1. Introduction
For teleosts and tetrapods, the activation of the melanocortin-2 receptor (MC2R) located on glucocorticoid-synthesizing cells (i.e., interrenal cells and adrenal cortex cells) involves formation of a heterodimer between the MC2R ortholog and the accessory protein, MRAP1 [
1,
2,
3]. The MC2R/MRAP1 heterodimer, not only facilitates the trafficking of MC2R from the endoplasmic reticulum to the plasma membrane, but also places the MC2R ortholog into the proper conformation to allow for binding of the pituitary hormone, ACTH (adrenocorticotropin; [
4,
5]); the only melanocortin ligand that can activate teleost and tetrapod MC2R orthologs [
6].
ACTH activation of MC2R orthologs requires the coordinated interaction between two functional motifs in the hormone (i.e., H6F7R8W9 and R/K15K16R17R18), and proposed corresponding binding sites on the MC2R ortholog [
7]. While all melanocortin peptides have the “melanocortin core” sequence, HFRW, that is required for activating all melanocortin receptors (i.e., MC1R, MC2R, MC3R, MC4R, MC5R), αMSH (N-acetyl-SYSMEHFRWGKPVamide) cannot activate either teleost or tetrapod MC2R orthologs [
7,
8] due to the absence of the tetrabasic motif (K/R15K16R17R18). Given these earlier observations by Schwyzer [
7] and Mountjoy et al. [
8], subsequent studies have focused on identifying first the common HFRW binding site on all melanocortin receptors, and then the unique site on MC2R orthologs required for binding the tetrabasic motif present in all vertebrate ACTH sequences [
9].
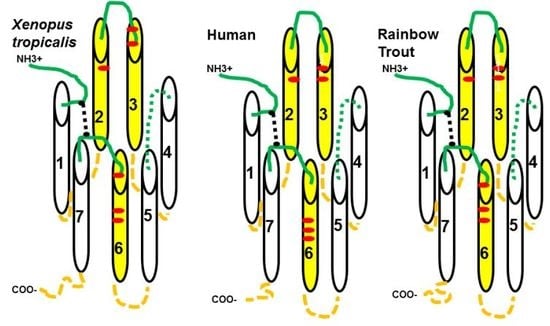
To localize the common HFRW binding site, Pogozheva et al. [
10], used a modeling strategy and a site directed mutagenesis paradigm to identify critical amino acid positions in transmembrane (TM) domains 2, 3, and 6 of human MC4R that are required for α-MSH binding, and activation of this receptor. The latter study also observed that several of the amino acid positions in the HFRW bind site of hMC4R, were also conserved in the sequence of human MC2R (
Figure 1; starred positions). Chen et al. [
11] used a site directed mutagenesis approach to confirm that several of the amino acid positions in the HFRW binding site for hMC4R were also required for activation of hMC2R (
Figure 1; bold red residues). These positions in hMC2R included, E80 (TM2), D103 and D107 (TM3), and F236 (TM6). In addition, Chen et al. [
11] hypothesized that phenylalanine residues either on an extracellular domain (EC) or in close proximity to the extracellular space on a TM might also be important for activation of MC2R. To this end, their site directed mutagenesis analysis found that F168 (EC2), and F178 (TM5) were also important for activation. Assuming that melanocortin receptors have a barrel conformation with TM1 and TM7 in close proximity to each other, these two phenylalanine residues are clearly outside the HFRW binding site (see Figure 5A). Hence, Chen et al. [
11] speculated that these residues could be in the KKRR binding site of hMC2R. Just one year later, Chung et al. [
12] reported that a spontaneous mutation at H170 (EC2;
Figure 1 bold green reside) resulted in a mutant form of MC2R that could traffic to the plasma membrane when expressed in a mammalian cell line, but could not be activated following stimulation with human ACTH. Collectively, these two studies pointed to the potential location of the proposed KKRR binding site in the TM4/EC2/TM5 domain of hMC2R. A more recent study of hMC2R used a single alanine substitution paradigm to evaluate the role of residues from G162 (TM4) to P183 (TM5) (
Figure 1, [
13]), and confirmed the importance of F168, H170, and F178 in the activation of hMC2R.
