylalanine concentration in patients with phenylketonuria: A phase III randomised placebo-controlled study. Lancet 2007, 370, 504–510. [Google Scholar] [CrossRef]
- Markham, A. Migalastat: First Global Approval. Drugs 2016, 76, 1147–1152. [Google Scholar] [CrossRef]
- Kjaergaard, S.; Schwartz, M.; Skovby, F. Congenital disorder of glycosylation type Ia (CDG-Ia): Phenotypic spectrum of the R141H/F119L genotype. Arch. Dis. Child. 2001, 85, 236–239. [Google Scholar] [CrossRef]
- Barone, R.; Carrozzi, M.; Parini, R.; Battini, R.; Martinelli, D.; Elia, M.; Spada, M.; Lilliu, F.; Ciana, G.; Burlina, A.; et al. A nationwide survey of PMM2-CDG in Italy: High frequency of a mild neurological variant associated with the L32R mutation. J. Neurol. 2015, 262, 154–164. [Google Scholar] [CrossRef]
- Citro, V.; Cimmaruta, C.; Monticelli, M.; Riccio, G.; Hay Mele, B.; Cubellis, M.V.; Andreotti, G. The Analysis of Variants in the General Population Reveals That PMM2 Is Extremely Tolerant to Missense Mutations and That Diagnosis of PMM2-CDG Can Benefit from the Identification of Modifiers. Int. J. Mol. Sci. 2018, 19, 2218. [Google Scholar] [CrossRef]
- Glycomine. Available online: http://glycomine.com (accessed on 12 August 2019).
- Dai, J.; Wang, L.; Allen, K.N.; Radstrom, P.; Dunaway-Mariano, D. Conformational cycling in beta-phosphoglucomutase catalysis: Reorientation of the beta-d-glucose 1,6-(Bis)phosphate intermediate. Biochemistry 2006, 45, 7818–7824. [Google Scholar] [CrossRef]
- PISA. Available online: https://www.ebi.ac.uk/pdbe/pisa/pi_link.html (accessed on 10 July 2019).
- Cammisa, M.; Correra, A.; Andreotti, G.; Cubellis, M.V. Identification and analysis of conserved pockets on protein surfaces. BMC Bioinform. 2013, 14 (Suppl. 7), S9. [Google Scholar] [CrossRef]
- PyMOL. Available online: https://pymol.org/2/ (accessed on 10 July 2019).
- Maestro. Schrödinger Release 2015–2: Maestro, Schrödinger; Maestro Molecular Modeling Interface, Tool “Ligand Interaction Diagram”; LLC: New York, NY, USA, 2015. [Google Scholar]
- Silvaggi, N.R.; Zhang, C.; Lu, Z.; Dai, J.; Dunaway-Mariano, D.; Allen, K.N. The X-ray crystal structures of human alpha-phosphomannomutase 1 reveal the structural basis of congenital disorder of glycosylation type 1a. J. Biol. Chem. 2006, 281, 14918–14926. [Google Scholar] [CrossRef]
- Andreotti, G.; Citro, V.; Correra, A.; Cubellis, M.V. A thermodynamic assay to test pharmacological chaperones for Fabry disease. Biochim. Biophys. Acta. 2014, 1840, 1214–1224. [Google Scholar] [CrossRef]
- Andreotti, G.; Monticelli, M.; Cubellis, M.V. Looking for protein stabilizing drugs with thermal shift assay. Drug Test. Anal. 2015, 7, 831–834. [Google Scholar] [CrossRef]
- D’Aniello, E.; Fellous, T.; Iannotti, F.A.; Gentile, A.; Allara, M.; Balestrieri, F.; Gray, R.; Amodeo, P.; Vitale, R.M.; Di Marzo, V.; et al. Identification and characterization of phytocannabinoids as novel dual PPARalpha/gamma agonists by a computational and in vitro experimental approach. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 586–597. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bisphosphate (145 μM) | IMP (170 μM) | Residual αG16P (%) | Residual βG16P (%) |
|---|---|---|---|
| αG16P | - | 54.2 ± 1.6 | - |
| βG16P | - | - | 62.5 ± 7.0 |
| αG16P + βG16P | - | 53.3 ± 1.3 | 93.6 ± 1.2 |
| αG16P | + | 0 | - |
| βG16P | + | - | 65.7 ± 11.4 |
| αG16P + βG16P | + | 0 | 74.6 ± 3.1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Monticelli, M.; Liguori, L.; Allocca, M.; Andreotti, G.; Cubellis, M.V. β-Glucose-1,6-Bisphosphate Stabilizes Pathological Phophomannomutase2 Mutants In Vitro and Represents a Lead Compound to Develop Pharmacological Chaperones for the Most Common Disorder of Glycosylation, PMM2-CDG. Int. J. Mol. Sci. 2019, 20, 4164. https://doi.org/10.3390/ijms20174164
Monticelli M, Liguori L, Allocca M, Andreotti G, Cubellis MV. β-Glucose-1,6-Bisphosphate Stabilizes Pathological Phophomannomutase2 Mutants In Vitro and Represents a Lead Compound to Develop Pharmacological Chaperones for the Most Common Disorder of Glycosylation, PMM2-CDG. International Journal of Molecular Sciences. 2019; 20(17):4164. https://doi.org/10.3390/ijms20174164
Chicago/Turabian StyleMonticelli, Maria, Ludovica Liguori, Mariateresa Allocca, Giuseppina Andreotti, and Maria Vittoria Cubellis. 2019. "β-Glucose-1,6-Bisphosphate Stabilizes Pathological Phophomannomutase2 Mutants In Vitro and Represents a Lead Compound to Develop Pharmacological Chaperones for the Most Common Disorder of Glycosylation, PMM2-CDG" International Journal of Molecular Sciences 20, no. 17: 4164. https://doi.org/10.3390/ijms20174164
APA StyleMonticelli, M., Liguori, L., Allocca, M., Andreotti, G., & Cubellis, M. V. (2019). β-Glucose-1,6-Bisphosphate Stabilizes Pathological Phophomannomutase2 Mutants In Vitro and Represents a Lead Compound to Develop Pharmacological Chaperones for the Most Common Disorder of Glycosylation, PMM2-CDG. International Journal of Molecular Sciences, 20(17), 4164. https://doi.org/10.3390/ijms20174164