Reaction of 1-propanol with Ozone in Aqueous Media


Abstract
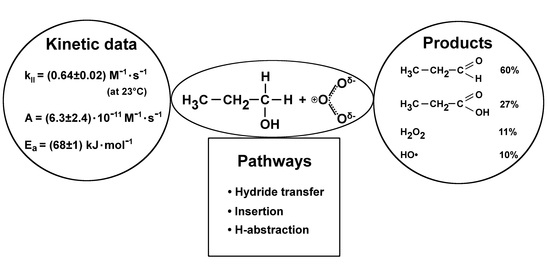
:
1. Introduction
2. Results
2.1. Products
2.1.1. Hydroxyl Radical
2.1.2. Aldehydes
2.1.3. Acids
2.1.4. Hydrogen Peroxide
2.1.5. Overview
2.2. Kinetic Data
3. Discussion
3.1. Mechanism Initiated by the Direct Reaction of O3 with 1-propanol
3.1.1. Hydride Transfer
3.1.2. Insertion
3.1.3. H-abstraction
- There is an acid-base equilibrium between HO2• and O2•− (reaction (18));
- At the circumneutral pH within our system, O2•− is the predominant species (pKa(HO2•) = (4.8 ± 0.1));
- Reaction between two HO2• moles or between one HO2• and one O2•− moles leads to the formation of H2O2 and HO2−, respectively (reactions (20) and (21));
- Given the large excess of substrate up against ozone within our system, reaction between O2•− and O3 is not important and it follows that O3•− (as a precursor of HO•) does not form practically via this pathway.
- Reform the α-hydroxyalkylperoxyl radical (reverse of reaction (29), ΔG = −94 kJ mol−1);
- Eliminate O2, thus forming two α-hydroxyalkyloxyl radicals (reaction (30), ΔG = −180 kJ mol−1) whose fate is described by the succession of reactions (12)–(17) at insertion mechanism;(C2H5)(H)(HO)C−O−O−O−O−C(OH)(H)(C2H5) → 2 (C2H5)(H)(HO)C−O• + O2
- Decay via Russel reaction that involves a transition state with a six-membered ring (reaction (31), ΔG = −583 kJ mol−1) [7,21]. Given that the diol formed in reaction (31) eliminates one water molecule intramolecularly according to reaction (32) (ΔG = −52 kJ mol−1), the products are propionaldehyde, propionic acid, and oxygen in a 1:1:1 ratio;
3.1.4. Electron Transfer
3.1.5. Overview
3.1.6. Comparison between 1-propanol/ozone and 2-propanol/ozone systems
- Carbonyl compounds with three carbon atoms formation yields with respect to ozone was very high for 2-propanol/ozone system (acetone formation yield was 87%) and decreased by 30% for the 1-propanol/ozone system (propionaldehyde formation yield was 60%). In the case of the 1-propanol/ozone system, propionic acid was also formed with a yield of 27%.
- Hydride transfer played a very important role for both systems, however the share of this mechanism was higher for the 2-propanol/ozone system (around 90% as opposed to maximum 60% for 1-propanol/ozone). The overall share of H-abstraction and insertion for 1-propanol/ozone system was higher or equal to 27% (propionic formation yield) and only a few percent for 2-propanol/ozone system.
- The second order rate constant of the reaction between 2-propanol and O3 at 23 °C, kII = (2.7 ± 0.1) M−1 s−1, was roughly four times higher than that between 1-propanol and O3, kII = (6.4 ± 0.2) × 10−1 M−1 s−1.
- Hydroxyl radicals formation yield was about four times higher for 1-propanol/ozone system, (9.8 ± 0.3)%, than that for 2-propanol/ozone, (2.4 ± 0.5)%. This information showed that the H-abstraction share was roughly four times higher in 1-propanol/ozone system than in 2-propanol/ozone (H-abstraction share was equal to HO• formation yield) and highlighted the increase of the importance of reactions between HO• and substrate for 1-propanol/ozone as against 2-propanol/ozone.