To corroborate the importance of the EC2 domain in the activation of vertebrate MC2R orthologs, a single alanine substitution study was done on the TM4/EC2/TM5 domain of a modern bony fish (Division Teleostei;
Oncyrhinchus mykiss,
rainbow trout (rt); [
14]). This study found that alanine substitution at F160 (EC2) in rtMC2R, which aligns with F168 in the EC2 domain of hMC2R (
Figure 1), disrupted activation of rtMC2R. However, since the primary sequence identity in the EC2 domain of rtMC2R and hMC2R (
Figure 1) is only 11%, a consensus sequence in the EC2 required for activation was not apparent.
Since teleosts and mammals are not closely related vertebrate groups, the current study was undertaken on a MC2R ortholog of a vertebrate group, the amphibians, which occupy a phylogenetic position between the modern bony fishes and mammals [
15]. The a priori assumption was that an amphibian MC2R ortholog might have features that would bridge teleost and mammalian MC2R orthologs. To this end, the current study was done on the MC2R ortholog of the amphibian,
Xenopus tropicalis (xt).
Initial studies on xtMC2R indicated that this MC2R ortholog may be very appropriate for this type of study [
16]. xtMC2R requires co-expression with the accessory protein, MRAP1, for functional expression in Chinese hamster ovary cells, and this MC2R ortholog could only be activated by ACTH, but not by any MSH-sized ligands [
16,
17]. However, as shown in
Figure 1, the amino acid identity between the three MC2R orthologs aligned in
Figure 1 is only 38% (black bold resides found at the same position in all three sequences). In addition, the amino acid sequence of the EC2 domain of xtMC2R has minimal primary sequence identity with either hMC2R or rtMC2R (
Figure 1), a point that will be discussed later in this study. Assuming that a contact site for the KKRR motif of ACTH might involve residues in either TM4 or TM5 that are close to the surface, as well as one or more residues in EC2, a single alanine-substitution paradigm was used beginning with G154 (TM4) and extending to L175 (TM5) of xtMC2R to evaluate the effect of alanine substitution on the activation of xtMC2R by using a cAMP reporter gene assay. The role of residues in TM4 and TM5 that could be involved in trafficking were analyzed using a Cell Surface ELISA protocol.
3. Discussion
MC2R is perhaps the most complex and enigmatic member of the melanocortin receptor gene family. This receptor plays a pivotal role in the hypothalamus/pituitary/adrenal/interrenal (HPA/HPI) Axis by inducing glucocorticoid synthesis from either adrenal cortex cells (mammals and reptiles) or interrenal cells (amphibians, bony fishes, cartilaginous fishes; [
19,
20]). As a result, this neuroendocrine circuit helps to re-establishing homeostasis after a chronic stress event in both teleosts and tetrapods [
19]. Given the importance of the HPA/HPI axis, it would be reasonable to predict that selection pressures would favor conservation of the primary sequences of ACTH, the ligand that binds to the glucocorticoid producing cells, and the ACTH receptor (i.e., MC2R). As predicted, the primary sequence of ACTH, within the minimal sequence required for full activation at target cells (i.e., ACTH(1-24); [
7]), is 89% for ACTH(1-24) sequences from a broad taxonomic spectrum (i.e., cartilaginous fishes to mammals; [
9]). All vertebrate ACTH sequences have the canonical motif, H6F7R8W9, that is required for activating all melanocortin receptors [
20], and the R/K15K16R17R18 motif that is required for facilitating the activation of all teleost and tetrapod MC2R orthologs [
7,
20,
21].
Given the observations for ACTH, the status of teleost and tetrapod MC2R orthologs is surprising. The primary sequence identity for the three bony vertebrate MC2R orthologs presented in
Figure 1 is only 38%. While this level of primary sequence conservation may seem reasonable for species of vertebrates that last shared a common ancestor over 400 million years ago [
22], a recent analysis of the primary sequence identity between a cartilaginous fish (stingray) MC5R ortholog and a mammalian (human) MC5R ortholog was found to be 55% [
23]. In mammals, MC5R plays a role in sebaceous gland secretion [
24]; whereas in non-mammalian vertebrates including stingrays, the function(s) and the selection pressures on MC5R orthologs are unknown. Clearly, melanocortin receptors paralogs have not been evolving at the same rate during the radiation of the gnathostomes. Given the physiological importance of the HPA/HPI axis, it seems remarkable that in spite of the accumulation of point mutations, teleost and tetrapod MC2R orthologs have retained functionality, and share a number of unique physiological features not seen in the other melanocortin receptors.
For example, the MC2R orthologs of teleosts and tetrapods, unlike the other paralogs in this gene family (i.e., MC1R, MC3R, MC4R, and MC5R), can only be activated by ACTH, and not by any of the MSH-sized melanocortin peptides derives from POMC [
7,
8,
21]. In fact, it appears that MSH-sized melanocortin peptides cannot even bind to the “ACTH” receptor (i.e., MC2R; [
25]). The inability to bind αMSH is an apparent enigma given that MC2R orthologs share an HFRW binding pocket in common with all the other teleost and tetrapod melanocortin receptors [
10,
26,
27]. In addition, teleost and tetrapod MC2R orthologs have intracellular trafficking restrictions which mandate that the receptor form a heterodimer with the accessory protein MRAP1 to facilitate unimpeded movement of the receptor from the endoplasmic reticulum to the plasma membrane [
4,
5]. Finally, for activation at the plasma membrane, MC2R must be in a complex with MRAP1for the ACTH binding event to occur [
1,
2]. Thus, the singular feature that sets teleost and tetrapod MC2R orthologs apart from the other members of the melanocortin receptor gene family is the obligatory interaction with MRAP1; an interaction that may have begun prior to the radiation of the ancestral gnathostomes [
13].
Since α-MSH lacks the R/KKRR motif, and as a result cannot activate teleost or tetrapod MC2R orthologs, it would appear that the activation mechanism for MC2R orthologs may involve a two-step process in which the R/KKRR motif of ACTH docks with the receptor, and this event causes a conformation change which exposes a site on the receptor for the binding of the HFRW motif of ACTH. It would be the later proposed binding event that leads to the activation of the G-protein associated with the receptor [
28]. Based on the predications made with the TMHMMServer, v. 2.0-DTU program on the location of transmembrane domains in the three MC2R orthologs in