3.2. Mechanism Initiated by the Reaction of HO• with 1-propanol
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hoigné, J.; Bader, H. Rate constants of reactions of ozone with organic and inorganic compounds in water—I. Non-dissociating organic compounds. Water Res. 1983, 17, 173–183. [Google Scholar] [CrossRef]
- Willson, R.L.; Greenstock, C.L.; Adams, G.E.; Wageman, R.; Dorfman, L.M. The standardization of hydroxyl radical rate data from radiation chemistry. Int. J. Radiat. Phys. Chem. 1971, 3, 211–220. [Google Scholar] [CrossRef]
- Adams, G.E.; Boag, J.W.; Michael, B.D. Reactions of the hydroxyl radical. Part 2. Determination of absolute rate constants. Trans. Faraday Soc. 1965, 61, 1417–1424. [Google Scholar] [CrossRef]
- Neta, P.; Schuler, R.H. Rate constants for the reaction of O•− radicals with organic substrates in aqueous solution. J. Phys. Chem. 1975, 79, 1–6. [Google Scholar] [CrossRef]
- Reisz, E.; von Sonntag, C.; Tekle-Röttering, A.; Naumov, S.; Schmidt, W.; Schmidt, T.C. Reaction of 2-propanol with ozone in aqueous media. Water Res. 2018, 128, 171–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reisz, E.; Fischbacher, A.; Naumov, S.; von Sonntag, C.; Schmidt, T.C. Hydride transfer: A dominating reaction of ozone with tertiary butanol and formate ion in aqueous solution. Ozone Sci. Eng. 2014, 36, 532–539. [Google Scholar] [CrossRef]
- Von Sonntag, C.; von Gunten, U. Chemistry of Ozone in Water and Wastewater Treatment: From Basic Principles to Applications; International Water Association Publishing: London, UK, 2012. [Google Scholar]
- Plesnicar, B.; Cerkovnik, J.; Tekavec, T.; Koller, J. On the mechanism of the ozonation of isopropyl alcohol: An experimental and density functional theoretical investigation. 17O NMR spectra of hydrogen trioxide (HOOOH) and the hydrotrioxide of isopropyl alcohol. J. Am. Chem. Soc. 1998, 120, 8005–8006. [Google Scholar] [CrossRef]
- Plesnicar, B.; Cerkovnik, J.; Tekavec, T.; Koller, J. 17O NMR spectroscopic characterisation and the mechanism of formation of alkyl hydrotrioxides (ROOOH) and hydrogen trioxide (HOOOH) in the low-temperature ozonation of isopropyl alcohol and isopropyl methyl ether: Water-assisted decomposition. Chem. Eur. J. 2000, 6, 809–819. [Google Scholar] [CrossRef]
- Rakovsky, S.K.; Anachkov, M.; Zaikov, G.E.; Stoyanov, O.V.; Sofina, S.Y. Reactions of ozone with alcohols, ketons, ethers and hydroxybenzenes. Part 1. Vestn. Kazan. Tekhnol. Univ. 2013, 16, 11–16. [Google Scholar]
- Wolfenden, B.S.; Willson, R.L. Radical-cations as reference chromogens in kinetic studies of one-electron transfer reactions: Pulse radiolysis studies of 2,2′-azinobis-(3-ethylbenzthiazoline-6-sulphonate). J. Chem. Soc. Perkin Trans. 1982, 2, 805–812. [Google Scholar] [CrossRef]
- Buxton, G.V.; Greenstock, C.L.; Helman, W.P.; Ross, A.B. Critical review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (HO•/O•−) in aqueous solution. J. Phys. Chem. Ref. Data 1988, 17, 513–886. [Google Scholar] [CrossRef]
- Plesnicar, B. Progress in the chemistry of dihydrogen trioxide (HOOOH). Acta Chim. Slov. 2005, 52, 1–12. [Google Scholar]
- Czapski, G.; Bielski, B.H.J. The formation and decay of H2O3 and HO2 in electron-irradiated aqueous solutions. J. Phys. Chem. 1963, 67, 2180–2184. [Google Scholar] [CrossRef]
- Bielski, B.H.J.; Schwarz, H.A. The absorption spectra and kinetics of hydrogen sesquioxide and the perhydroxyl radical. J. Phys. Chem. 1968, 72, 3836–3841. [Google Scholar] [CrossRef]