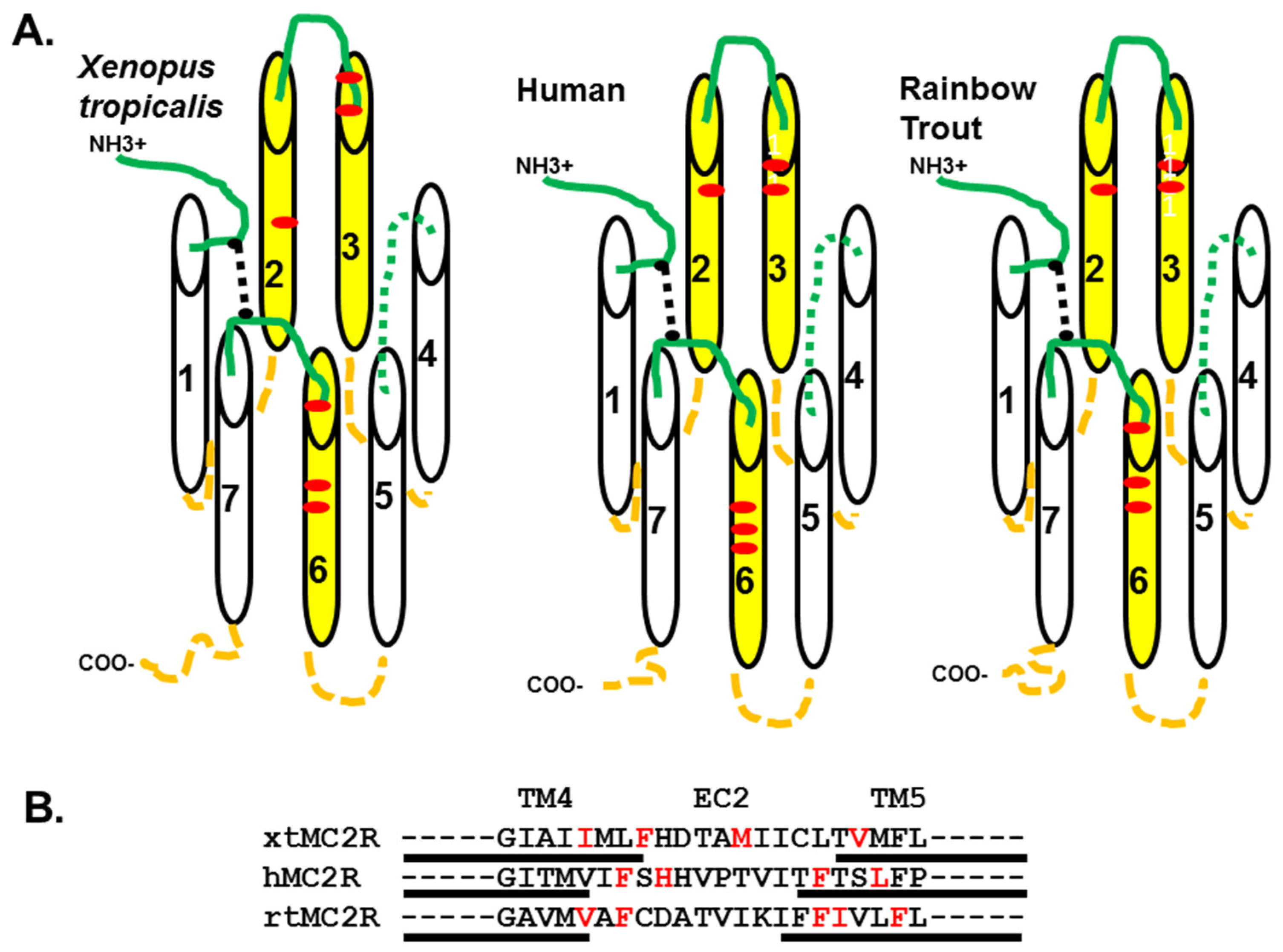
Figure 1, a schematic representation of the three receptors is presented in
Figure 5A. Note that six of the critical residues that formed the HFRW binding site in hMC4R [
10] are in nearly the same position in or near the TM2 and TM3 domain, and in or near the TM6 domain of the three MC2R orthologs. Malik et al. [
29] observed that alanine substitution at E80 rendered the mutant hMC2R inactive to ACTH stimulation, but had no effect on the trafficking of the mutant MC2R when co-expressed with MRAP1. The same outcome would be expected for E172 in xtMC2R and E172 in rtMC2R. While site-directed mutagenesis experiments are needed to verify the role of E172 in the activation of xtMC2R, the models presented in
Figure 5A point to EC2 as another potential contact site for ACTH interaction with MC2R as previously proposed by Chen et al. [
11].
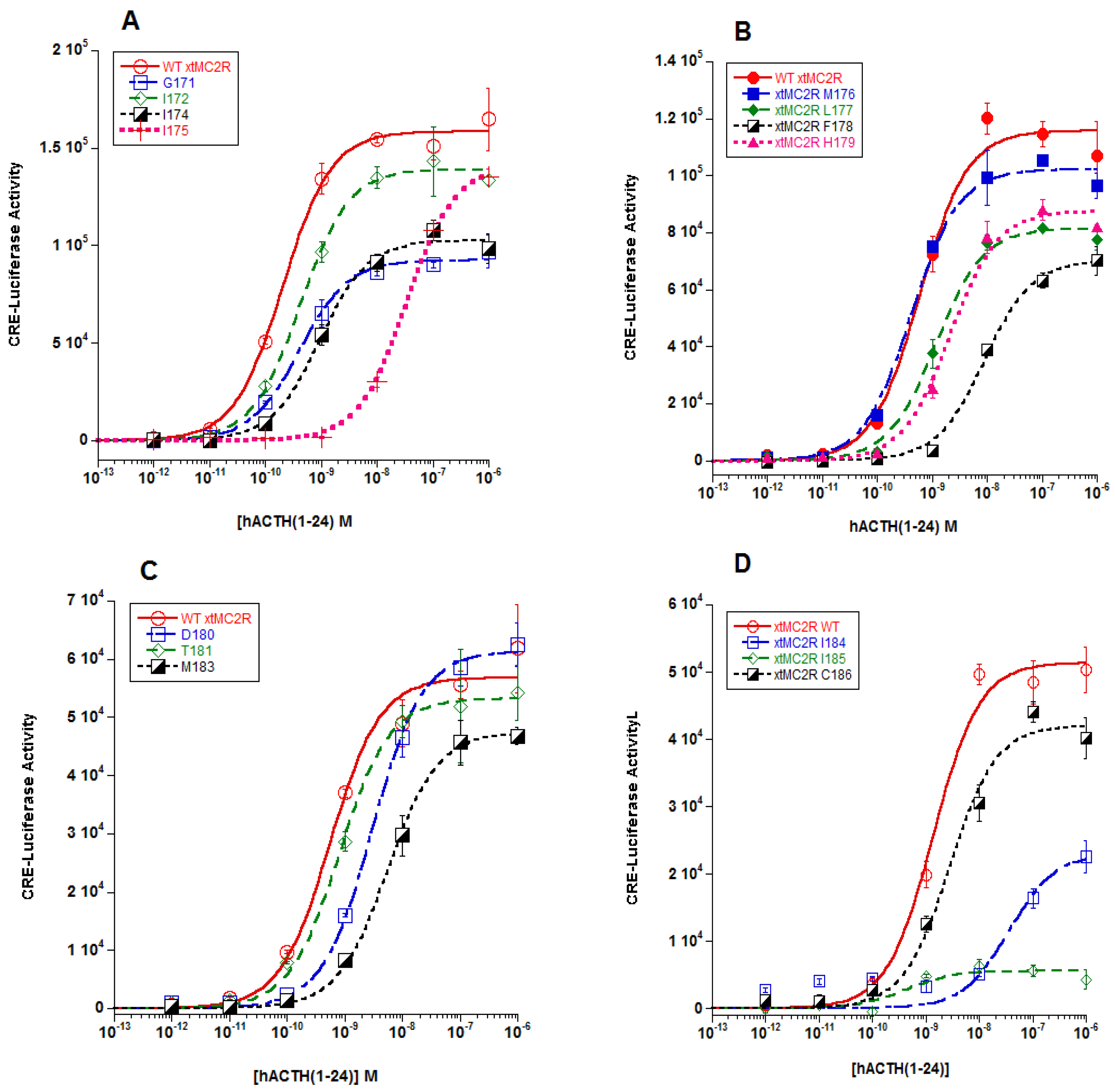
The focus, then, of this study was to evaluate the role that residues in the TM4/EC2/TM5 domain of xtMC2R may play in the activation of this receptor. The results from previous studies using a single-alanine substitution paradigm for hMC2R [
13] and rtMC2R [
14] are shown in
Figure 5B. The current study was done on the corresponding region of xtMC2R with the following results. Single alanine substitution at I158, F161, two residues in TM4 located close to extracellular space, M166 in EC2, and V172 in TM5 also located near the extracellular space all resulted in a drop in sensitivity to stimulation by hACTH(1-24) of 10 fold or more in the cAMP reporter gene assay analysis (
Figure 2;
Table 1;
Figure 5B). The location of these residues would be consistent with a shallow hydrophilic pocket at the TM4/EC2/TM5 domain of xtMC2R. These results are comparable to our earlier studies on the TM4/EC2/TM5 domain of hMC2R [
13] and the TM4/EC2/TM5 domain of rtMC2R ([
14];
Figure 5B). For all three species, a limited number of amino acid positions (4 to 5) were negatively impacted by single alanine substitution. What is perplexing about these results (
Figure 5B) is that a consensus sequence for modulating activation of the three MC2R orthologs within this domain is not apparent. The assumption is that the EC2 domain and perhaps a portion of TM5 form the putative R/RKRR binding site for ACTH. Hence, it might be expected that this site would be rich in acidic amino acids that presumably would ionically interact with the R/KKRR motif of ACTH, but this was not the case. However, there are a few aromatic amino acids in this region such as phenylalanine, and alanine substitution at these residues consistently lowered sensitivity to stimulation by ACTH(1-24).
In the alanine substitution study for rtMC2R [
14], a cell surface ELISA analysis indicated that V158, F171, and F175 (highlighted in
Figure 5B) did not have any negative effect on trafficking. The same analysis was done for I158, F161 in TM4 of xtMC2R, and V172 and M173in TM5 of xtMC2R (
Figure 3). The residues in TM5 had no negative effect on trafficking (
Figure 3B), and neither did residue F161 in TM4 (
Figure 3A). However, alanine substitution at residue I158 in TM4 apparently reduced trafficking of the receptor to the plasma membrane by nearly 50%. While the later observation is preliminary, it appears that TM4 could be the contact site for the TM of MRAP1; an interaction that would facilitate trafficking. Now an in-depth analysis of the other residues in TM4 will need to be done to clarify the role of TM4 in trafficking. Since the three studies summarized in
Figure 5B, all required co-expression with an MRAP1 ortholog, perhaps the critical question with respect to the role of TM4, EC2, and TM5 in the activation of MC2R should refocus on establishing the actual contact site between MRAP1 and MC2R when the MC2R/MRAP1 heterodimer forms at the ER [
3,
4].