- Bothe, E.; Behrens, G.; Schulte-Frohlinde, D. Mechanism of the first order decay of 2-hydroxy-propyl-2-peroxyl radicals and of O2•− formation in aqueous solution. Z. Naturforsch. 1977, 32, 886–889. [Google Scholar] [CrossRef]
- Bothe, E.; Schulte-Frohlinde, D. The bimolecular decay of the α-hydroxymethylperoxyl radicals in aqueous solution. Z. Naturforsch. 1978, 33, 786–788. [Google Scholar] [CrossRef]
- Bothe, E.; Schuchmann, M.N.; Schulte-Frohlinde, D.; von Sonntag, C. Hydroxyl radical-induced oxidation of ethanol in oxygenated aqueous solutions. A pulse radiolysis and product study. Z. Naturforsch. 1983, 38, 212–219. [Google Scholar] [CrossRef]
- Von Sonntag, C.; Schuchmann, H.-P. Peroxyl radicals in aqueous solutions. In Peroxyl Radicals; Alfassi, Z.B., Ed.; John Wiley & Sons Ltd.: Chichester, UK, 1997; pp. 173–234. [Google Scholar]
- Bielski, B.H.J.; Cabelli, D.E.; Arudi, R.L.; Ross, A.B. Reactivity of HO2/O2− radicals in aqueous solution. J. Phys. Chem. Ref. Data 1985, 14, 1041–1100. [Google Scholar] [CrossRef]
- Russel, G.A. Deuterium-isotope effects in the autoxidation of aralkyl hydrocarbons. Mechanism of the interaction of peroxyl radicals. J. Am. Chem. Soc. 1957, 79, 3871–3877. [Google Scholar] [CrossRef]
- Bennett, J.E.; Summers, R. Product studies of the mutual termination reactions of sec-alkylperoxy radicals: Evidence for non-cyclic termination. Can. J. Chem. 1974, 52, 1377–1379. [Google Scholar] [CrossRef]
- Naumov, S.; von Sonntag, C. The reaction HO• with O2, the decay of O3•− and the pKa of HO3•—Interrelated questions in aqueous free-radical chemistry. J. Phys. Org. Chem. 2011, 24, 600–602. [Google Scholar] [CrossRef]
- Sein, M.M.; Golloch, A.; Schmidt, T.C.; von Sonntag, C. No marked kinetic isotope effect in the peroxone (H2O2/D2O2 + O3) reaction: Mechanistic consequences. ChemPhysChem 2007, 8, 2065–2067. [Google Scholar] [CrossRef]
- Merényi, G.; Lind, J.; Naumov, S.; von Sonntag, C. Reaction of ozone with hydrogen peroxide (peroxone process): A revision of current mechanistic concepts based on thermokinetic and quantum-chemical considerations. Environ. Sci. Technol. 2010, 44, 3505–3507. [Google Scholar] [CrossRef]
- Merényi, G.; Lind, J.; Naumov, S.; von Sonntag, C. The reaction of ozone with the hydroxide ion: Mechanistic considerations based on thermokinetic and quantum chemical calculations and the role of HO4− in superoxide dismutation. Chem. Eur. J. 2010, 16, 1372–1377. [Google Scholar] [CrossRef]
- Elliot, A.J.; McCracken, D.R. Effect of temperature on O2•− reactions and equilibria: A pulse radiolysis study. Radiat. Phys. Chem. 1989, 33, 69–74. [Google Scholar]
- Thomas, J.K. Rates of reaction of the hydroxyl radical. Trans. Faraday Soc. 1965, 61, 702–707. [Google Scholar] [CrossRef]
- Asmus, K.-D.; Möckel, H.; Henglein, A. Pulse radiolytic study of the site of HO• radical attack on aliphatic alcohols in aqueous solution. J. Phys. Chem. 1973, 77, 1218–1221. [Google Scholar] [CrossRef]
- Lipari, F.; Swarin, S.J. Determination of formaldehyde and other aldehydes in automobile exhaust with an improved 2,4-dinitrophenyl-hydrazine method. J. Chrom. 1982, 247, 297–306. [Google Scholar] [CrossRef]
- Nash, T. The colorimetric estimation of formaldehyde by means of the Hantzsch reaction. Biochem. J. 1953, 55, 416–421. [Google Scholar] [CrossRef] [Green Version]
- Schuchmann, M.N.; von Sonntag, C. Hydroxyl radical-induced oxidation of 2-methyl-2-propanol in oxygenated aqueous solution. A product and pulse radiolysis study. J. Phys. Chem. 1979, 83, 780–784. [Google Scholar] [CrossRef]
- Flyunt, R.; Leitzke, A.; Mark, G.; Mvula, E.; Reisz, E.; Schick, R.; von Sonntag, C. Determination of HO•, O2•−, and hydroperoxide yields in ozone reactions in aqueous solution. J. Phys. Chem. B 2003, 107, 7242–7253. [Google Scholar] [CrossRef]
- Dowideit, P.; von Sonntag, C. Reaction of ozone with ethene and its methyl- and chlorine-substituted derivatives in aqueous solution. Environ. Sci. Technol. 1998, 32, 1112–1119. [Google Scholar] [CrossRef]
- Sänger, D.; von Sonntag, C. UV-photolysis (λ = 185 nm) of liquid t-butanol. Tetrahedron 1970, 26, 5489–5501. [Google Scholar] [CrossRef]
- Allen, A.O.; Hochanadel, C.J.; Ghormley, J.A.; Davis, T.W. Decomposition of water and aqueous solutions under mixed fast neutron and gamma radiation. J. Phys. Chem. 1952, 56, 575–586. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Jaguar. Jaguar 9.6; Schrodinger Inc.: New York, NY, USA, 2017. [Google Scholar]
- Tannor, D.J.; Marten, B.; Murphy, R.; Friesner, R.A.; Sitkoff, D.; Nicholls, A.; Ringnalda, M.; Goddard, W.A.; Honig, B. Accurate first principles calculation of molecular charge distributions and solvation energies from ab initio quantum mechanics and continuum dielectric theory. J. Am. Chem. Soc. 1994, 116, 11875–11882. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Oxidising Agent | K (M−1 s−1) | Working Conditions | References |
|---|---|---|---|
| O3 | (0.37 ± 0.04) | pH = 2 t = (20 ± 0.5) °C | [1] |
| (0.64 ± 0.02) | pH = 7 t = 23 °C | this work | |
| HO• | 2.8 × 109 | pH = 7 | [2] |
| 1.5 × 109 | pH = 10.7 | [3] | |
| 1.5 × 109 | pH = 7 | [3] | |
| 2.7 × 109 | [4] | ||
| O•− | 1.5 × 109 | pH = 14 | [4] |
| Product | Propion-Aldehyde | Propionic Acid | Acet-Aldehyde | Acetic Acid | Form-Aldehyde | Formic Acid | Hydroxyl Radical | Hydrogen Peroxyde |
|---|---|---|---|---|---|---|---|---|
| Yield (%) | 60 ± 3 | 27.4 ± 1.0 | 4.9 ± 0.3 | 0.3 ± 0.1 | 1.0 ± 0.1 | 4.6 ± 0.3 | 9.8 ± 0.3 | 11.1 ± 0.3 |
| C–H Bond Type | Alcohol | kII(S + O3) | |
|---|---|---|---|
| Value (M−1 s−1) | Reference | ||
| RH2C−H | (H3C)2(HO)C H2C−H (tert-Butanol) | 1.1 × 10−3 | [6] * |
| 3 × 10−3 | [1] ** | ||
| (HO)H2C−H (Methanol) | 2.4 × 10−2 | [1] ** | |
| R(HO)HC−H | (H3C)(HO)HC−H (Ethanol) | (3.7±0.4) × 10−1 | [1] ** |
| (H5C2)(HO)HC−H (1-Propanol) | (6.4 ± 0.2) × 10−1 | this work * [1] ** | |
| (3.7 ± 0.4) × 10−1 | |||
| (H7C3)(HO)HC−H (1-Butanol) | (5.8 ± 0.6) × 10−1 | [1] ** | |
| (H15C7)(HO)HC−H (1-Octanol) | ˂8 × 10−1 | [1] ** | |
| R2(HO)C−H | (H3C)2(HO)C−H (2-Propanol) | (2.7 ± 0.1) | [5] * [1] ** |
| (1.9 ± 0.2) | |||
| (H2C)4(HO)C−H (Cyclopentanol) | (2 ± 0.2) | [1] ** | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reisz, E.; Tekle-Röttering, A.; Naumov, S.; Schmidt, W.; Schmidt, T.C. Reaction of 1-propanol with Ozone in Aqueous Media. Int. J. Mol. Sci. 2019, 20, 4165. https://doi.org/10.3390/ijms20174165
Reisz E, Tekle-Röttering A, Naumov S, Schmidt W, Schmidt TC. Reaction of 1-propanol with Ozone in Aqueous Media. International Journal of Molecular Sciences. 2019; 20(17):4165. https://doi.org/10.3390/ijms20174165
Chicago/Turabian StyleReisz, Erika, Agnes Tekle-Röttering, Sergej Naumov, Winfried Schmidt, and Torsten C. Schmidt. 2019. "Reaction of 1-propanol with Ozone in Aqueous Media" International Journal of Molecular Sciences 20, no. 17: 4165. https://doi.org/10.3390/ijms20174165
APA StyleReisz, E., Tekle-Röttering, A., Naumov, S., Schmidt, W., & Schmidt, T. C. (2019). Reaction of 1-propanol with Ozone in Aqueous Media. International Journal of Molecular Sciences, 20(17), 4165. https://doi.org/10.3390/ijms20174165