As noted previously, the interaction between MRAP1 and MC2R involves both trafficking of the receptor, and activation of the receptor [
3,
4]. The trafficking function for MRAP1 requires that the TM of MRAP1 make contact with some TM domain in MC2R [
2]. The activation function associated with MRAP1 resides in an amino acid motif (activation motif) in the N-terminal of the MRAP1 homodimer [
3,
4]. Neither corresponding domains in hMC2R (trafficking or activation) had been identified prior to 2010. Initial attempts at resolving these issues have used a chimeric receptor paradigm [
30,
31], and collectively these studies point to a role for TM4 in trafficking of hMC2R. In addition, the results from both studies could be explained by assuming that formation of the heterodimer between MRAP1 and MC2R results in a conformation change in the receptor which places the R/KKRR binding site (presumably EC2) in the proper conformation for the activation process to begin.
In a more recent study, Malik et al. [
29] found definitive evidence that at the plasma membrane, the activation motif [
3,
4] on the N-terminal domain of MRAP1 that is facing the extracellular space makes contact with an extracellular domain on hMC2R to facilitate activation. Since MRAP1 is a homodimer with reverse topology [
2], when at the plasma membrane the homodimer would have a N-terminal domain of one monomer in the homodimer facing the extracellular space and one monomer with the N-terminal domain facing the cytosol. Since both N-terminal domains carry the activation motif, interaction with MC2R potentially could be through either an extracellular loop, or an intracellular loop. The later study eliminated the intracellular loops as the contact site, but did not identify the extracellular domain making contact with the activation motif of MRAP1. The implications of the Malik et al. [
29] study are very relevant to the current study. While the current study, and the previous alanine-substitution studies [
11,
13,
14] have focused on the role of EC2 in the activation process, it may be more expedient to first identify the extracellular domain in MC2R that makes contact with the activation motif on the N-terminal of MRAP1. That contact site should then point to the adjacent transmembrane domain(s) in MC2R making contact with the transmembrane domain of MRAP1 to facilitate trafficking. With that information in hand the role of EC2 in the activation process may be clarified.
4. Material and Methods
4.1. DNA Constructs
All cDNA constructs of xtMC2R were synthesized by GenScript (Piscataway, NJ, USA). Each construct was designed with an N-terminal V5 epitope tag, and each construct was inserted into a pcDNA3.1+ vector. The xtMC2R cDNA constructs included wild-type xtMC2R (accession number: XP002936118.1) and single alanine mutants of xtMC2R (
Figure 1: G154 to L175 excluding positions 156, and 165 where alanine residues were located. GenScript also synthesized the Gallus gallus (chicken; c) cMRAP1 (accession number: XR_001470382.1) cDNA construct, and this construct was also inserted into a pcDNA3.1+ vector. The cAMP reporter gene construct CRE-Luciferase [
33] was provided by Dr. Patricia Hinkle (University of Rochester, NY, USA).
4.2. Tissue Culture
For this project, the cDNAs were expressed in Chinese hamster ovary (CHO) cells (ATCC, Manassas, VA, USA) as described in Liang et al. [
16]. The CHO cells were grown to confluence in Kaighn’s Modification of Ham’s F12K media (ATCC). The incubation media was supplemented with 10% fetal bovine serum, 100 unit/mL penicillin, 100 μg/mL streptomycin, 100 μg/mL normocin, and maintained in a humidified incubator with 95% air and 5% CO
2 at 37 °C.
4.3. ACTH Analog Peptide
Human (h) ACTH(1-24) was purchased from New England Peptides (Gardiner, MA, USA). For the cAMP-reporter gene assay, hACTH(1-24) was used to stimulate transfected cells at concentrations ranging from 10–12 M to 10–6 M.
4.4. cAMP-Reporter Gene Assay
For the cAMP-reporter gene assay, 3.0 × 10
6 CHO cells were transiently transfected with either the xtMCR cDNA construct or one of the single alanine xtMC2R mutants, a cMRAP1 cDNA, and the CRE-Luciferase cDNA construct following the protocol described in Liang et al., [
16]. The transfections were performed using the Amaxa Cell Line Nucleofector II system (Lonza, Portsmouth, NH, USA), and utilized the Solution T transfection kit. Transfected cells were seeded in a white 96-well plate at a final density of 1 × 10
5 cells/well. Stimulation with concentrations of hACTH(1-24) in serum-free CHO media was done 2 days after transfection and plating, and the hormone stimulation period was 4 hours at 37 °C. The hormone stimulation was terminated by adding Bright GLO (Promega, Madison, WI, USA), the luciferase substrate reagent. After a 5-minute incubation period at room temperature, luminescence was immediately measured using a Bio-Tek Synergy HTX plate reader (Winooski, VT, USA). To determine the background levels of cAMP production, a set of transfected CHO cells were stimulated with serum free CHO media only (no ACTH) for the four-hour incubation period, and the average background luminescence reading for these control wells was subtracted from the ligand-stimulated luminescence readings. The dose response curves for the stimulated cells were analyzed using the Michaelis–Menton equation to obtain EC50 values. All assays were done in triplicate. The data were plotted using Kaleidograph software (
www.synergy.com).
4.5. Cell Surface ELISA
CHO cells were transfected with 0.1 μg/mL wildtype or single alanine mutant xtMC2R and 0.1 μg/mL cMRAP1 using jetPRIME transfection reagent (Polyplus transfection SA, Illkirch, France). Cells were grown for 48 h at 37 °C and then incubated on ice. Cells were surface-labeled with anti-V5 antibody (1:1000, Rockland Immunochemicals, Limerick, PA, USA) and then fixed in 4% paraformaldehyde, pH 7.2 (Sigma, St. Louis, MO, USA). Fixed cells were washed in 1x phosphate-buffered saline and incubated at room temperature with HRP-conjugated goat-anti-rabbit secondary antibody (Sigma, St. Louis, MO, USA). Cells were then washed and treated with 1-step ABTS (Thermo-Fisher Scientific, Watham, MA, USA). Absorbance was measured at 405 nm using a Bio-Tek Synergy HTX plate reader (Winooski, VT, USA). The analyses were done in triplicate. For each analysis the negative control was xtMC2R transfected alone, and the positive control xtMC2R co-transfected with cMRAP1.
4.6. Western Blot Protocol
CHO cells were separately transfected with either wild-type xtMC2R or xtMC2R mutant I158/A, F161/A (TM4), V172/A (TM5) or M166/A (EC2) and each can was co-expressed with cMRAP for 5 days in advance of cell lysis. Cells were homogenized in RIPA buffer and sonicated to collect total protein. Sample protein levels were then measured by nano-spectrophotometry and analyzed by poly-acrylamide gel electrophoresis. A 4% to 20% Mini-PROTEAN® TGX™ Precast Protein Acrylamide Gels (Biorad, Hercules, CA, USA), and then samples were transferred to a nitrocellulose membrane (Genesee Scientific; San Diego, CA, USA). Membrane was blotted with anti-V5 antibody (Rockland Immunochemicals, Limerick, PA, USA) at a final concentration of 1:500, and anti-alpha-tubulin from Developmental Studies Hybridoma Bank (University of Iowa) at a final concentration of 1:500. Membrane was scanned using a Protein Simple FluorChem system (San Jose, CA, USA) and analyzed using Fiji; an open-source platform for biological-image analysis. The Western analysis was done in triplicate.
4.7. Statistical Analysis
Data points are expressed as the mean + standard error of the mean (n = 3). For the cAMP reporter gene assay, statistical differences between the EC50 value of the xtMCR positive control and the xtMCR single alanine mutants were evaluated using one-way ANOVA followed by Tukey’s multi-comparison test using GraphPad Prism 2 (GraphPad Software Inc, La Jolla, CA, USA) for equal variance. Significance was set at p ≤ 0.05. For the Cell Surface ELISA assay, statistical differences were also evaluated using on-way ANOVA followed by Tukey’s multi-comparison test (GraphPAD). The western blot analysis was done in triplicate, and the intensity of the blots was analysis using a one-way ANOVA.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}